
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antibody fragments as nanoparticle targeting ligands: a step in the right direction
Daniel A.
Richards
*,
Antoine
Maruani
 and
Vijay
Chudasama
*
and
Vijay
Chudasama
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: daniel.richards.11@ucl.ac.uk; v.chudasama@ucl.ac.uk; Tel: +44 (0)207 679 2077
First published on 16th September 2016
Abstract
Recent advances in nanomedicine have shown that dramatic improvements in nanoparticle therapeutics and diagnostics can be achieved through the use of disease specific targeting ligands. Although immunoglobulins have successfully been employed for the generation of actively targeted nanoparticles, their use is often hampered by the suboptimal characteristics of the resulting complexes. Emerging data suggest that a switch in focus from full antibodies to antibody derived fragments could help to alleviate these problems and expand the potential of antibody–nanoparticle conjugates as biomedical tools. This review aims to highlight how antibody derived fragments have been utilised to overcome both fundamental and practical issues encountered during the design and application of antibody–targeted nanoparticles.
 Daniel A. Richards | Dr Daniel Richards began his studies at the University Of York, obtaining an MChem in 2011 with his thesis focused on the study of metal–halogen exchange reactions in nitrogen containing heterocycles. He subsequently joined the lab of Dr James Baker at University College London (UCL) to study for his PhD, focusing on the development and application of novel biocompatible photochemical reactions. He is currently working as a postdoctoral fellow in the group of Dr Vijay Chudasama, developing novel methods for the selective functionalisation of nanoparticles. |
 Antoine Maruani | Dr Antoine Maruani obtained his Master's degree in Chemistry from École Normale Supérieure of Lyon (France) in 2011. He then joined Prof. Stephen Caddick's group at University College London (UK) where he obtained his PhD in 2015 with his thesis focusing on the site-selective dual modification of proteins. He is currently working as a postdoctoral fellow under the supervision of Dr Vijay Chudasama on the development of novel methodologies for bioconjugation. |
 Vijay Chudasama | Dr Vijay Chudasama obtained his MSci degree and PhD from University College London in 2008 and 2011, respectively. Following post-doctoral studies under the supervision of Prof. Stephen Caddick, Vijay obtained a Ramsay Memorial Fellowship. During this time, he was made Technical Director of a biotechnology spin-out (ThioLogics). In April 2015, he was awarded a lectureship at UCL for him to focus on the research areas of aerobic C–H activation and various aspects of Chemical Biology. Vijay's research has recently been highlighted by Forbes, Scientific American, CNN, Nature Chemistry and the Royal Society of Chemistry. |
1. Introduction
The last twenty years have seen a rapid, and accelerating, increase in the use of nanoparticles for biomedical applications. From a conceptual standpoint it is not difficult to understand why; various nanoparticles are now at a stage of being tuneable, functionalisable and biocompatible vehicles that can safely transport large quantities of cargo through the body. This enables the delivery of entities at concentrations significantly higher than traditional methods.1 This factor, in combination with the ease in which the surface of nanoparticles can be decorated with high affinity disease-specific targeting ligands to enhance selective delivery, means that they have a plethora of downstream therapeutic and diagnostic applications. A large variety of chemical and biological molecules have been explored for this enhanced targeting purpose, including: novel small molecules, sugars, fatty acids, proteins, peptides, antibodies, and aptamers.1–7 Of these, antibody based targeting ligands have become incredibly popular due to their unique in vivo properties and high target specificities.8–11 Whilst the contributions of other targeting ligands should not be ignored, this review focuses on the use of antibodies, or more specifically their associated fragments, as targeting ligands for nanoparticle-based therapeutic and diagnostic tools. To ensure broad accessibility of the review content, a brief overview of common nanoparticle (Section 2.1) and antibody (Section 2.2) scaffolds used in this context will be given.2. Antibody–decorated nanoparticles
2.1 Nanoparticle structure
When designing nanoparticle–antibody conjugates for biomedical applications several considerations regarding the structure of the nanoparticle are important. The nanoparticle must be biologically inert, stable under physiological conditions, move freely through the body, securely encapsulate chemical entities (where applicable), and contain a surface which is easily conjugated to the desired targeting antibody. In the case of therapeutics, it is also important to consider the mechanism by which the nanoparticle vehicle will release cargo and whether this will be compatible with other aspects of the overall construct. The most successful approaches strike a delicate balance between the properties of the nanoparticle, the targeting antibody, and where appropriate the encapsulated cargo. Fortunately, a great deal of research has been done on the design and modification of nanoparticles over the last 20 years, providing a rich pool of work from which suitable vehicles can be selected for antibody conjugation. Nanocarriers can be broadly categorised as organic or inorganic,† and each of these will be discussed in turn (Fig. 1, Table 1).4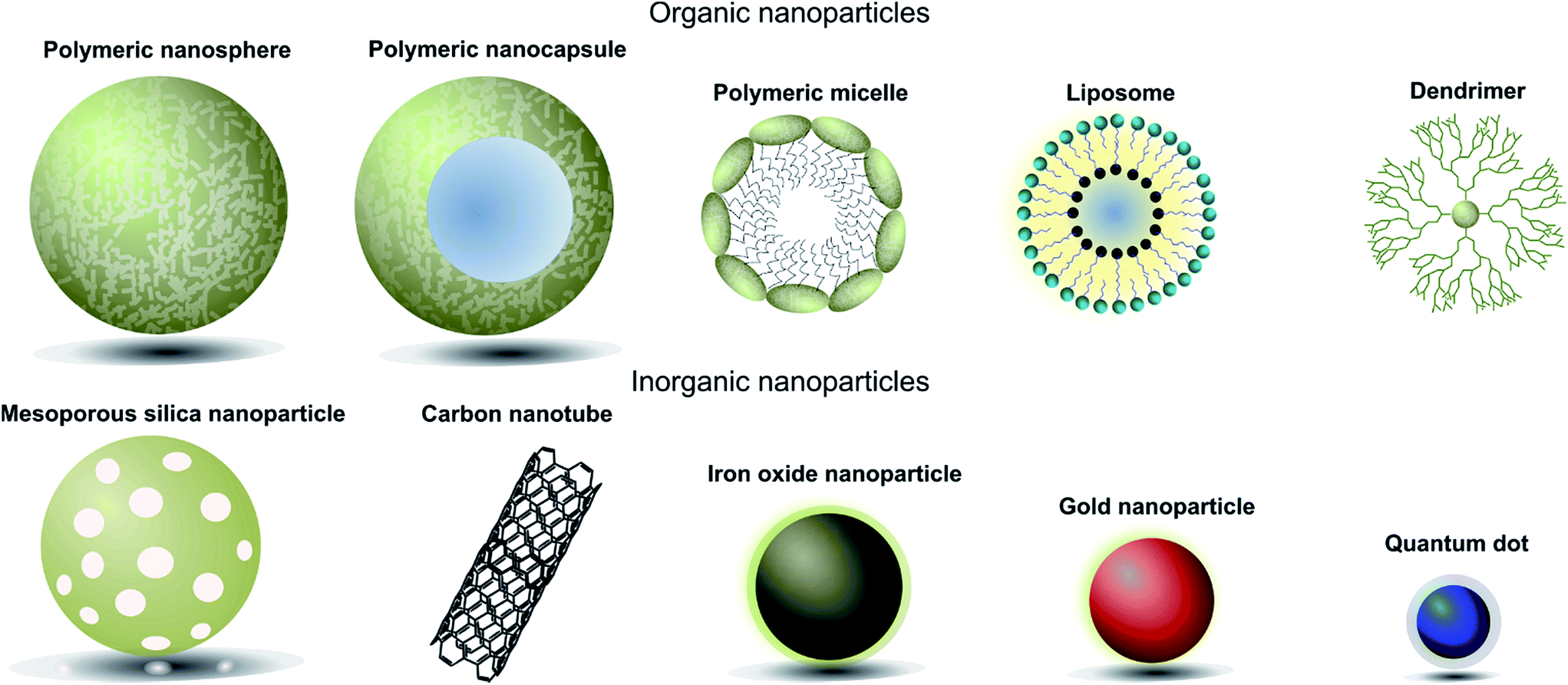 | ||
| Fig. 1 Pictorial representation of different types of nanoparticles used in biomedical applications. | ||
| Nanoparticle | Material(s) | Cargo attachment | Advantages | Disadvantages |
|---|---|---|---|---|
| Liposomes | Self-assembling lipid bilayer | Encapsulated within the hydrophilic core | Easily synthesised, biocompatible, high internal loading | Highly sensitive to structural changes and nature of payload |
| Polymeric micelles | Hydrophobic polymer core surrounded by hydrophilic polymeric chains | Encapsulated within the hydrophobic core | Small, biocompatible, able to incorporate highly hydrophobic cargo | Highly sensitive to structural changes, poor release profiles |
| Polymeric nanospheres/nanocapsules | Solid hydrophobic polymer matrix with optional aqueous core (nanocapsule) | Embedded in the polymer matrix or within the core | High loading capacity, flexible loading capabilities, reliable release profiles | Difficult to purify and poor store properties |
| Dendrimers | Highly branched polymer matrix | Embedded in the polymer branches | Highly soluble, non-immunogenic, high loading capacity, controlled synthesis | Lacking data on toxicity and biocompatibility |
| Iron oxide nanoparticles | Iron oxide core surrounded by biocompatible coating | Attached to the surface/surface coating | Innate magnetic properties | No internal loading capacity |
| Gold nanoparticles | Solid gold particles typically coated with PEG chains | Attached to the surface/surface coating | Innate optical and photothermal properties | No internal loading capacity, poor biocompatibility and biodegradability |
| Mesoporous silica nanoparticles | Mesopores surrounded by a silica framework | Encapsulated within the mesopores | High loading capacity, good biodegradability | Issues with physiological stability, rapid clearance rates |
| Carbon nanoparticles | Graphite arranged in either a sheet or cylindrical conformation | Attached to the carbon backbone | Innate optical and electrical properties, high surface loading capacities | Poor biodegradability, organ accumulation |
| Quantum dots | Typically a cadmium selenide core with a zinc selenide cap | Attached to the surface/surface coating | Innate optical properties, high extinction coefficients | No internal loading capacity, potential toxicity issues |
Liposomes. Liposomal nanoparticles were first developed near the genesis of nanomedicine and have since become one of the most widely utilised vehicles for encapsulating chemical payloads, with several formulations having gained FDA approval.12 They comprise natural lipids with polar and non-polar components which self-assemble into colloidal particles. Whilst early liposomal nanoparticles suffered from issues of stability and rapid clearance, the introduction of surface ligands such as polyethylene glycol (PEG) chains has helped to address these drawbacks.12,13 The main advantages of liposomal nanoparticles created from state-of-the-art technologies lie in their excellent biocompatibility, ease of synthesis/functionalisation, and their ability to safely encapsulate a variety of small molecules.4,6,14 However, they are limited by a high level of sensitivity to structural change(s) and have demonstrated highly specific cargo-dependency, thus decreasing their universal appeal and broad applicability.6,14
Polymeric micelles. Polymeric micelles consist of a core of aggregated hydrophobic polymers surrounded by hydrophilic polymeric chains. Their small size and hydrophilic nature allow them to avoid uptake by the reticuloendothelial system, significantly increasing their circulation time.15 Their hydrophilic exterior also allows polymeric micelles to effectively and safely encapsulate very hydrophobic drugs for safe transport through the body.16 As with liposomal nanoparticles, polymeric micelles also demonstrate excellent biocompatibility.17 However, poorly controlled release profiles of encapsulated cargo, and a high sensitivity to structural change(s), mean that there is still significant scope for improvement.4
Polymeric nanoparticles. Polymeric nanoparticles can be further categorised as either nanospheres or nanocapsules. Nanospheres consist of a solid polymer matrix which is able to encapsulate hydrophobic drugs, whilst nanocapsules contain an aqueous hydrophilic core that is more amenable to the loading of hydrophilic payloads such as DNA/RNA.10 This payload flexibility increases the versatility of polymeric nanoparticles, making them attractive candidates as nanocarriers. Additionally, it has been shown that the release rates of encapsulated payloads are constant and proceed on clinically relevant time scales.6 Nonetheless, despite these favourable characteristics, polymeric nanoparticles are not simple to purify and do not store well, making them a poor choice for applications that require large scale production.18
Dendrimers. Dendrimers are branched polymer complexes generated through highly controlled successive polymerisation steps. This leads to a nanoparticle which consists of an initiator core contained within branched polymer chains. These polymer chains are generally synthetic, although examples that employ natural polymers such as sugars and amino acids have been reported.19 Their highly regulated synthesis enables excellent control over shape and size – important parameters for medical applications.20 They also display excellent solubility and have been shown to be non-immunogenic.21 Whilst dendrimers have several excellent qualities, research into their use in the biomedical field is still early stage. Further studies to establish their biocompatibility and toxicity are ongoing and will be pivotal to their further application.
Iron oxide nanoparticles. Iron oxide nanoparticles generally consist of an iron oxide (typically Fe3O4) core surrounded by a dextran coating to improve the physical properties of the complex. The application of these nanoparticles commonly centres on their innate magnetic properties, which allow them to act as excellent MRI contrast agents and tools for therapeutic magnetic hypothermia.22,23 This dual functionality has led to superparamagnetic iron oxide nanoparticles (SPIONS) being used as theranostic tools, i.e. chemical entities which display both therapeutic and diagnostic properties. However, the lack of a spacious “core” or any porous space leads to low loading volumes,24 an issue for many applications. Whilst the generation of hybrid iron oxide/polymer-based nanoparticles has gone some way towards addressing these issues, the current situation is not ideal.23
Gold nanoparticles. Gold nanoparticles have been extensively studied for use in biomedical applications due to their interesting size dependent physicochemical and optical properties. For example, their ability to produce heat upon absorbance of near-infrared light has been explored for use in photothermal therapy, whilst the ability to enhance optical processes such as absorbance and fluorescence has led to widespread use in the field of biosensors and imaging agents.25,26 However, their non-hollow structure precludes internal loading,4 and they also tend to suffer from poor biodegradation and questionable biocompatibility.27–29
Mesoporous silica nanoparticles. Mesoporous silica nanoparticles (MSNs) consist of mesopores (2–50 nm pores) surrounded by a silica framework. These nanoparticles have a high surface area to volume ratio which affords them a large loading capacity. MSNs have also demonstrated good biocompatibility and biodegradability, desirable features for biomedical purposes.30,31 However, stability issues and rapid clearance rates significantly restrict the use of MSNs from certain applications.32–34
Carbon nanoparticles. Carbon nanoparticles, such as carbon nanotubes, comprise a single layer of graphite in either a sheet or cylindrical conformation. Excellent loading capacities, unique optical and electrical properties, and low synthetic costs make them promising candidates for several applications, especially imaging and diagnostics.35,36 Unfortunately, issues of poor biodegradability,37 pulmonary damage,29,38 and undesirable organ accumulation29,39,40 have hindered the adoption of carbon based nanoparticles for in vivo applications.
Quantum dots. Quantum dots (QDs) most commonly consist of a cadmium selenide core with a zinc selenide cap, although many other combinations exist. QDs emit bright colours and also display size dependent optical properties, making them ideal for imaging or biosensing technologies.41 Whilst potential toxicity issues have to-date limited their utility in vivo, recent advances are helping to overcome these remaining hurdles.41–43
2.2 Antibody structure and function
Antibodies, or immunoglobulins (Ig), are large glycoproteins found in all vertebrate life forms. These essential proteins are involved in several key processes within the immune system including complement dependent cytotoxicity (CDC), opsonisation, phagocytosis, and antibody-dependent cytotoxicity (ADCC). To date, five major classes of immunoglobulin have been discovered, IgA, IgD, IgE, IgG and IgM, each characterised by unique structural characteristics. IgGs represent the dominant class of human immunoglobulins and can be further divided into four sub-types; IgG1, IgG2, IgG3 and IgG4. Although the IgG sub-types show significant sequence variation in key regions, they share a common overall structure. IgG antibodies consist of four protein chains; two identical ca. 25 kDa light chains (i.e. L-subscript) and two identical ca. 50 kDa heavy (i.e. H-subscript) chains. These chains contain multiple domains which are characterised by their degree of sequence variability. The N-termini of the chains converge in the variable domain (V) to form the antigen-binding region. Further from the terminus, the structure becomes more conserved, leading to the area being designated the constant region (C). The heavy and light chains are held together by several interchain disulfide bonds and considerable non-covalent interactions to form a Y-shaped structure. The overall structure can be broadly divided into two distinct segments; the fragment antigen-binding (Fab) region and the fragment crystallisable (Fc) section. Fabs can be further divided into variable (Fv, VH/L) and constant (CH/L) regions (Fig. 2).44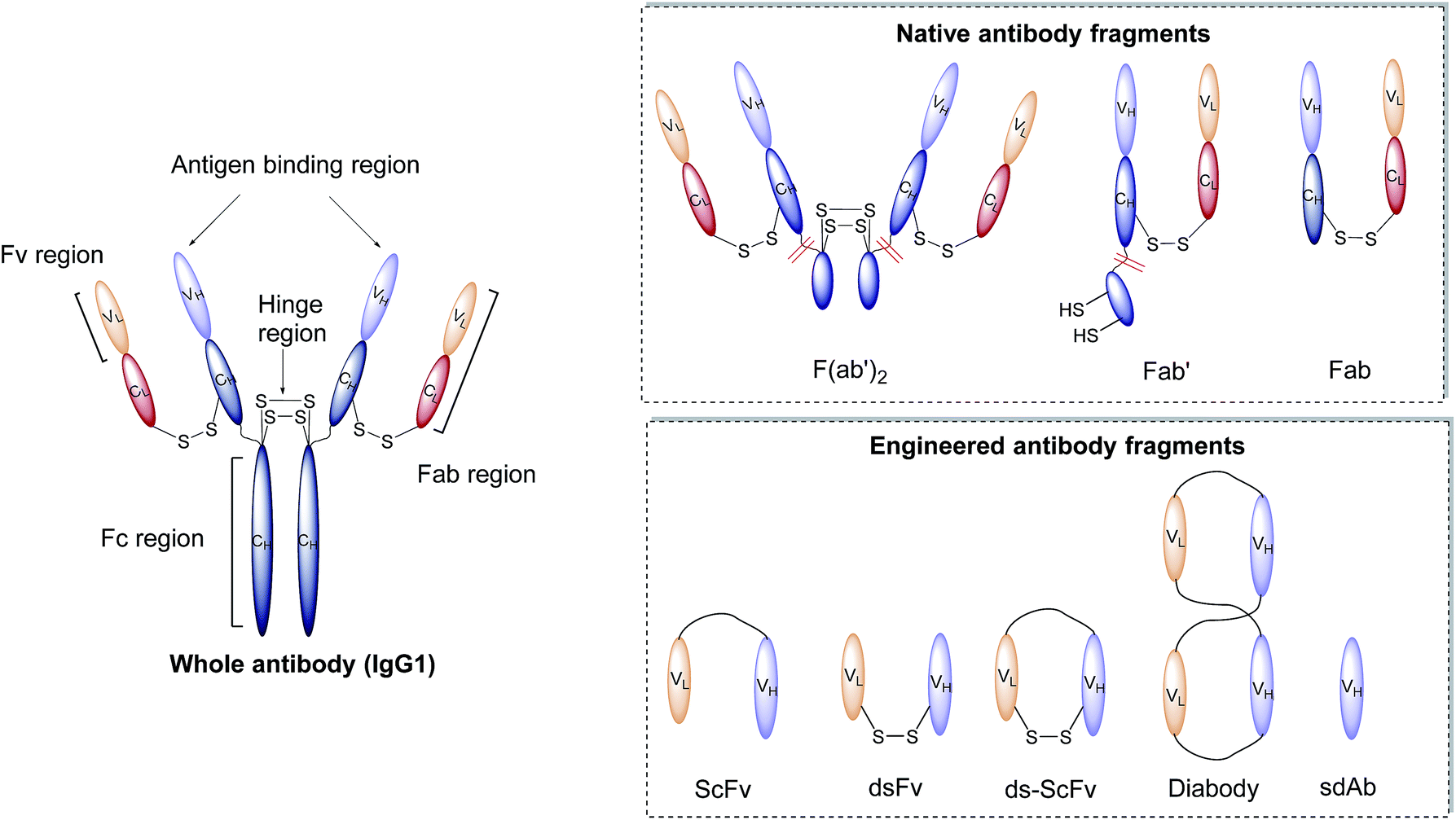 | ||
| Fig. 2 Graphic representations of whole antibody (IgG1) and various fragments. | ||
2.3 Nanoparticle–antibody fragment conjugates
Antibodies function by targeting specific antigens that are expressed only on the surface of diseased cells, or heavily overexpressed on these cells relative to healthy cells. As these antigens are present solely, or majorly, on the surface of the target diseased cells, antibodies can conceptually be exploited to courier nanoparticles (and also their cargo) through the body and enable selective delivery/targeting. Whilst this approach was first conceptualised in the early 1980's, practical and theoretical limitations at the time (e.g. insufficient methods for generating and evaluating antibody–decorated nanoparticles) prevented significant progress in the area. Advancements in both antibody expression techniques and nanoparticle design over the past few decades have enabled a more thorough exploration of nanoparticle–antibody conjugates, which has resulted in a rapid expansion of the field. Early developments focused almost entirely on using full antibodies as targeting ligands, primarily due to the wealth of available information on both their generation and modification. However several issues associated with the use of full antibody ligands, such as immunogenicity,9 rapid elimination,58 poor stability,59,60 and lower than expected efficacy,1,6,8,61 soon came to light and these are being increasingly emphasised/supported by emerging data. A significant amount research has now been published on the use of antibody fragments to address both fundamental and practical issues encountered during the use of whole immunoglobulins. In addition to being less immunogenic, the small size of antibody fragments allows for higher loading capacities and superior orientation of targeting ligands, leading to overall improvements in efficacy (Fig. 3).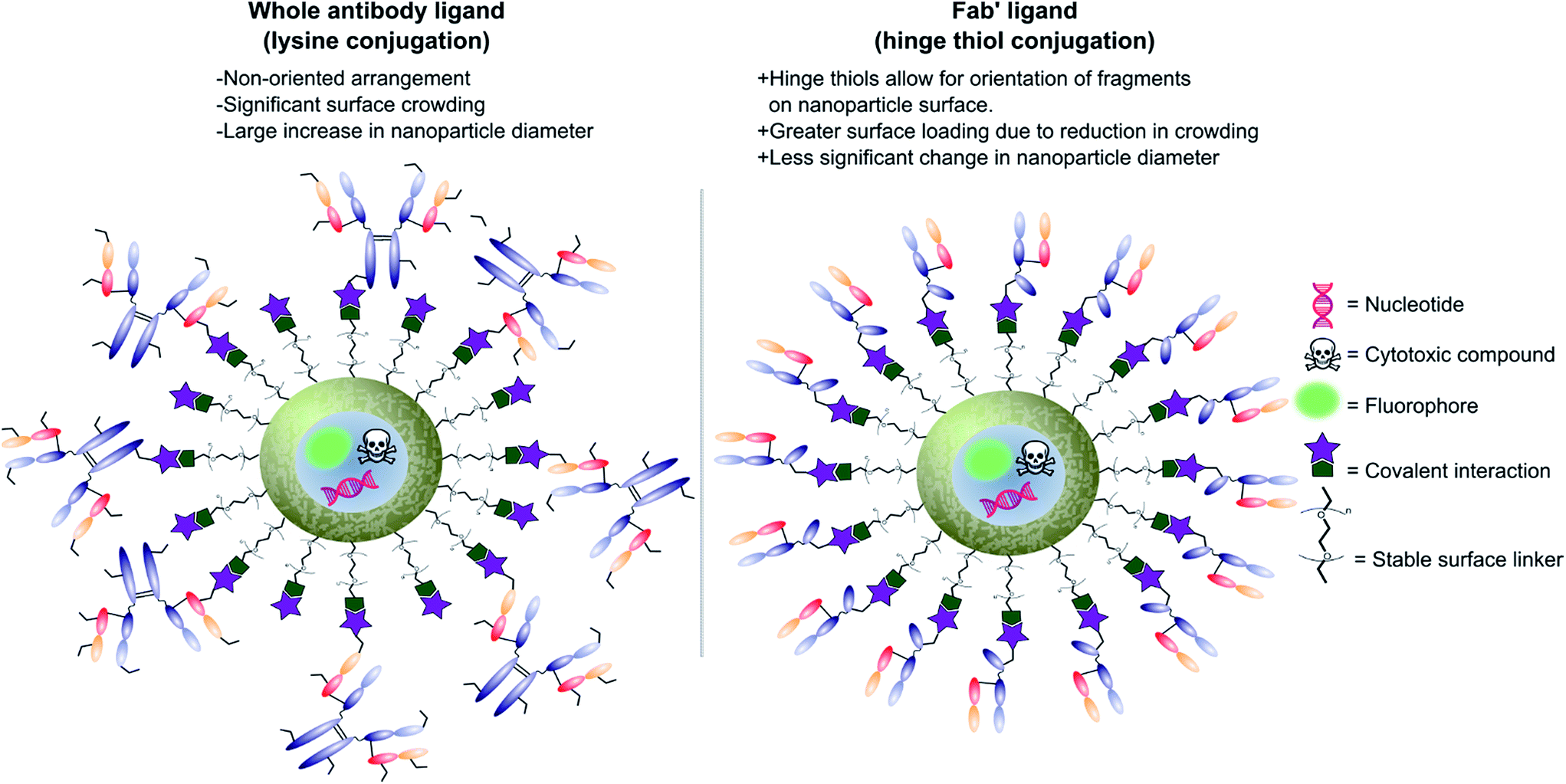 | ||
| Fig. 3 Graphic representations comparing whole antibody and antibody fragment (Fab′) targeting ligands for nanocarriers. | ||
In view of the above advantages, it is anticipated that the use of antibody fragments as directing ligands for nanoparticle targeting will increase significantly over the next few years.9 Whilst several excellent reviews have been written on the use of targeted nanoparticles in biomedicine, with a few focusing on the subject of antibodies as targeting ligands,8,9 very few specifically highlight and accurately detail work on nanoparticle–antibody fragment conjugates. This short review aims to introduce the area, with particular emphasis on recent developments in the generation and application of nanoparticle–antibody fragment conjugates for biomedical uses.
3. Generating nanoparticle–antibody fragment complexes
During the design of nanoparticle–antibody fragment complexes important consideration must be given to the method by which the two entities are attached. The antibody fragment needs to be conjugated to the nanoparticle in a way that causes minimal perturbation to the shape, size, and functionality of both the nanoparticle and the antibody fragment itself. Additionally, the linker between the two should be stable, biocompatible, non-toxic, and facile to install. Fortunately, a great deal of work on the installation of functional chemical moieties on both nanoparticles and antibody fragments has been carried out. Moreover, attempts to utilise these chemistries to functionalise nanoparticles with antibody fragments have been largely successful, as will be discussed in more detail below.3.1 Modification of antibody fragments
Modifications of antibody fragments largely centre on exploiting the innate chemical reactivity of the natural amino acids on the backbone of each protein. The amino acids most commonly used as sites for modification include lysine, cysteine, and glutamic/aspartic acid, as they can be functionalised using well-established chemistries. Initially, lysine was a popular target for modification as it could be readily conjugated, however, the high abundance of this amino acid on the surface of many proteins means that it is hard to control conjugation, resulting in random functionalisation and a heterogeneous mixture of antibody fragment products post-conjugation. More recently, site-selective methods which exploit the natural structure of antibody fragments, such as the hinge thiols of Fab′ fragments, or utilise amino acids incorporated through site-directed mutagenesis, have been successfully employed; this has resulted in far more homogeneous and better characterised conjugates. Antibody modification (including antibody fragments) has maintained a healthy research focus for several decades now, largely due to the rapid development of the antibody–drug conjugate field. This has resulted in a rich toolbox of chemical reactions which enable facile, site-selective modification whilst avoiding negative effects on the function of the protein. Several excellent reviews have been written on this subject, so it will not be covered in depth here.62–65 However, Fig. 4 highlights some of the most common methods employed for functionalising antibody fragments for subsequent attachment to nanoparticles.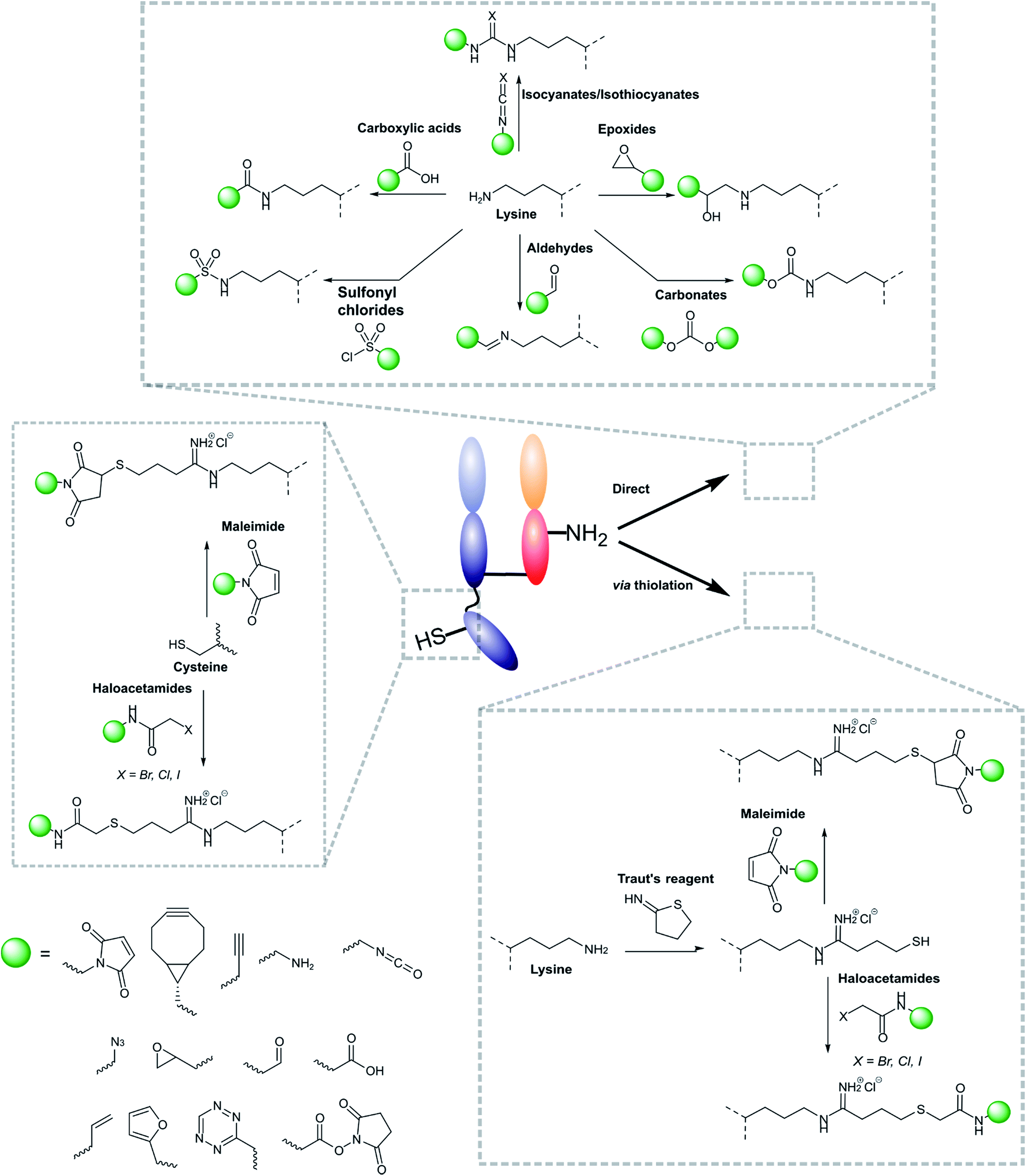 | ||
| Fig. 4 Schematic representations of common ways in which antibody fragments are modified. | ||
3.2 Modification of nanoparticle surfaces
Nanoparticle surface modification techniques can be broadly separated in two main categories: (i) covalent and (ii) non-covalent. Covalent modifications involve the incorporation of a chemical functional group that can subsequently attach covalently to a targeting ligand. In contrast, non-covalent technologies involve the incorporation of a functionality that can interact either (i) intermolecularly or (ii) by physisorption with a ligand. For decorating nanoparticles with antibodies, covalent methods are preferred as they provide greater in vivo stability.8 Moreover, covalent methods also allow for greater control over the position and orientation of the attached antibody fragment, especially when combined with a site-selectively modified antibody fragment itself. Methods for incorporating an assortment of functional groups onto the surfaces of various nanoparticles have been reported, including amines, carboxylic acids, alcohols, thiols, azides, alkynes, aldehydes, and maleimides. Subsequent modification of these groups can further expand the reactivity profile of the nanoparticle, leading to a large selection of functional handles which can be paired with complimentary groups on the desired antibody ligand (Fig. 5). Several reviews have been written on the incorporation and utilisation of chemical functionality on nanoparticles,4,8,18,66,67 including a comprehensive overview by Sapsford et al.68 Despite these advances, non-specific interactions of antibody ligands with nanoparticle surfaces remains an issue, and methods for distinguishing specific interactions from non-specific interactions are lacking. These issues can be particularly problematic when site-specific or oriented conjugation of an antibody fragment is desired.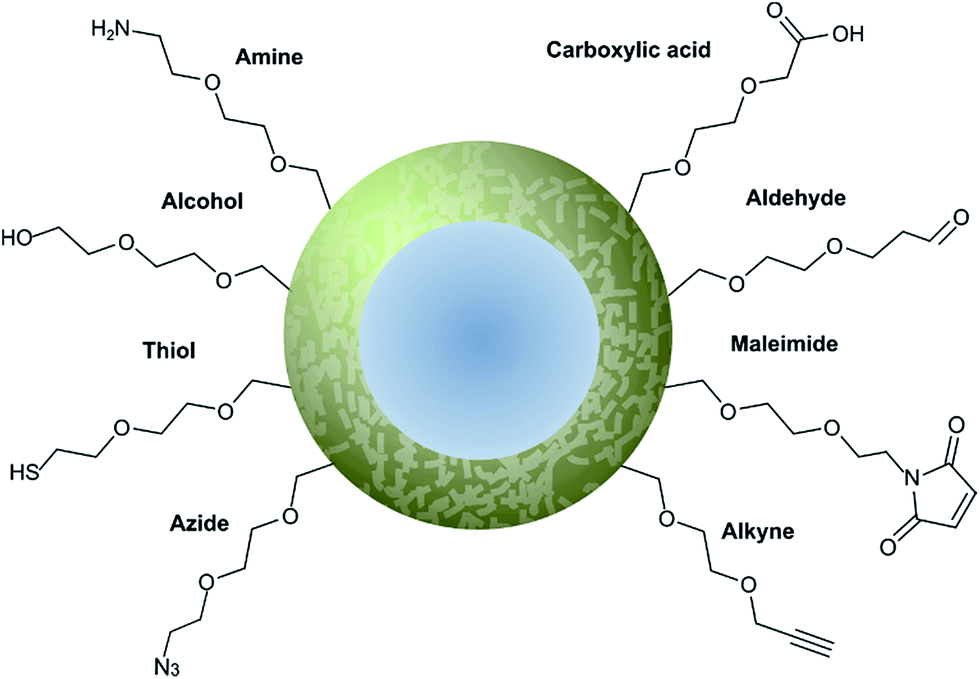 | ||
| Fig. 5 Graphical representation of common functional ligands attached to the surface of a nanoparticle. | ||
4. Nanoparticle–antibody fragment conjugates in biomedicine
4.1 As therapeutic agents
The ability to safely encapsulate a cocktail of toxic chemicals and deliver them selectively remains a long standing goal for medicine. To this end nanoparticle–antibody conjugates have shown great potential and indeed several promising candidates have entered clinical trials (Table 2). Interestingly, the majority of these candidates utilise antibody fragments as the targeting ligand, highlighting a preference over full-length antibodies for therapeutic applications. This preference is indicative of the advantages provided by the use of smaller, less immunogenic antibody-derived targeting ligands. However, it is important to note that in the cases exemplified in Table 2, side-by-side comparisons to whole immunoglobulins were not made, or at least the data was not published.| Name | NP type | Target | Ligand | Bioactive compound | Indication | Phase |
|---|---|---|---|---|---|---|
| SGT-53 | Lipid | Transferrin receptor | Anti-transferrin receptor ScFv | p53 DNA | Solid tumours | Ib |
| SGT-94 | Lipid | Transferrin receptor | Anti-transferrin receptor ScFv | RB94 DNA | Solid tumours | I |
| C225-ILS-Dox | Lipid | EGFR | Cetuximab Fab | Doxorubicin | Solid tumours | I |
| Erbitux-EDVspac | Bacterially derived mini-cell | EGFR | Bispecific monoclonal antibody (mAb) | Paclitaxel | Solid tumours | II |
| MM-302 | Lipid | HER2 | Anti-HER ScFv | Doxorubicin | Breast cancer | I |
| Lipovaxin-MM | Lipid | Dendritic cell CD209 | dAb | Melanoma antigens + IFNγ | Melanoma vaccine | I |
| MCC-465 | Lipid | Uncharacterised (GAH) | Anti-GAH F(ab′)2 | Doxorubicin | Metastatic stomach cancer | I |
| Anti-EGFR ILs-Dox | Lipid | EGFR | Cetuximab Fab | Doxorubicin | Solid tumours | I |
Nonetheless, a lack of clarity regarding the advantages and disadvantages of whole mAb compared with antibody fragments for therapeutic purposes was, at least to some extent, addressed by Cheng and Allen.69 During the design of liposomes which could selectively target B-cell malignancies with encapsulated doxorubicin (Stealth® immunoliposomes, SIL), they compared the in vivo effectiveness of doxorubicin bearing liposomes targeted with HD-37 mAb, HD-37-Fab′ and a HD-37-ScFv against the B-cell antigen CD19.69 The targeting ligands were attached to the protein using maleimide–thiol conjugation techniques, natively in the case of the Fab′ and ScFv and via lysine thiolation in the case of the whole antibody. In vitro binding assays revealed no significant difference in CD19 binding between HD-37-mAb and HD-37-ScFv targeted liposomes, however, a steep improvement in binding was observed for HD-37-Fab′. Interestingly the HD-37-ScFv targeted liposome proved the most selective for CD19+ over CD19− cells with the mAb being the worst performer over both studies. Drastic differences were also noticed in vivo, with the HD-37-mAb targeted liposome being rapidly cleared (0.41 mL h−1) due to Fc-mediated uptake into the liver and spleen in comparison to the fragment conjugates (0.10 mL h−1 for the Fab and 0.12 mL h−1 for the ScFv). Of the fragment-decorated liposomes HD-37-ScFv cleared slightly quicker, possibly due to His-tag/c-myc tag mediated uptake into the liver. The culmination of these effects is an improved mean survival rate of mice treated with HD-37-Fab′ targeted doxorubicin liposomes when compared to HD-37-mAb and HD-37-ScFv targeted doxorubicin liposomes. Although the presence of the His and c-myc tags caveat the results of the HD-37 ScFv targeted liposome due to increased clearance rates, this work clearly demonstrated the differences between using full mAb and antibody fragments as targeting ligands for nanoparticles. It also provided early evidence for advantages in using smaller fragments that do not contain the Fc region. These results corroborated previous work by Allen which showed that a Fab′ conjugated liposome outperformed a full mAb conjugated liposome due to increased circulation time.70
Whilst the majority of nanoparticle–antibody fragment drug delivery systems utilise lipid-based nanoparticles (Table 2), the last few years have seen an increased exploration of non-liposomal nanoparticle–antibody conjugates for cytotoxic drug delivery. Work by Ahn et al. showed that anti-tissue factor (TF) Fab′ targeted polymeric micelles loaded with dichloro(1,2-diaminocyclohexane)platinum(II) displayed greater selectivity for the cellular target, increased internalisation rate, and afforded significant retardation of tumour growth when compared to non-targeted polymeric micelles or free drug alone.74 By utilising a selective maleimide–thiol reaction to attach their Fab′ ligand, the group were able to exert delicate control over the conjugation and introduce a single Fab′ per micelle. This allowed for the installation of targeting capabilities whilst causing minimal perturbation to the nanoparticle properties, an advantage for moving forward into the clinic.
Further to this, Xiangbao et al. successfully used an anti-VEGFR ScFv targeted polyethylene glycol–polylactic acid (PEG–PLA) polymersome containing As2O3 as the cytotoxic payload.75 Despite the use of suboptimal non-specific lysine–NHS ester conjugation techniques to attach the ScFv ligand, their approach yielded improved selectivity and decreased tumour volume, resulting in far greater mean survival rates when compared to the non-targeted nanoparticles and free drug controls. It is expected that controlled orientation of the ScFvs would yield even better results.
Proof of principle research by Quarta et al. has demonstrated the tumour targeting capability of iron oxide nanoparticles conjugated to anti-folate receptor antibody (AFRA) Fab fragments.76 The group chose the Fab fragment over the full antibody in order to minimise any increase in the diameter of the resulting conjugate and thus increase internalisation rate and stability. The ARFA Fab had been previously expressed to contain a hinge region with a single glutathione protected cysteine residue that could be used to conjugate to the maleimide coated nanoparticle after reductive deprotection. Interestingly, the group employed TCEP for the deprotection, a reducing agent known to cleave the interchain heavy-light disulfide bond of the Fab fragment. This would enable cysteine residues on both chains to react independently with the nanoparticle, potentially decreasing the control offered through the specific introduction of the hinge cysteine, although this it is appreciated that all liberated thiols are distal from the binding site. Whilst no cytotoxic compounds were delivered in this preliminary study, the group did demonstrate excellent in vivo stability, along with dramatically increased selectivity for αFR-expressing tumours when compared to non-targeted controls. Thus, whilst this approach is still in its relative infancy, it shows promise as a way of utilising inorganic iron oxide nanoparticles to deliver cytotoxic payloads for the treatment of ovarian cancer.
Other early stage research has explored the use of bispecific ScFv and SdAb targeted liposomes, and have demonstrated a clear advantage in the use of both bispecific ScFv and SdAb fragments as targeting ligands for liposomal nanoparticles.77,78
Recently, Katakowski et al. showed that liposomes containing small interfering RNA (siRNA) could be targeted at dendritic cells using anti-DEC205 ScFv fragments, with in vivo results demonstrating improved gene silencing.82 Their targeting ScFv was conjugated to the nanoparticle via a C-terminal cysteine introduced using site-directed mutagenesis, allowing conjugation to occur distal to the binding region so as to minimise any deleterious effects on binding. The authors note that in unpublished preliminary data they were unable to utilise full anti-DEC205 antibody for the same purpose, and highlight the risks of proceeding to the clinic with full mAb targeted nanoparticles.
In addition to this, early in vitro work by Okamoto et al. suggests siRNA containing liposomes targeted to heparin-binding epidermal growth factor (HB-EGF) using anti-HB-EGF Fab′ fragments could provide effective treatment for breast cancer.83 Similarly, Laroui et al. demonstrated effective treatment of colitis through the delivery of TNF-α siRNA encapsulated within F4/80 Fab′ targeted PEG–PLA polymersomes.84 The group found that Fab′ targeted TNF-α siRNA containing nanoparticles granted a greater reduction in all symptoms of colonic inflammation when compared to the non-targeted controls. In both studies the Fab′ fragment was site-specifically conjugated to the nanoparticle via the hinge region using maleimide–thiol chemistry, highlighting the emerging prevalence of this approach for conjugating antibody fragments to nanoparticles.
Further to the above examples, work carried out at Sun Yat-sen University has pioneered the use of ScFv targeted superparamagnetic iron oxide nanoparticles (SPIONS) as MRI visible siRNA delivery vectors.85,86 One study demonstrated the applicability of this approach towards the treatment of neuroblastoma tumours, with in vivo data suggesting significant gene silencing and subsequent tumour suppression.85 Early data suggests a similar approach could be utilised for the treatment and imaging of gastric cancer.86 These studies show that delivery of nucleotides is not limited to organic nanoparticles, and that the innate physical properties of inorganic nanoparticles can grant significant benefits.
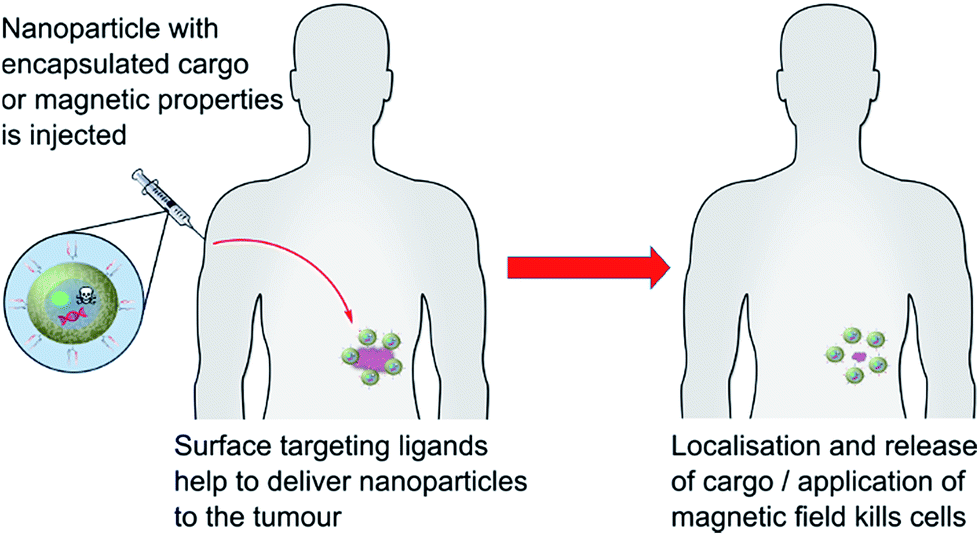 | ||
| Fig. 6 A graphical representation of actively targeted nanoparticle therapeutics. | ||
Towards the end of their studies into magnetic field therapy, Gerald and Sally DeNardo published work in which the full mAb was abandoned in favour of a di-ScFv ligand, which was attached in a highly oriented fashion via a carefully introduced cysteine residue.98 Whilst the SPION-ScFv showed greatly increased accumulation at the tumour site in vivo, efficacy of the hyperthermic properties of the nanoparticle was not explored. Similar work by Yang et al. showed that magnetic iron oxide nanoparticles can be selectively targeted towards the EGFR using an anti-EGFR ScFv ligand, showing promise as a treatment for various EGFR presenting cancers.99
The results discussed above clearly demonstrate the advanced capabilities of nanoparticle–antibody fragment conjugates for chemotherapy. It is anticipated that the trend of using antibody fragments could also provide benefits in other areas of nanomedicine, e.g. targeted immunotherapy through the activation of cell receptors such as Death Receptor 5 (DR-5).100,101
4.2 As imaging agents
Conceptually, targeted nanoparticles provide a myriad of benefits for in vivo imaging of cellular targets. The generous loading capacity of most particles enables the site-selective delivery of large quantities of imaging agent, increasing signal-to-noise ratio, and/or the nanoparticle surface itself can often be tailored to provide intrinsic imaging functionality, as is the case with SPIONs, gold nanoparticles or quantum dots. Early work in the use of antibody–decorated nanoparticles for imaging applications encountered problems due to specific accumulation, with the limiting step found to be extravasation of the nanoparticles from the vasculature, rather than cell binding.1,4,7,41,93,102 Whilst this is also a problem for therapeutic nanoparticles, it is more apparent for imaging applications where the utility is highly dependent on achieving high resolution between the target site and the background. In an attempt to tackle this problem, recent work has focused on the use of smaller antibody fragments as targeting ligands. By utilising smaller antibody fragments, which do not contain the Fc region, overall circulation times and subsequent tumour accumulation rates can be increased greatly.Another excellent example is provided by the work of Rüger et al. who used a self-quenching near-infrared dye incorporated inside a ScFv–decorated liposome to image fibroblast activation protein alpha (FAP) expressing cells. Application of a self-quenching fluorophore ensured significant fluorescence was only observed after intra-cellular degradation of the liposome post-FAP cell binding. This approach led to a significant increase in the signal-to-noise ratio of the ScFv–decorated liposomes when compared to the non-targeted controls in vivo. The authors specify their decision to utilise an ScFv rather than a whole mAb was driven by potential immunogenic concerns.104
Exploiting inherently fluorescent nanoparticles such as gold nanoparticles or quantum dots is more widely utilised, likely due to their relatively large extinction coefficients and resistance to photobleaching. Several excellent examples of antibody fragment-decorated approaches exist. As way of an example, Xu et al. showed that anti-GRP78 ScFv-conjugated quantum dots can be tracked in vivo using fluorescence imaging.105 A similar approach was used by Balalaeva et al. to image breast cancer in vivo.106 Other groups are currently exploring the use of an anti-CEA sdAb conjugated quantum dot for imaging CEA expressing cancer cells, with initial results showing great promise.107–109 Use of an sdAb allowed for highly orientated attachment of the targeting ligand through an engineered cysteine residue, greatly increasing avidity. The superiority of their sdAb is supported by recent results comparing the sdAb ligand with a full antibody analogue; the study demonstrated a dramatic increase in sensitivity when the smaller targeting ligand was employed.110 In both cases lysine residues on the targeting antibody ligands were modified with D-biotin using NHS ester chemistry, allowing the ligands to be attached to the quantum dot using the highly stable biotin–streptavidin interaction. Whilst this non-covalent approach is not ideal, it allowed the researchers to utilise the same coupling strategy for both ligands and thus gain a fairer comparison of their sdAb against the full antibody.
Chen et al. utilised a highly functionalised mesoporous silica nanoparticle (MSN) to successfully image tumour vasculature in vivo using a multimodal approach which employed both PET and optical imaging techniques.117 To target the nanoparticles, the group attached a Fab fragment targeted against CD10, a vascular-specific marker for tumour angiogenesis, and demonstrated a significant improvement in both PET and fluorescence imaging resolution in vivo compared to non-targeted controls.
Work by Hoang et al. has utilised 111In-labelled block copolymer micelles conjugated to trastuzumab Fab to image HER2 positive cell lines in vitro using SPECT/CT.118 In addition to the trastuzumab Fab targeting ligand the group incorporated nuclear localisation signal (NLS) peptides onto the surface of their nanoparticle, leading to effective nuclear translocalisation after initial HER2 mediated internalisation. More recently, this approach was demonstrated in vivo, with significant benefits in tumour accumulation, cellular uptake, and nuclear uptake being reported, when compared to non-targeted controls. Tumour uptake studies indicate the nanoparticles functionalised with both extra-cellular (trastuzumab Fab) and intra-cellular (NLS peptides) targeting ligands outperformed the nanoparticles targeted using trastuzumab Fab alone, indicating post-internalisation nuclear translocation could be beneficial.119
4.3 As immunoassays
The impact of nanoparticles on biomedicine is perhaps most pronounced in the field of immunoassays and diagnostics. The varied optical, physical, and electrochemical properties of nanoparticles present a wide range of observable outputs which can be exploited for the detection of disease biomarkers. The in vitro nature of diagnostic tools eliminates the negative impact of the suboptimal in vivo properties found with many inorganic nanoparticles (e.g. toxicity, bioaccumulation), allowing their full potential to be more readily realised. To date, nanoparticle–full antibody conjugates have found use in immunoassays based on fluorescence,124 Förster resonance energy transfer (FRET),125 catalytic redox reactions,126,127 surface plasmon resonance (SPR),128 surface-enhanced Raman (SER),128,129 and surface electrochemistry,130,131 amongst many others (Fig. 7).124,132,133 Examples of nanoparticle–antibody fragment conjugates are less abundant; this may be as a result of the relative infancy of the field and mAb immunogenicity no longer being an issue. However, recent reports suggest that significant gains can still be obtained through a switch in focus from full antibodies to antibody fragments, some of which are described below.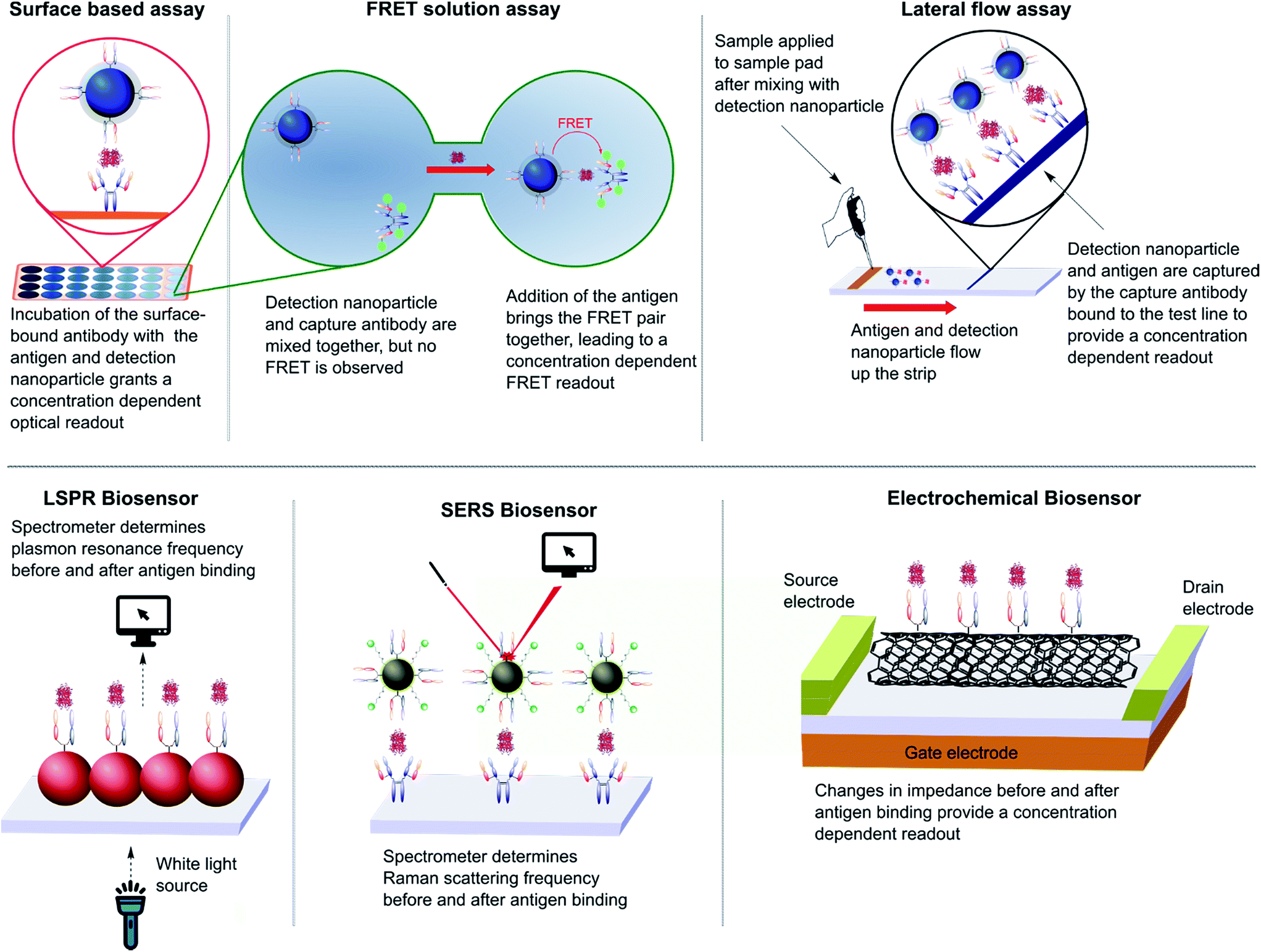 | ||
| Fig. 7 Various designs of immunoassay ranging from surface based, FRET and lateral flow assays to LSPR, SERS and electrochemical biosensing. | ||
Further to the above, Wegner et al. have employed the FRET capabilities of QDs in their sandwich immunoassays to great effect.135 Their assays rely on an antigen-mediated FRET coupling between a QD conjugated reporter antibody and a terbium-labelled capture antibody. The group compared full antibody, F(ab′)2, and Fab fragments as targeting ligands for their QD–antibody conjugates in an immunoassay for prostate specific antigen (PSA). In each case, non-specific conjugation techniques were employed. It was found that the QD–Fab significantly outperformed the QD–full antibody, achieving a 5-fold increase in sensitivity for PSA in serum samples. The authors attributed this to a combination of decreased distance between the FRET pairs and improved orientation of the Fab on the surface of the QD. The group utilised a similar assay for the detection of EGFR in serum, achieving comparable success when employing a QD–nanobody construct as their reporter molecule.136 These solution based assays hold advantages over the more traditional surface based assays as they do not require immobilisation of the capture antibody onto a surface. This increases efficiency and practicality, whilst eliminating potential inaccuracies brought about by non-specific sticking of the nanoparticles to the plate.
Lo et al. employed the use of an electrochemical immunoassay for the detection of CEA. By immobilising an anti-CEA ScFv onto the surface of nickel coated carbon nanotubes the group were able to demonstrate a quantifiable difference in electrical conductivity before and after incubation with the disease marker.144 This approach provided a detection limit of 10 ng mL−1, a 10-fold increase in sensitivity compared to a near identical study where a full antibody against CEA was employed.145 The authors attribute this increased sensitivity to the smaller size of the ScFv and its orientation on the nanoparticle through a selective interaction between the nickel coating and the His tag on the ScFv. When this selectivity was removed through the introduction of multiple chelating sites, a nullification of the activity was observed, thus demonstrating the importance of oriented immobilisation. More recently, Lerner et al. utilised a carbon nanotube to design an immunoassay for the detection of osteopontin (OPN), a disease marker for prostate cancer.146 The group attached an anti-OPN ScFv to a carbon nanotube and were able to detect OPN in serum samples at concentrations as low as 1 pg mL−1, a detection limit three orders of magnitude lower than commercial ELISA assays against the same marker.
5. Conclusions and future outlook
Whilst traditional nanoparticle–full antibody conjugates have proven to be effective tools for both therapeutic and research purposes, limitations resulting from the use of whole immunoglobulins briefly plateaued progress in the area. However, a switch in focus to antibody-based fragments, both natural and engineered, is leading to a positive step-shift in progress. It is clear from the evidence presented in this review that antibody fragments have great potential as targeting ligands for nanoparticle based therapeutics, diagnostics and bioassays, with the resulting constructs demonstrating greater selectivity, superior antigen binding, and more favourable pharmacokinetic properties.It seems we are now at a stage where we are fine-tuning how the antibody fragment is specifically connected to the nanoparticle; as exemplified, the choice of conjugation technique plays an important role in the properties of the resulting nanoparticle–antibody fragment conjugate with more controlled chemistries consistently providing superior results. The marriage of site-selective conjugation strategies with the unique properties and smaller size of antibody fragments allows for the installation of highly oriented targeting ligands, a clear advantage for selectivity, in vivo tolerance and binding affinity. We predict that the future in this field will see a continuation in the trend towards antibody fragment based targeting ligands being installed via increasingly selective and controlled chemistries; potentially providing access to hitherto unexplored applications for antibody targeted nanoparticles.
Acknowledgements
We gratefully acknowledge EPSRC (EP/M01792X/1) and i-sense EPSRC IRC in Early Warning Sensing Systems for Infectious Diseases (EP/K031953/1) for funding AM and DAR, respectively. Certain images in Fig. 3, 6 and 7 were obtained from http://www.freepik.com.Notes and references
- A. Annapragada, Annu. Rev. Med., 2015, 66, 177–193 CrossRef CAS PubMed.
- W. C. Chen, A. X. Zhang and S.-D. Li, Eur. J. Nanomed., 2012, 4, 89–93 Search PubMed.
- G. T. Noble, et al. , Trends Biotechnol., 2014, 32, 32–45 CrossRef CAS PubMed.
- V. J. Yao, et al. , J. Controlled Release, 2016 DOI:10.1016/j.jconrel.2016.01.002.
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2012, 64, 206–212 CrossRef.
- S. D. Steichen, M. Caldorera-Moore and N. A. Peppas, Eur. J. Pharm. Sci., 2013, 48, 416–427 CrossRef CAS PubMed.
- S. J. Shin, J. R. Beech and K. A. Kelly, Integr. Biol., 2012, 5, 29–42 RSC.
- M. Arruebo, M. Valladares and Á. González-Fernández, J. Nanomater., 2009, 439389 Search PubMed.
- V. H. Shargh, H. Hondermarck and M. Liang, Nanomedicine, 2016, 11, 63–79 CrossRef CAS PubMed.
- F. Fay and C. J. Scott, Immunotherapy, 2011, 3, 381–394 CrossRef CAS PubMed.
- S. Goodall, M. L. Jones and S. Mahler, J. Chem. Technol. Biotechnol., 2015, 90, 1169–1176 CrossRef CAS.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- E. C. Wang and A. Z. Wang, Integr. Biol., 2014, 6, 9–26 RSC.
- A. Lavasanifar, J. Samuel and G. S. Kwon, Adv. Drug Delivery Rev., 2002, 54, 169–190 CrossRef CAS PubMed.
- K. Kawano, et al. , J. Controlled Release, 2006, 112, 329–332 CrossRef CAS PubMed.
- Z. Ahmad, et al. , RSC Adv., 2014, 4, 17028–17038 RSC.
- K. Werengowska-Ciecwierz, et al. , Adv. Condens. Matter Phys., 2015, 198175 Search PubMed.
- S. H. Medina and M. E. H. El-Sayed, Chem. Rev., 2009, 109, 3141–3157 CrossRef CAS PubMed.
- D. A. Tomalia, L. A. Reyna and S. Svenson, Biochem. Soc. Trans., 2007, 35, 61–67 CrossRef CAS PubMed.
- R. Shukla, et al. , Bioconjugate Chem., 2006, 17, 1109–1115 CrossRef CAS PubMed.
- Z. Bakhtiary, et al. , Nanomedicine, 2016, 12, 287–307 CAS.
- W. Wu, et al. , Sci. Technol. Adv. Mater., 2015, 16, 023501 CrossRef.
- L. H. Reddy, et al. , Chem. Rev., 2012, 112, 5818–5878 CrossRef CAS PubMed.
- X. Yang, et al. , Chem. Rev., 2015, 115, 10410–10488 CrossRef CAS PubMed.
- E. C. Dreaden, et al. , Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- C. Carnovale, et al. , Prog. Mater. Sci., 2016, 83, 152–190 CrossRef CAS.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2011, 40, 1647–1671 RSC.
- A. Kunzmann, et al. , Biochim. Biophys. Acta, 2011, 1810, 361–373 CrossRef CAS PubMed.
- I. I. Slowing, et al. , Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- C. Bharti, et al. , Int. J. Pharm. Invest., 2015, 5, 124–133 CrossRef CAS PubMed.
- C. Fu, et al. , Biomaterials, 2013, 34, 2565–2575 CrossRef CAS PubMed.
- Q. He, et al. , Small, 2011, 7, 271–280 CrossRef CAS PubMed.
- D. Tarn, et al. , Acc. Chem. Res., 2013, 46, 792–801 CrossRef CAS PubMed.
- C. J. Serpell, K. Kostarelos and B. G. Davis, ACS Cent. Sci., 2016, 2, 190–200 CrossRef CAS PubMed.
- M. F. L. De Volder, et al. , Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- A. B. Seabra, et al. , Chem. Res. Toxicol., 2014, 27, 159–168 CrossRef CAS PubMed.
- C.-C. Chou, et al. , Nano Lett., 2008, 8, 437–445 CrossRef CAS PubMed.
- J.-H. Liu, et al. , ACS Appl. Mater. Interfaces, 2014, 6, 14672–14678 CAS.
- R. Li, et al. , Nanotechnology, 2014, 25, 495102 CrossRef PubMed.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- F. M. Winnik and D. Maysinger, Acc. Chem. Res., 2013, 46, 672–680 CrossRef CAS PubMed.
- B. A. Rzigalinski and J. S. Strobl, Toxicol. Appl. Pharmacol., 2009, 238, 280–288 CrossRef CAS PubMed.
- H. W. Schroeder and L. Cavacini, J. Allergy Clin. Immunol., 2010, 125, S41–S52 CrossRef PubMed.
- A. L. Nelson, mAbs, 2010, 2, 77–83 CrossRef PubMed.
- J.-X. Zhao, et al. , Int. J. Mol. Sci., 2010, 12, 1–11 CrossRef PubMed.
- M. M. Harmsen and H. J. De Haard, Appl. Microbiol. Biotechnol., 2007, 77, 13–22 CrossRef CAS PubMed.
- S. Muyldermans, Annu. Rev. Biochem., 2013, 82, 775–797 CrossRef CAS PubMed.
- J. Wesolowski, et al. , Med. Microbiol. Immunol., 2009, 198, 157–174 CrossRef CAS PubMed.
- P. Holliger and P. J. Hudson, Nat. Biotechnol., 2005, 23, 1126–1136 CrossRef CAS PubMed.
- R. Huang, et al. , Antibodies, 2016, 5, 11 CrossRef.
- K. Groff, J. Brown and A. J. Clippinger, Biotechnol. Adv., 2015, 33, 1787–1798 CrossRef CAS PubMed.
- N. Nuñez-Prado, et al. , Drug Discovery Today, 2015, 20, 588–594 CrossRef PubMed.
- G. Rodrigo, M. Gruvegård and J. Van Alstine, Antibodies, 2015, 4, 259–277 CrossRef.
- S. Steeland, R. E. Vandenbroucke and C. Libert, Drug Discovery Today, 2016, 21, 1076–1113 CrossRef CAS PubMed.
- O. Y. Dmitriev, S. Lutsenko and S. Muyldermans, J. Biol. Chem., 2016, 291, 3767–3775 CrossRef CAS PubMed.
- S. Krah, et al. , Immunopharmacol. Immunotoxicol., 2016, 38, 21–28 CrossRef CAS PubMed.
- R. E. Kontermann, Curr. Opin. Mol. Ther., 2006, 8, 39 CAS.
- Y. Wang, et al. , Nanomedicine, 2008, 3, 475–483 CrossRef CAS PubMed.
- S. Shanehsazzadeh, et al. , Contrast Media Mol. Imaging, 2015, 10, 225–236 CrossRef CAS PubMed.
- J. M. Montenegro, et al. , Adv. Drug Delivery Rev., 2013, 65, 677–688 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- N. Krall, et al. , Nat. Chem., 2015, 8, 103–113 Search PubMed.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- R. Mout, et al. , Chem. Soc. Rev., 2012, 41, 2539–2544 RSC.
- J.-H. Oh, et al. , Anal. Bioanal. Chem., 2015, 407, 8627–8645 CrossRef CAS PubMed.
- K. E. Sapsford, et al. , Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- W. W. K. Cheng and T. M. Allen, J. Controlled Release, 2008, 126, 50–58 CrossRef CAS PubMed.
- P. Sapra, et al. , Clin. Cancer Res., 2004, 10, 1100–1111 CrossRef CAS PubMed.
- W. W. Cheng and T. M. Allen, Expert Opin. Drug Delivery, 2010, 7, 461–478 CrossRef CAS PubMed.
- A. S. Manjappa, et al. , J. Drug Targeting, 2014, 22, 698–711 CrossRef CAS PubMed.
- R. van der Meel, et al. , Adv. Drug Delivery Rev., 2013, 65, 1284–1298 CrossRef CAS PubMed.
- J. Ahn, et al. , Biomaterials, 2015, 39, 23–30 CrossRef CAS PubMed.
- Y. Xiangbao, et al. , Biomed. Pharmacother., 2014, 68, 597–602 CrossRef PubMed.
- A. Quarta, et al. , Nanoscale, 2015, 7, 2336–2351 RSC.
- K. Mack, et al. , Antibodies, 2012, 1, 199–214 CrossRef CAS.
- M. Rabenhold, et al. , J. Controlled Release, 2015, 201, 56–67 CrossRef CAS PubMed.
- Y. Zhang, A. Satterlee and L. Huang, Mol. Ther., 2012, 20, 1298–1304 CrossRef CAS PubMed.
- J. Conde, et al. , Mater. Today, 2015, 19, 29–43 CrossRef.
- Q. Li, et al., Gene Therapy - Tools and Potential Applications, InTech, Rijaka, Croatia, 2013 Search PubMed.
- J. A. Katakowski, et al. , Mol. Ther., 2015, 24, 146–155 CrossRef PubMed.
- A. Okamoto, et al. , Biochem. Biophys. Res. Commun., 2014, 449, 460–465 CrossRef CAS PubMed.
- H. Laroui, et al. , J. Controlled Release, 2014, 186, 41–53 CrossRef CAS PubMed.
- M. Shen, et al. , Int. J. Nanomed., 2012, 7, 3319–3332 CrossRef CAS PubMed.
- Y. Chen, et al. , Int. J. Nanomed., 2012, 7, 359–368 CAS.
- M. Bañobre-López, A. Teijeiro and J. Rivas, Rep. Practical Oncol. Radiother., 2013, 18, 397–400 CrossRef PubMed.
- S. J. DeNardo, et al. , Clin. Cancer Res., 2005, 11, 7087s–7092s CrossRef CAS PubMed.
- R. Ivkov, et al. , Clin. Cancer Res., 2005, 11, 7093s–7103s CrossRef CAS PubMed.
- S. J. DeNardo, et al. , J. Nucl. Med., 2007, 48, 437–444 CrossRef CAS PubMed.
- G. L. DeNardo and S. J. DeNardo, Cancer Biother. Radiopharm., 2008, 23, 671–679 CrossRef PubMed.
- A. K. Hauser, et al. , J. Controlled Release, 2015, 219, 76–94 CrossRef CAS PubMed.
- J. Verma, S. Lal and C. J. F. Van Noorden, Eur. J. Nanomed., 2015, 7, 271–287 Search PubMed.
- B. Kozissnik, et al. , Int. J. Hyperthermia, 2013, 29, 706–714 CrossRef PubMed.
- T. Kobayashi, Biotechnol. J., 2011, 6, 1342–1347 CrossRef CAS PubMed.
- M. Shinkai, et al. , Jpn. J. Cancer Res., 2001, 92, 1138–1145 CrossRef CAS PubMed.
- D. Cui, et al. , Nano Biomed. Eng., 2009, 1, 94–112 CAS.
- A. Natarajan, et al. , Cancer Biother. Radiopharm., 2008, 23, 82–91 CrossRef CAS PubMed.
- L. Yang, et al. , Small, 2009, 5, 235–243 CrossRef CAS PubMed.
- F. Fay, et al. , Biomaterials, 2011, 32, 8645–8653 CrossRef CAS PubMed.
- D. Schmid, et al. , Mol. Ther., 2014, 22, 2083–2092 CrossRef CAS PubMed.
- A. B. Chinen, et al. , Chem. Rev., 2015, 115, 10530–10574 CrossRef CAS PubMed.
- L. Fiandra, et al. , ACS Nano, 2013, 7, 6092–6102 CrossRef CAS PubMed.
- R. Rüger, et al. , J. Controlled Release, 2014, 186, 1–10 CrossRef PubMed.
- W. Xu, et al. , Molecules, 2012, 17, 796–808 CrossRef CAS PubMed.
- I. V. Balalaeva, et al. , J. Biophotonics, 2012, 5, 860–867 CrossRef CAS PubMed.
- A. Sukhanova, et al. , Nanomedicine, 2012, 8, 516–525 CAS.
- T. Y. Rakovich, et al. , ACS Nano, 2014, 8, 5682–5695 CrossRef CAS PubMed.
- R. Bilan, et al. , Phys. Procedia, 2015, 73, 228–234 CrossRef CAS.
- G. Rousserie, et al. , Anal. Biochem., 2015, 478, 26–32 CrossRef CAS PubMed.
- Y. Xing, et al. , Austin Journal of Nanomedicine & Nanotechnology, 2014, 2, 1016 Search PubMed.
- Y. Xing, et al. , Theranostics, 2014, 4, 290–306 CrossRef PubMed.
- F. Chen, E. B. Ehlerding and W. Cai, J. Nucl. Med., 2014, 55, 1919–1922 CrossRef CAS PubMed.
- N. Ahmed, H. Fessi and A. Elaissari, Drug Discovery Today, 2012, 17, 928–934 CrossRef CAS PubMed.
- T. Aweda, D. Sultan and Y. Liu, Nanotechnology for Biomedical Imaging and Diagnostics: From Nanoparticle Design to Clinical Applications, John Wiley & Sons, Inc., First, 2015 Search PubMed.
- Y. Liu and M. J. Welch, Bioconjugate Chem., 2012, 23, 671–682 CrossRef CAS PubMed.
- F. Chen, et al. , Mol. Pharm., 2014, 11, 4007–4014 CrossRef CAS PubMed.
- B. Hoang, R. M. Reilly and C. Allen, Biomacromolecules, 2012, 13, 455–465 CrossRef CAS PubMed.
- B. Hoang, et al. , Mol. Pharm., 2013, 10, 4229–4241 CrossRef CAS PubMed.
- M. A. Busquets, J. Estelrich and M. J. Sánchez-Martín, Int. J. Nanomed., 2015, 10, 1727–1741 CrossRef PubMed.
- C. Zhang, et al. , Asia–Pac. J. Clin. Oncol., 2016, 12, 13–21 CrossRef PubMed.
- K. L. Vigor, et al. , Biomaterials, 2010, 31, 1307–1315 CrossRef CAS PubMed.
- C. Alric, et al. , RSC Adv., 2016, 6, 37099–37109 RSC.
- M. R. Kierny, T. D. Cunningham and B. K. Kay, Nano Rev., 2012, 3, 17240 CrossRef CAS PubMed.
- D. Geißler and N. Hildebrandt, Anal. Bioanal. Chem., 2016, 408, 4475–4483 CrossRef PubMed.
- H. Y. Shin, T. J. Park and M. Il Kim, J. Nanobiotechnol., 2015, 756278 Search PubMed.
- H. Wei and E. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- P. D. Howes, S. Rana and M. M. Stevens, Chem. Soc. Rev., 2014, 43, 3835–3853 RSC.
- J. H. Granger, et al. , Chem. Soc. Rev., 2016, 45, 3865–3882 RSC.
- K. Dawson and A. O'Riordan, Annu. Rev. Anal. Chem., 2014, 7, 163–181 CrossRef CAS PubMed.
- X. Luo and J. J. Davis, Chem. Soc. Rev., 2013, 42, 5944–5962 RSC.
- J.-J. Xu, et al. , Chem. Soc. Rev., 2014, 43, 1601–1611 RSC.
- K. S. McKeating, A. Aubé and J.-F. Masson, Analyst, 2016, 141, 429–449 RSC.
- G. P. Anderson, et al. , Anal. Chim. Acta, 2013, 786, 132–138 CrossRef CAS PubMed.
- K. D. Wegner, et al. , ACS Nano, 2013, 7, 7411–7419 CrossRef CAS PubMed.
- K. D. Wegner, et al. , Small, 2014, 10, 734–740 CrossRef CAS PubMed.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS PubMed.
- J.-Y. Byun, et al. , Chem. Commun., 2013, 49, 9497–9499 RSC.
- P. L. Stiles, et al. , Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- X.-M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- S. W. Bishnoi, et al. , Anal. Chem., 2011, 83, 4053–4060 CrossRef CAS PubMed.
- X. Qian, et al. , Nat. Biotechnol., 2008, 26, 83–90 CrossRef CAS PubMed.
- B.-Y. Chang and S.-M. Park, Annu. Rev. Anal. Chem., 2010, 3, 207–229 CrossRef CAS PubMed.
- Y.-S. Lo, et al. , ACS Nano, 2009, 3, 3649–3655 CrossRef CAS PubMed.
- D.-W. Park, et al. , J. Nanosci. Nanotechnol., 2006, 6, 3499–3502 CrossRef CAS PubMed.
- M. B. Lerner, et al. , ACS Nano, 2012, 6, 5143–5149 CrossRef CAS PubMed.
Footnote |
| † Hybrid organic–inorganic particles will not be focused on in this review. |
| This journal is © The Royal Society of Chemistry 2017 |