
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-selective Suzuki–Miyaura coupling of heteroaryl halides – understanding the trends for pharmaceutically important classes†
Joshua
Almond-Thynne
a,
David C.
Blakemore
b,
David C.
Pryde
b and
Alan C.
Spivey
 *a
*a
aDepartment of Chemistry, Imperial College London, South Kensington Campus, London, SW& 2AZ, UK. E-mail: a.c.spivey@imperial.ac.uk; Fax: +44 (0)1474 813219
bPfizer Worldwide Medicinal Chemistry, The Portway Building, Granta Park, Great Abington, Cambridge, CB21 6GS, UK
First published on 9th August 2016
Abstract
Suzuki–Miyaura cross-coupling reactions of heteroaryl polyhalides with aryl boronates are surveyed. Drawing on data from literature sources as well as bespoke searches of Pfizer's global chemistry RKB and CAS Scifinder® databases, the factors that determine the site-selectivity of these reactions are discussed with a view to rationalising the trends found.
1. Introduction
Substituted heteroarenes form the core of numerous pharmacologically active agents and drug substances,1 as well as agrochemical products, ligands, secondary metabolites, polymers and electronic materials.2 Notwithstanding widespread recent advances in transition metal-catalysed C–H bond activation processes,3 Pd-catalysed Suzuki–Miyaura coupling (SMC)4 of (pseudo)halogenated5 heteroarenes with (hetero)aryl boronic acids/esters constitutes the most widely used approach to heteroarene elaboration with C–C bond formation particularly in a pharmaceutical discovery chemistry setting.6 This reflects the wide palette of methods available for preparation of both reaction partners, the versatility and functional group compatibility of these methods, the general stability, low toxicity, ease of handling and commercial availability of the reaction partners, the relatively environmentally benign conditions of the SMC reactions themselves (e.g. high efficiencies, low catalyst loadings etc.), as well as the opportunities the SMC disconnection affords for rapid parallel exploration of structural diversity and chemical space.4b,6When multiple SMC reactions are to be choreographed to occur sequentially, this can sometimes be achieved by judicious site-selective introduction of different types of halogen into a substrate. However, particularly for heteroaryl substrates, the intrinsic polarities of the ring carbons also strongly influence site-selectivity and this factor is critical when coupling substrates containing two or more of the same type of halogen. These latter substrates are often the preferred precursors on cost and availability grounds and are the main focus of this review. Underscoring not only the importance of site-selective cross-coupling reactions of heteroaryl halides from a synthesis perspective, but also highlighting the challenges associated with predicting the outcome of such reactions, there have been several excellent reviews compiling and classifying published examples of these reactions including notable contributions by Bach (heteroarenes),7 Stanetty (azoles),8 Handy (heteroarenes),9 Fairlamb (heteroarenes),10 Manabe (polyhalides),11 Rossi (heteroarenes)12 and Langer (bis-triflates).13
Notwithstanding these previous compilations, we considered that systematic interrogation of reaction databases would reveal patterns of selectivity that could reinforce and extend our understanding of the factors that affect site-selectivity in SMC reactions of heteroarenes and improve our ability to predict outcomes for new substrates. In particular, we envisioned that the in-house Pfizer global chemistry Reaction Knowledge Base (RKB)14 would constitute a rich source of reaction data that would extend and compliment data mined from the CAS Scifinder®15 database. To this end, in this review we provide a concise overview of the factors determining the site-selectivity of SMC reactions of heteroaryl halides (Section 3) and then a summary of the results of some database searches of structures of potential medicinal interest (Section 4). The overview draws on data both from the literature and from the structure-by-structure database searches.
2. Data gathering and analysis
Based on our perception of their relevance as scaffolds and/or intermediates in drug discovery programs,6 the heteroarene ring systems that were selected for investigation were: pyridines, pyrimidines, pyrazines, pyridazines, pyrroles, furans, thiophenes, imidazoles, pyrazoles, (is)oxazoles, (iso)thiazoles, (iso)quinolines, benzodiazines, indoles, benzoxazoles, benzothiazoles, benzodiazoles, benz(is)oxazoles, benz(is)othiazoles and aza(iso)quinolones (naphthyridines). Parallel searches were carried out on the CAS Scifinder® and RKB reaction databases using as similar search queries as their respective interfaces would allow (see ESI†). Only reactions with (hetero)aromatic boronic acid and ester coupling partners were retrieved;16,17 alkenyl and alkynyl congeners were excluded since these motifs occur much less frequently in pharmaceuticals. Alkyl coupling partners,18 stereoselective processes,19 and non-Pd-mediated processes20 were also excluded.4c Reactions involving substrates having two (or more) of the same halide substituents were systematically retrieved; specifically, di-chlorides, di-bromides and di-iodides although selected examples containing two different halides were also noted where these enable complementary site-selectivities to be achieved.3. Factors affecting the site-selectivity of SMC reactions of heteroryl halides
As indicated above, we have divided our analysis into two sections. In this section factors that determine site-selectivity in SMC reactions based both on published studies and our database searches are presented. In Section 4, the data from the CAS Scifinder® and RKB database searches are summarised by class of heterocycle. Hopefully, this structure will help readers both to predict the outcome of reactions on new heterocyclic systems per se and also to quickly locate relevant prior-art on key heterocyclic systems of interest. In the Schemes and Figures, the halide highlighted with a blue disk is the preferred site of reaction.When discussing SMC reactions, it is generally accepted that the oxidative addition (OA) step is rate-determining and irreversible, and that the rate of OA is largely controlled by the bond dissociation energies (BDEs) of the C–Hal bond such that usually Ar–I > Ar–Br > Ar–Cl > Ar–F.21 Although cases where OA is not rate limiting in SMC reactions have been proposed,22 and OA can be reversible under high steric stress,23 this assumption is probably accurate for most catalytic reaction situations. The BDE is however by no means the exclusive arbiter of ease of OA because other structural features and the reaction conditions (particularly: solvent, pre-catalyst, ligand, base, additive etc.) are also influential. When different halides are present in a substrate, the site-selectivity of SMC reactions is strongly influenced by the intrinsic propensity of each halide to undergo OA (Section 3d). However, we will start by discussing the key factors that influence site-selectivity in heteroarenes containing two or more of the same type of halogen. We will see that for these cases the ‘molecular environment/electrophilicity’ of the carbon atom to which the halide is bound is a key factor and that this is reasonably predictable for a given heteroarene core (Sections 3a–c).
3a. Influence of the intrinsic relative electrophilicities of different ring carbons
For heteroarenes containing two or more of the same type of halogen (e.g. di-chlorides, tri-bromides etc.), several indicators based on experimental data have been identified to help predict the intrinsically most reactive positions for OA. Since the OA step in SMC reactions and the addition step in SNAr reactions have mechanistic similarities, both are generally favoured at the more electrophilic carbon when two identical halogen substituents are in competition. Consequently, experimental SNAr site-selectivity data has been used to predict SMC reactivity.7,24 Others have drawn the analogy with propensity to undergo lithium–halogen exchange, which generally favours the position that results in the most stable resulting aryl lithium derivative.2513C NMR chemical shift values (δC) can similarly provide insight into the relative electrophilicities of carbons bearing halogens.26 Most notably, Handy and Zhang have advocated analysis of the 1H NMR chemical shift values (δH) of the parent non-halogenated heteroarenes as a guide for predicting the site of cross-coupling reactions;9 with the position of the most deshielded proton being the favoured site for SMC. Although this has the appeal of simplicity, it is not fail-safe, particularly in cases where ΔδH < 0.3 ppm.25,27 For example, although the method was accurate for several polysubstituted pyrroles, for the case of 3-arylpyrrole 5, where ΔδH was just 0.02 ppm, the site of SMC could be switched from C2 to C4 simply by changing the solvent from DMF to ethanol–toluene (Scheme 1).9,28 | ||
| Scheme 1 The Handy and Zhang method for site-selectivity prediction based on the 1H NMR δH values for the corresponding non-halogenated heteroarenes – as applied to (a) 2,3-, (b) 3,4-, and (c) 2,4-dibromopyrroles, the last of which displays solvent dependent selectivity.9,28 | ||
Computation has also been used to predict the order of susceptibility to OA in heteroaryl polyhalides on a case-by-case basis.26 Computation can in principle not only dissect out the heteroaryl electronic components but also account for steric factors and directing effects from adjacent functional groups during the OA process. Studies that draw out trends rather than focus on isolated examples are of particular interest. Houk et al. have noted that computed BDEs cannot account for all observed reaction selectivities and have used a DFT-based ‘distortion–interaction’ model (sometime referred to as an ‘activation-strain’ model) to better understand the origins of selectivity in Pd(0)-catalysed cross-coupling reactions of heteroaryl polychlorides and polybromides including isoquinolines, pyridines, benzofurans and furans.27a,29 Using Pd(PH3)2 as a model di-ligated complex, the energies required to distort isolated reactants to the OA transition state geometries (the distortion energy, ΔEdist) were computed along with the energy of interaction between these distorted reactants (the interaction energy, ΔEint). It was concluded that ΔEdist closely tracks the BDE and that ΔEint is dominated by a favourable back-bonding (dxy → π*) secondary frontier molecular orbital (FMO) interaction as the bent PdL2 moiety approaches the C–Hal bond η2-fashion (i.e. side-on).27a The ΔEdist contribution is therefore relatively invariant when one type of halogen is involved although they note that in general BDE values are (i) lower in 6-membered compared to 5-membered rings,30 and (ii) lowered by the presence of a sulfur atom in the ring or when the halogen is an iminoyl halide.29 The stabilising ΔEint term is dependent on the π* LUMO coefficient31 which is generally increased for positions adjacent to ring heteroatoms (Fig. 1).29,32
 | ||
| Fig. 1 Houk's ‘distortion–interaction’ DFT approach to computationally predicting the most favourable position for OA by bis-ligated Pd-catalysts in heteroaryl polyhalides.27a | ||
Thus for each of the three systems 8–10 shown below, the experimentally observed site for SMC reaction is not the one predicted on the basis of having the lowest calculated BDE value but the one with the lowest activation barrier (ΔE). The larger the δΔE value, the more selective a reaction can be expected to be (Fig. 2).25
 | ||
| Fig. 2 The Houk ‘distortion–interaction’ DFT approach to site-selectivity prediction – as applied to (a) benzofuran 8, (b) furan 9 and (c) isothiazole 10.27a | ||
Computational studies have also thrown significant light on how the nature of the phosphine ligands, the ligation state of the Pd and complexation of a pre-catalyst with the substrate33 can all influence OA activation energies, but this will be discussed later in the context of the influence of reaction conditions (Section 3c).
In general, for heteroaryl polyhalides containing a single type of halogen, the intrinsic relative electrophilicities of different ring carbons is a critical factor controlling SMC site-selectivity. In the case of otherwise unsubstituted substrates, the electronic distribution is controlled by the position of the halides in the ring-system relative to the ring heteroatoms. In cases where the heteroaryl polyhalide contains other substituents, these substituents provide additional electronic and steric perturbations but it appears that the intrinsic heterocycle polarity is usually dominant (Section 3b). These generalisations are strongly supported by the data from our database searches which show that the position at which SMC reactions occur are characteristic of the particular heterocycle and largely independent of substituents and the nature of the boronic acid/ester coupling partner (Section 4).
3b. Influence of ring substituents
The influence of substituents on site-selectivities in heteroarene SMC reactions appears to be surprisingly limited, with significant perturbations to the intrinsic directing influence of the ring-system generally being restricted to situations where the heterocycle itself is not strongly polarised and/or where substituents are strongly electron withdrawing and/or are sufficiently Lewis basic to coordinate to the catalyst and promote reaction via a palladacycle. Steric factors can sometimes be decisive but these generally appear to be of secondary importance.To illustrate this, consider first the case of 4-substituted 3,6-dichloropyridazines 11a–c and 13a–c (Scheme 2).
 | ||
| Scheme 2 The site-selectivity of SMC reactions can be determined by substituents: e.g. (a) 3,6-dichloropyrimidines containing 1°, 2° or 3° amine substituents at C4 (11a–c) generally react at C3, but (b) when the C4 substituent is non-basic (13a–c) reaction is at C6 presumably for steric reasons.34a | ||
Blaise et al.34a have investigated a range of heteroatom-based substituents at the 4-position of 3,6-dichloropyridazines and found that 1°, 2° and 3° amines (11a–c) promote SMC reaction at C3 (i.e. proximal to the amine) using Pd(PPh3)4/Na2CO3/toluene/EtOH/H2O but that the reactivity and selectivity of these substrates decreases with increasing bulk of the amine substituents. An N-MeBoc group at C4 (13a) however promotes SMC reaction at C6 (i.e. distal to the amine); similarly, OMe and OBn groups at C4 (13b and 13c) promote SMC reactions at C6 in 60% and 50% yields respectively, implicating coordination of the Pd to a Lewis basic amine group as facilitating reaction at C3 (Scheme 2, above).34a
The reactivity of 2,6-dichloro nicotinic acid 15 and its derivatives is also instructive and demonstrates the role that catalyst coordination to Lewis basic functional groups can have on the site-selectivity of SMC reactions (Scheme 3).35
 | ||
| Scheme 3 Carboxylic ester, -amide and -acid modulation of site-selectivity: e.g. 2,6-dichloro nicotinic acid (18) and its derivatives can undergo SMC reactions at C2 or C6 selectively depending on the conditions (a–d).35 | ||
With the methyl ester derivative (15, R = OMe), Yang et al.35a found that Pd(PPh3)4 promoted SMC reactions at C6 (→ 17), but that Li's PXPd2 pre-catalyst [Pd(t-Bu2Cl)2Cl2]2 (ref. 36) promoted SMC reactions at C2 (→ 16a). Yang et al. hypothesised that the latter, more electron rich and coordinatively unsaturated complex was able to coordinate to the ester carbonyl thereby overcoming the inherent steric bias of the substrate. To corroborate this, they showed that a more Lewis basic amide congener gave even greater selectivity for SMC reaction at C2 (→ 16b, Scheme 3, above). This notion was extended to the case of the free acid 18 by Ma et al.35b and by Houpis et al.35c who found that Pd(PPh3)4 and Pd(OAc)2/PPh3 promoted SMC reactions at C6 (→ 20), but that phosphine-free Pd [i.e. Pd2(dba)3·CHCl3] resulted in high levels of selectivity for SMC reaction at C2 (→ 19), presumably by virtue of its ability to coordinate to the carboxylate.
The divergent behaviour of methyl 1,4-ditrifloxy phenyl-2-carboxylate (21)37 and phenyl 1,4-ditrifloxynaphthalene-2-carboxylate (23)38 with respect to their SMC site-selectivity is also revealing. Although not heteroarenes, a comparison of their behaviour demonstrates how the subtle interplay between steric and electronic effects imparted by a substituent can be critical in controlling SMC reactions when intrinsic ring polarity effects are weak (Scheme 4).
 | ||
| Scheme 4 A subtle interplay of steric and electronic factors can control SMC reaction site-selectivity: e.g. (a) methyl 1,4-ditrifloxy phenyl-2-carboxylate (21) and (b) phenyl 1,4-ditrifloxynaphthalene-2-carboxylate (23) undergo SMC at C4 and C1 respectively.37,38 | ||
Phenyl ditriflate 21 undergoes SMC reactions at C4 (→ 22) whereas naphthyl ditriflate 23 undergoes SMC reactions at C1 (→ 24). In both cases, the steric crowding at C1 is essentially equivalent and so the divergent behaviour is presumably electronic in origin: i.e. C1 is sufficiently electrophilic in naphthyl derivative 23 to override the steric crowding due to the ester but insufficiently electrophilic in phenyl derivative 21 to do likewise. Langer et al. have proposed that this is consistent with the naphthalene having significant diene character and being relatively easily polarised in its substituted ring thus allowing the ester substituent to impart greater electrophilicity to the proximal C1 position than is possible for the phenyl system without incurring a concomitant energetic penalty from loss of aromaticity.38 Langer has studied several additional ditriflate-containing substrates in a systematic fashion and similar conclusions regarding the delicate balance of steric vs. electronic factors emerge.13
Notwithstanding the above studies, it is perhaps surprising how limited the influence of ring substituents is in controlling site-selectivity in SMC coupling reactions. As emphasised previously, this allows the outcome of most reactions to be predicted simply on the basis of the position of the halides in a given heteroarene. A contributory factor towards this situation is that a large proportion of the available data both in the CAS Scifinder® and the Pfizer RKB databases relates to reactions using ‘standard conditions’ (e.g. Pd(dppf)Cl2 or Pd(PPh3)4 with Na2CO3 or NaHCO3 or K2CO3 in DME–H2O or THF–H2O or 1,4-dioxane–H2O).14 The predominance of these conditions reflect the low cost and high convenience of these conditions and also their wide substrate scope. However, there are of course SMC reactions of heteroaryl polyhalides where the choice of reaction conditions, particularly the choice of ligand and solvent, can be decisive in dictating site-selectivity (Section 3c). This is the case for substrates containing a single type of halogen and even more so for those containing mixed halides.
3c. Influence of the reaction conditions – particularly the Pd pre-catalyst/ligand
The specific reaction conditions used for a SMC reaction on a heteroaryl polyhalide can sometimes strongly influence the outcome in terms of site-selectivity of coupling. Due to the mechanistic complexity of these reactions, interpretation let alone prediction of these effects is difficult, but a number of studies which have documented such reactions and sought to rationalise them have been published.An investigation by Dai et al. examined the effect of different phosphines on the site-selectivity of SMC reactions of 3,5-dichloropyridazine 25.39 They found that chelation and electron density played key roles and specifically that electron deficient bidentate ligands (such as dppf) favoured SMC reactions at C3 over C5 (i.e. → 26) whereas electron rich monodentate ligands (such as Qphos) favoured C5 over C3 (i.e. → 27). Electron rich bidentate ligand dtbpf also promoted reactions at C5 over C3, although this was interpreted as indicating that steric effects as well as electronic effects play a role in determining site-selectivity (Scheme 5).39
 | ||
| Scheme 5 Ligand-dependent site-selectivity: e.g. 3,5-dichloropyridazine 25 undergoes SMC (a) at C3 with Pd(OAc)2/dppf, (→ 26) and (b) at C5 with Pd(OAc)2/Qphos (→ 27).39 | ||
The effect of different phosphines on the site-selectivity of SMC reactions of various diiodo- and dibromo-oxazoles, -imidazoles and -thiazoles has been studied by Strotman et al.25 and their findings are summarised below (Scheme 6).
 | ||
| Scheme 6 Ligand-dependent site-selectivity: (a) 2,4-diiodooxazole, (b) 2,5-dibromoimidazole, (c) 2,4-dibromoimidazole, and (d) 2,4- and 2,5-dibromothiazoles.25 | ||
Handy and Zhang's 1H NMR analysis on the parent non-halogenated heteroaryls predicts SMC reactions should occur at C2 for oxazole 28 and N-methylimidazoles 31 and 34 (cf.Scheme 1). It was found experimentally however that under most conditions 2,4-diiodooxazole (28) underwent SMC at C4 but often with poor selectivity over C2 and with high levels of bis-arylation. After screening ∼200 achiral phosphines, Xantphos® was found to be uniquely capable of mediating highly selective mono-SMC reactions at C4 (→ 29) and 1,3,5-triaza-7-phospha-adamantane in MeCN gave high selectivity for mono-SMC reactions at C2 (→ 30). N-Methyl-2,5-dibromoimidazole (31, and its diiodo-congener) behaved very similarly: all phosphines except the phospha-adamantane in MeCN gave C5 selectivity (→ 32). Intriguingly however, N-methyl-2,4-diiodoimidazole (34) showed no appreciable reactivity at C4 for any of the ligands screened and the most selective conditions in terms of minimising bis-arylation involved the use of tri-(p-fluorophenyl)phosphine to give the C2 product 35. Similarly, both 2,4- and 2,5-dibromothiazoles gave almost exclusive mono-SMC reactions at C2 irrespective of the conditions employed. As for the case of the dichloropyridazines, it appears that the electron density, ability to chelate and steric demand of the ligand system play key roles in determining selectivity with particularly electron rich and/or sterically demanding ligands being prevalent among ligands which promote unusual selectivities.
Our understanding of the basis of some of these ligand effects has been significantly enhanced by observations made on mixed halide-containing, non-heteroaryl substrates and associated computational studies. Hayashi made the seminal observations on ligand-dependent regiodivergent Pd-catalysed Kumada couplings of 4-trifloxybromobenzene in 1997,40 which Brown in 2007 showed to be replicated for Stille and Negishi type couplings but interestingly not for SMC reactions.41 In 2000, Fu et al. reported that the site-selective SMC reaction of 4-trifloxychlorobenzene (36) occurred selectively at the chloride (i.e. C1, → 37) when using Pd2(dba)3/P(t-Bu)3 in THF (as expected on the basis of BDE), but selectively at the triflate (i.e. C4, → 38) when using Pd2(dba)3/PCy3 in THF42 (Scheme 7).
 | ||
| Scheme 7 Control of site-selectivity in the SMC reaction of 4-trifloxychlorobenzene (36) according to the conditions: (a) ligand,42,43 and (b) solvent44 control. | ||
Subsequent theoretical and experimental studies concluded that the steric bulk of P(t-Bu)3 generally favours formation of mono-ligated, 12 electron Pd complexes (i.e. PdL) whereas the less sterically demanding PCy3 generally stabilises di-ligated, 14 electron complexes (i.e. PdL2),45 and that this difference accounts for their divergent behaviour. This hypothesis was tested computationally by Houk and Schoenebeck using the aforementioned ‘distortion–interaction’ DFT analysis (see Fig. 1, above).43 Unsurprisingly, the computed activation energies (ΔE) were found to be highly sensitive to the ligation state of the Pd: e.g. PdL2vs. [PdL2X]−vs. PdL vs. [PdLX]−.43,46,47 Specifically, it was shown that for PdL complexes, the computed activation energies (ΔEs) were dominated by the ΔEdist (substrate) term whereas for PdL2 complexes the ΔE values were dominated by the interaction energy (ΔEint). This situation, combined with the aforementioned expectation that the highly bulky ligand P(t-Bu)3 would favour a mono-ligated PdP(t-Bu)3 complex in THF whereas the less bulky PCy3 would favour a di-ligated Pd(PCy3)2 complex, explained the observed site-divergent behaviour. Decisively, the lower BDE of the chloride cf. the triflate minimised ΔE for insertion of the mono-ligated PdP(t-Bu)3 complex into the C–Cl bond whereas the strong dxy → π* interaction between the highly nucleophilic di-ligated PdPCy3 and the distorted vinyl triflate group minimised ΔE for insertion into the C–OTf bond.43 Subsequent higher level DFT computational studies have corroborated these conclusions and furnish reaction energy profiles for PdL and PdL2 pathways that mirror experiment provided dispersion terms are incorporated in the calculations.48
Schoenebeck et al. showed experimentally that if a polar solvent like MeCN was used in place of THF for the SMC reaction of 4-trifloxychlorobenzene (36) then the Pd2(dba)3/P(t-Bu)3 conditions promote selective SMC coupling at the C4 triflate (→ 38 in 74% yield) like Fu's Pd2(dba)3/PCy3 conditions (see Scheme 7, above).49 Schoenebeck also performed calculations to demonstrate that this experimental outcome was consistent with the formation of an anionic [PdLX]− complex under these conditions (where X was either F or ArBO2H).44 Subsequent studies demonstrated that the Pd(I) dimer complex [BrPdP(t-Bu)3]2, also promotes these reactions and favours reaction at C1 (→ 37) in THF and at C4 (→ 38) in MeCN.50 The behaviour of the dimer in these reactions was attributed to its in situ conversion to PdP(t-Bu)3 induced by the base acting as a nucleophile;51 the bromine-bridged Pd(I) dimer is more labile in this respect than the corresponding iodide-bridged one, although with an appropriately nucleophilic base both can act as precursors to catalytically active Pd(0) species.52
Schoenebeck has also introduced the P(i-Pr)(t-Bu)2 ligand, which has a Tolman cone angle53 (175°) intermediate between that of P(t-Bu)3 (182°) and PCy3 (170°), and which imparts P(t-Bu)3-like behaviour (OA at C1 → 37) when added 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 relative to Pd [i.e. favouring monoligated PdP(i-Pr)(t-Bu)2], but PCy3-like behaviour (OA at C4 → 38) when added in excess (e.g. 10:1 relative to Pd).54 Sigman has also recorded concentration-dependent selectivity for other phosphines in this reaction.55
:1 relative to Pd [i.e. favouring monoligated PdP(i-Pr)(t-Bu)2], but PCy3-like behaviour (OA at C4 → 38) when added in excess (e.g. 10:1 relative to Pd).54 Sigman has also recorded concentration-dependent selectivity for other phosphines in this reaction.55
More generally, there is increasing evidence for phosphine-free Pd (nanoparticles) being active catalytic species in SMC reactions (i.e. heterogeneous catalysis).56 The likelihood of nanoparticulate Pd being the catalytically active species is minimal for SMC reactions carried out at ambient temperature using chlorides, but significant for high temperature reactions using e.g. bromides.57 Given that adventitious Pd(0) contaminants can be active at levels as low as 50 ppb,58 caution must be applied when trying to rationalise switches in site-selectivities as a function of changes of conditions as the observed products may not arise from the ligated species expected.
Notwithstanding these caveats when interpreting changes in site-selectivity in SMC reactions, the aforementioned studies highlight how the steric and electronic characteristics of phosphines affect the ligation state of the Pd and consequently reaction outcomes. Although this has been most intensively studied for 4-trifloxychlorobenzene (36, Scheme 7), this applies in all SMC reactions and particularly those of substrates containing mixed halides (Section 3d). These compounds are frequently investigated with a view to overriding the ‘intrinsic’ site-selectivity of the parent heterocycle.
3d. Influence of the nature of the halide
Arguably the most conceptually straightforward method to ensure that site-selective sequential SMC reactions take place in a required order is to anticipate the relative reactivity of different types of carbon–halogen bonds towards the initial OA step, by varying the halides present in the substrate. As noted previously, this prediction is based on the generalisation that OA in SMC reactions is usually rate-determining, irreversible, and strongly affected by the relative BDEs which in turn vary predictably as a function of the halide: Ar–I > Ar–Br > Ar–Cl > Ar–F.24,59 Although this is certainly the case for relatively unpolarised carboaromatic ring systems, how well does it hold for more intrinsically polarised systems of pharmaceutical interest? Often this can be a successful tactic, but for strongly polarised positions in heteroarenes it can be difficult to overturn the intrinsic site-selectivity trends discussed above (Sections 3a–c). The reactivity of mixed halide–triflates in particular are rather difficult to predict in this context – a discussion of these is provided in the ESI.†5-Bromo-2-chloropyridine (39) and 2-bromo-3-iodopyridine (41) are illustrative of heteroarenes that undergo SMC reactions with aryl boronic acids at C5 and C3 respectively despite the fact that the C2 position is intrinsically ‘activated’ in both cases vide infra (Scheme 8).60
 | ||
| Scheme 8 The BDE of the C–Hal bond clearly influences the site of SMC reaction for pyridine derivatives: e.g. (a) 5-bromo-2-chloropyridine (39), and (b) 2-bromo-3-iodopyridine (41) undergo SMC at C5 (→ 40) and C3 (→ 42) respectively.60 | ||
In both cases, OA takes place at the position bearing the more reactive halide as predicted on the basis of average C–Hal BDEs. Additional examples where judicious use of mixed halides can successfully allow the intrinsic electronic bias of a particular ring-system to be overturned are highlighted in Section 4 (i.e.Schemes 16, 19, 20 and 25).
By contrast, 6-bromo-2-chloroquinoxaline (43)61 and 6-bromo-2-chloro-8-fluoroquinazoline (45)62 both react in SMC reactions first at the chlorides at C2 in preference to the bromides at C6 (Scheme 9).
 | ||
| Scheme 9 The high intrinsic electrophilicity of certain ring positions (e.g. C2 in quinoxalines and quinazolines) can perturb the BDE sufficiently to override the usual ArBr > ArCl order of reactivity: e.g. (a) 6-bromo-2-chloroquinoxaline (43), and (b) 6-bromo-2-chloro-8-fluoroquinazoline (45) undergo SMC at C2.61,62 | ||
Apparently, for these ring systems the intrinsic, strong electrophilicity at C2 (Section 4c) can facilitate OA to a greater extent than can be ‘compensated for’ by the normally lower BDE of C–Br relative to C–Cl.
Reactions involving isoquinolines and quinolones (Section 4c), containing halides at C1 and C2 respectively, constitute an intermediate situation between these contrasting pyridine and quinoxaline/quinazoline cases. For these substrates, a chloride substituent at these intrinsically electrophilic positions sometimes reacts in preference to a bromide elsewhere in the heteroarene but not always (Schemes 10 and 11). For example, 1-chloro-5-bromoisoquinoline reacts at C1 (47 → 48),63 as does a 1,3-dichloro-6-bromoisoquinoline (49 → 50),64 but 1-chloro-3-tert-butyl-6-bromoisoquinoline reacts at C6 (51 → 52)65 and 1-chloro-7-bromoisoquinoline and 1,4-dichloro-7-bromoisoquinoline react at C7 (53a/b → 54a/b)66 (Scheme 10).
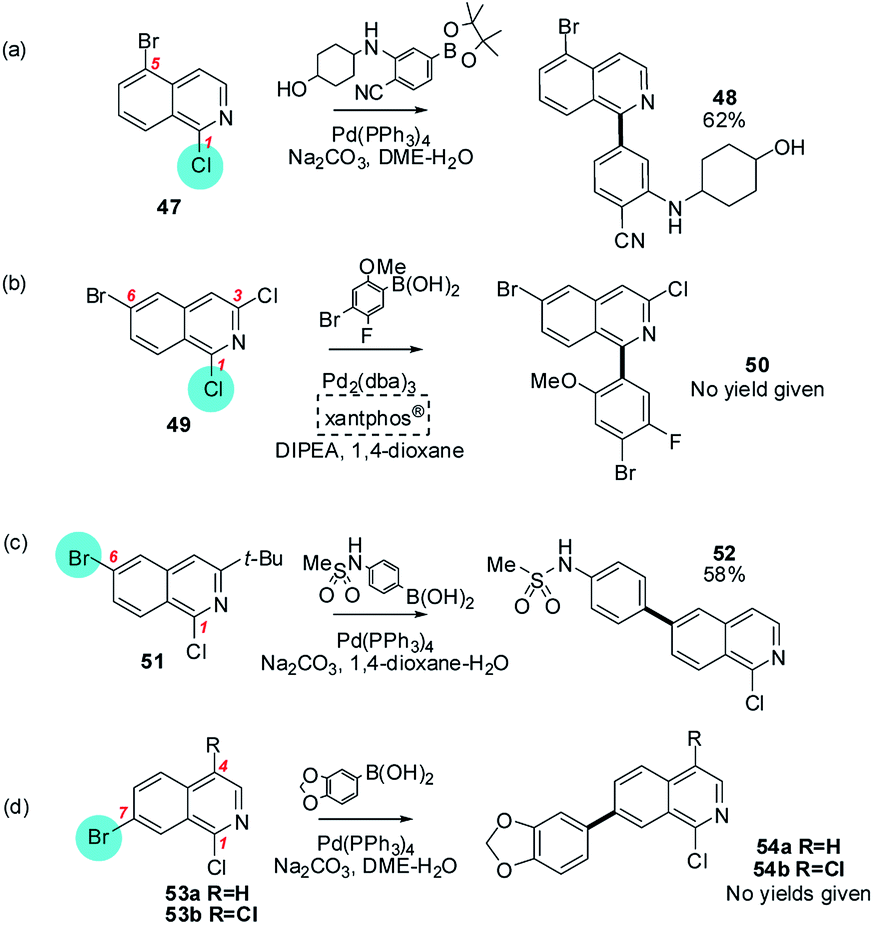 | ||
| Scheme 10 The intrinsic electrophilicity of C1 in isoquinolines is sufficient to override the usual ArBr > ArCl order of halide reactivity for (a) 1-chloro-5-bromoisoquinoline (47),63 and (b) 1,3-dichloro-6-bromoisoquinoline (49),64 but not for (c) 1-chloro-3-tert-butyl-6-bromoisoquinoline (51), or (d) 1-chloro-7-bromoisoquinoline (53a) or 1,4-dichloro-7-bromoisoquinoline (53b).66 | ||
 | ||
| Scheme 11 The intrinsic electrophilicity of C2 in quinolines is sufficient to override the usual ArBr > ArCl order of halide reactivity e.g. for (a) 2,4-dichloro-8-bromo-7-methoxyquinoline (55),67 but 2-chloro-6-bromoquinoline (57) can react (b) at C2 (→ 58) using Pd(PPh3)4,68 or (c) at C6 (→ 59) using Pd(dppf)Cl2,69 and 2-chloro-7-bromo-5-isopropylquinoline reacts at C7 (60 → 61).70 | ||
It is not clear what features of these molecules and/or the conditions employed are responsible for this site-divergent behaviour but it presumably reflects the fact that the opposing influences on the BDE elicited by the ring polarisation and the change of halogen are of similar magnitude, making both positions similarly reactive towards SMC.
Similarly, a chloride at C2 in quinolines can sometimes react in preference to a non-activated bromide elsewhere in the heterocycle but not always. For example, 2,4-dichloro-8-bromo-7-methoxyquinoline reacts at C2 (55 → 56),67 but 2-chloro-6-bromoquinoline reacts at C2 (57 → 58) using Pd(PPh3)468 or at C6 (57 → 59) using Pd(dppf)Cl2,69 and 2-chloro-7-bromo-5-isopropylquinoline reacts at C7 (60 → 61)70 (Scheme 11).
Another particularly finely balanced case is that of 2-(4-bromophenyl)-5-chloropyrazine (62).71 For this substrate, the pyrazine chloride at C2 is electronically activated but it undergoes SMC reactions in preference to the bromide only with certain pre-catalysts: Pd(Xantphos®)Cl2 gives high site-selectivity for the chloride (→ 63) but most other pre-catalysts and particularly Pd(Qphos)2 favour the bromide (→ 64, Scheme 12).71
 | ||
| Scheme 12 The site-selectivity for SMC reactions of 2-(4-bromophenyl)-5-chloropyrazine (62) are ligand-dependent: it undergoes SMC (a) at C2 with Pd(Xantphos®)Cl2 (→ 63) and (b) at C4′ with Pd(Qphos)2 (→ 64).61 | ||
The authors attempted to correlate this ligand-dependent divergence of behaviour with a suite of physicochemical parameters which characterise phosphines (e.g. Tolman cone angle) but without success, perhaps implicating a change in ligation state as being responsible, as discussed above. However, the nature of the nucleophile, the base, additives (e.g. halide salts), and the solvent can also influence the energetics of OA.21,33,47b,72
The foregoing discussion illustrates how the tactic of deploying different halogens to control site-selectivity in SMC reactions is often an effective strategy, but that the expected order of reactivity based on average C–Hal BDEs can be subverted for heteroarenes with strong intrinsic electronic bias and so allowance for this should be made in synthetic planning.
The foregoing survey of factors that control the site-selectivity of SMC reactions of heteroaryl halides can be summarised as:
• For substrates containing two or more of the same halide: selectivity is primarily controlled by the intrinsic relative electrophilicities of the different ring carbons but this can be tempered by the electronic (and to lesser extent steric) influence of ring substituents.
• For substrates containing more than one kind of halide: selectivity can be controlled by the nature of the halide but the intrinsic relative electrophilicities of different ring-carbons can subvert this order in strongly polarised systems.
In both scenarios, the influence of the reaction conditions and particularly the nature of the Pd pre-catalyst/ligand can be decisive but this is generally only observed when using significantly more sterically hindered and/or electron-rich phosphines (e.g. QPhos, P(t-Bu)3, amphos, dtbpf) than the ‘standard’ phosphines employed for most SMC reactions (e.g. PPh3, dppf).14 These differences likely often reflect the ligation state of the Pd as these ‘non-standard’ ligands are prone to adopting low-coordination complexes and ligation state is an important factor in determining the ease of OA.
These features are consistent with and reinforced by the data we retrieved from our database searches which are summarised below (Section 4).
4. Key heteroarene ring-systems on a case-by-case basis
In this section we summarise on a heterocycle-by-heterocycle basis the results of a series of searches of the Pfizer RKB and CAS Scifinder® reaction databases as detailed in Section 2. For each ring type, a brief summary of published site-selectivity trends for the otherwise unsubstituted (i.e. unbiased ‘parent’) core molecule having two (or more) of the same halide substituents is presented. Preferred site-selectivity inferences based on published substituted cases are only mentioned if data on unsubstituted cases have not been published.73 Subsequent discussion of substituted derivatives is restricted to cases where substituents and/or conditions apparently induce a change in the intrinsic selectivity and to cases where a single substituent dictates the site-selectivity of systems for which the parent is symmetrical.74 Cases when the symmetry of the parent system make site-selectivity redundant (in the absence of additional substituents) are enclosed in hatched boxes; the number indicated below each of these structures indicates the number of reactions of this type found. Examples of selectivity in these reactions which arise from substituent effects are discussed as are some selected reactions which fall outside the scope of the searches but where intrinsic site-selectivities have been reversed by deploying two different halides.As in Section 3, the data is depicted in the Schemes and Figures such that the halide highlighted with a blue disk is the preferred site of reaction (or yellow if there is no actual data but the site is predicted on the basis of expected ring C electrophilicity) and, where relevant, the numbers below indicate the number of hits conforming to that selectivity and, in parenthesis, the number of exceptions. The hits from the Pfizer RKB and the CAS Scifinder® searches are separated and reported in blue and black text respectively. The figures in this section are reproduced the in ESI† with footnotes added giving further details of the hits retrieved (Fig. 3S–11S†).
4a. Pyridines, pyridazines, pyrimidines & pyrazines
Our data, which incorporate additionally substituted cases, corroborate these trends (Fig. 3).
 | ||
| Fig. 3 Coupling outcomes for pyridines. | ||
The greater electrophilicities of the C2 and C4 positions relative to C3 is expected from simple resonance analysis of the intrinsic polarisation of the pyridine ring-system. The retrieved exceptions have either no yield or evidence for assignment or are minor isomers (>17% yield). Ligand dependent selectivity for coupling 2,4-dichloropyridine with phenyl boronic acid at C4 over C2 (2.4:1) can be achieved albeit with a modest yield of 36% with Pd(OAc)2/Q-Phos/KF/toluene–H2O.39 Moreover, the C4 coupled product predominates when coupling methyl-4,6-dichloropyridine-2-carboxylate with a biaryl pinnacolato boronate ester using Pd(dppf)Cl2/TBAF/THF (28% yield, cf. 23% at C2),83 and when coupling 3-cyano-2,4-dichloropyridine with 4-aminophenyl pinnacolato boronate using PdCl2(dppf)/Na2CO3/DME–H2O (no yield given but C4:C2 ratio ∼2:1).84 No useful selectivity for SMC at C4 over C3/C5 could be achieved when using symmetrical 2,6-diaryl-3,4,5-trichloropyridine substrates.79
Inversion of the intrinsic selectivity trends can be engineered by deploying mixed halide substrates in which a halide more susceptible to OA is placed at the intrinsically less reactive position,12e.g. 2-bromo-3-iodopyridine reacts at C3 (41 → 65)60b,85 and 2-chloro-3,4-diiodopyridine reacts at C4 then C3 then C2 (66 → 67)86 (Scheme 13).
 | ||
| Scheme 13 (a) 2-Bromo-3-iodopyridine undergoes SMC reactions at C3 (41 → 65), and (b) 2-chloro-3,4-diiodopyridine reacts at C4 then C3 then C2 (66 → 67).60b,85,86 | ||
Symmetrical 2,6-75e,76f,77t,87 and 3,5-dibromopyridines76f,87d,88 can undergo efficient sequential SMC reactions. For unsymmetrical 2,6-dichloropyridines, an ester or amide group at C3, as discussed earlier (cf.Scheme 3, Section 3b), promotes reaction at C6 over C2 (5:1) using Pd(PPh3)4/K2CO3/THF but at C2 over C6 (2.5:1) using PdCl2(dppf)/K2CO3/MeOH. The behaviour of the PdCl2(dppf) was suggested to be as the result of chelation between the ester/amide carbonyl and the coordinatively unsaturated Pd(0).35a Similarly, a carboxylic acid group at C3 promotes reaction at C6 using Pd(OAc)2/PPh3/Na2CO3/MeOH35c [or Pd(Ph3)4/Na2CO3/1,4-dioxane–H2O]35b but at C2 using Pd2dba3·CHCl3/K2CO3/EtOH.35c A CF3 group at C3 of 2,6-dichloropyridine promotes reaction at C2 using Pd(OAc)2/K3PO4/DMF–H2O) (68 → 69);89 interestingly, this contrasts with the behaviour of the phenyl analogue, 2,4-dichloro-1-trifluoromethylbenzene, which couples at C4 under identical conditions (70 → 71)90 (Scheme 14).
 | ||
| Scheme 14 (a) A 3-CF3 group directs OA of 2,6-dichloropyridine to C2 (68 → 69),89 whereas (b) reaction occurs at C4 in the benzene analogue (70 → 71).90 | ||
For unsymmetrical 3,5-dibromopyridines, a pyridinium aminide (–N−N+C5H5),91 a methylamine,92 or a piperazine93 substituent at C2 promotes reaction at C3, presumably by coordination to Pd(0).
 | ||
| Fig. 4 Coupling outcomes for pyridazines, pyrimidines and pyrazines. | ||
An example from the Pfizer RKB in which C3 selectivity is observed for a SMC reaction of 3,5-dichloropyridazine (25) is shown below27a (Scheme 15).
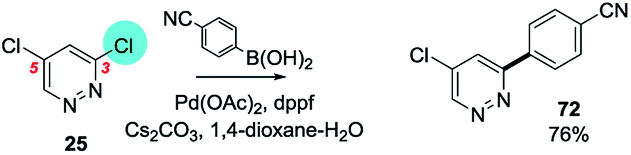 | ||
| Scheme 15 Example of SMC reaction of 3,5-dichloropyridazine at C3, (25 → 72) from Pfizer RKB.27a | ||
Similarly, 3,5-dichloropyridazine reacts with 2-fluoro-5-bromo-3-pyridine boronic acid using Pd(PPh3)4/Na2CO3/1,4-dioxane to give the C3 substituted product as the major isomer.94b Other cases for which C3 coupling has been observed include cases where 4-amino-3,5-dichloropyridazine95 reacts with 2-fluoro-4-trifluoromethylboronic acid using PdCl2(PPh3)2/Na2CO3/1,4-dioxane–H2O to give the C3 substituted product in 67% yield and where 6-methyl-3,5-dichloropyridazine96 reacts with a complex 4-substituted phenyl pinnacolato boronate using PdCl2(PPh3)2/Cs2CO3/1,4-dioxane to give the C3 coupled product as the major isomer. However, as discussed earlier (Scheme 5, Section 3c), site selectivity for SMC reactions on 3,5-dichloropyridazine are ligand-dependent. This was highlighted by Dai et al.39 who screened 20 ligands for its coupling with phenyl boronic acid: e.g. Pd(OAc)2/dppf/Cs2CO3/1,4-dioxane–H2O gave C3 selectivity whereas Pd(OAc)2/Q-Phos/KF/toluene–H2O gave C5 selectivity. The SMC reaction of 3,5-dibromopyridazine with a complex aryl boronic acid using Pd(PPh3)2C12/K3PO4/DMF also occurred selectively at C5,94a although caution should be associated with attributing these selectivity differences solely to the ligand given the concomitant changes in reaction conditions.
Mixed halide substrates can be employed to reverse the inherent bias of 3,5-dihalopyridazines for SMC reactions at C3: e.g. 3-chloro-5-bromo-6-phenylpyridazine, which reacts at C5 (73 → 74, Scheme 16).97
 | ||
| Scheme 16 3-Chloro-5-bromo-6-phenylpyridazine undergoes SMC reaction at C5 (73 → 74).97 | ||
5-Amino-3,4-dichloropyridazines react preferentially at C3 over C4 (84% combined yield, C3:C4 = 8:1) using Pd(PPh3)4/Na2CO3/toluene/EtOH/H2O.34a
Symmetrical 3,6-dibromo-98 and 3,6-dichloropyridazines99 can undergo efficient mono-SMC reactions; analogous reactions with symmetrical 4,5-dichloropyridazines are rare. SMC reactions of unsymmetrical 4-substituted-3,6-dichloropyridazines usually result in reaction predominantly at C6, i.e. distal to alkyl, aryl,34b–f carbamate and alkoxy groups,34a but for basic amine substituents at C4, reaction is promoted at C3 as discussed earlier (cf.Scheme 2, Section 3b).34a
2,4-Dihalopyrimidines which give anomalous selectivity include a case where 2,4-dibromopyrimidine reacts with 2,4-di-tert-butoxypyrimidine-5-boronic acid using Pd(PPh3)4/NaHCO3/DME to give the C2 substituted product in 58% yield.87b The other examples involve 2,4-dichloropyrimidines which additionally contain an amine substituent at C6.103 An alkyl,104 ether,105 thioether105 or amino106 substituent at C5 also appears to disfavour SMC reactions at C4, resulting in reaction at C2, presumably, mainly for steric reasons.
2,5-Dihalopyrimidines which give anomalous selectivity include a case where tetrachloropyrimidine reacts with 3-chloro-6-methoxyphenyl boronic acid using Pd(OAc)2/PPh3/K3PO4/MeCN–H2O to give the C5 substituted product.107 The other examples involve 2,5-dichloropyrimidines which additionally contain an amine108 substituent at C4 which appears to promote SMC reaction at C5. Inversion of the intrinsic selectivity trends can be engineered by deploying mixed halide substrates, e.g. 5-bromo-2-chloropyrimidine which reacts at C5 (75 → 76, Scheme 17).100g
 | ||
| Scheme 17 5-Bromo-2-chloropyrimidine undergoes SMC at C5 (75 → 76).100g | ||
All 4,6-dihalopyrimidines are symmetric and can undergo efficient mono-SMC reactions under appropriate conditions109 and 2,4,5,6-tetrachloropyrimidines also react selectively at C4/6.82,110
 | ||
| Scheme 18 A 3-OMe group directs OA of 2,5-dibromopyrazine to C2 (77 → 78).113 | ||
2,3-Dichloropyrazine itself can undergo efficient mono-SMC reactions.114 Only two unsymmetrical variants were retrieved, one with a C5 (ref. 115) amino substituent and the other with a C6 (ref. 116) substituent; both gave SMC coupling at C3. Parent 2,6-dibromo-117 and 2,6-dichloropyrazines118 can undergo efficient mono-SMC reactions. 3-Amino-119 and 3-pyridinium aminide (–N−N+C5H5)91,120 substituted 2,6-dibromopyrazines couple at C2 whereas interestingly 3-imide-substituted 2,6-dibromopyrazines couple at C6, albeit in low yields.121 Similarly, 3-acetyl-, 3-cyano- and 3-formyl-2,6-dichloropyrazines couple at C6.122
4b. Pyrroles, furans, thiophenes, imidazoles, pyrazoles, (is)oxazoles & (iso)thiazoles
 | ||
| Fig. 5 Coupling outcomes for pyrroles, furans and thiophenes. | ||
No SMC reactions displaying anomalous selectivity were retrieved; it appears that the presence of various additional substituents does not overcome the inherent bias of the pyrrole ring system. Symmetrical 3,4-dihalopyrroles,125 2,3,4,5-tetrabromopyrroles124 and to a lesser extent 2,5-dihalopyrroles126 can undergo efficient mono-SMC reactions. Unsymmetrical cases include N-methyl-2-cyano-,127 2-methoxycarbonyl-128 and N-methoxycarbonyl-3,4-dibromopyrrole-2-methyl ester (79)128 reacting at the proximal C3 position (→ 80, Scheme 19).128
 | ||
| Scheme 19 A methyl ester at C2 directs OA of 3,4-dibromopyrrole to C3 (79 → 80).128 | ||
Additional unsymmetrical cases include the aforementioned N-methyl-2,5-dibromopyrroles with an additional bromine substituent at C3 which undergo SMC reactions at the distal C5 position,123 and an N-methyl-2,5-dichloro-3-amidopyrrole which also reacts at C5.129
 | ||
| Scheme 20 A 3-ethoxycarbonyl group directs OA of 3,4-dibromofuran to C5 (81 → 82).133 | ||
 | ||
| Scheme 21 A 3-alkyl group directs OA of 2,5-dibromothiophene to C5 (83 → 84).138p | ||
Cases of unsymmetrical 3,4-dibromothiophenes undergoing selective SMC reactions include ones with 2-aryl substituents which couple at C4;136 these substrates are often intermediates in sequential bis-SMC reactions of 2,3,4-tribromothiophenes. 2,5-Diaryl-3,4-dibromothiophenes appear to couple distal to most sterically demanding aryl group with good selectivity.140a 2-Formyl-3,4-dibromothiophenes couple at C3.145
 | ||
| Fig. 6 Coupling outcomes for imidazoles, pyrazoles, (is)oxazoles and (iso)thiazoles. | ||
The retrieved exceptions include the case of N-methyl-2,5-dibromoimidazole for which SMC reaction at C5 was favoured when using Pd(OAc)2 with either XPhos, 1,3,5-triaza-7-phospha-adamantane, dppf or tris(4-trifluoromethylphenyl)phosphine but at C2 (i.e. as ‘normal’) when using Pd(OAc)2/Xantphos®.25 The reason for the anomalous behaviour with these particular ligands is not apparent. One patent also reports a 2-amino- and a 2-aryl-4,5-dibromo-N-SEM-imidazole undergoing SMC coupling at C4 (i.e. proximal to the pyridyl nitrogen) when using Pd(PPh3)4/Na2CO3/DME–H2O,148 but the SEM group is also present in two cases149 where normal C5 selectivity is observed so this does not appear to be the critical factor in the site-selectivity.
4c. (Iso)quinolines & benzodiazines
 | ||
| Fig. 7 Coupling outcomes for quinolines. | ||
No exceptions were retrieved although inversion of the intrinsic selectivity trends can be engineered by deploying mixed halide substrates, e.g. 2-bromo-4-iodoquinoline which reacts at C4 (85 → 86)169 and 3,4-dichloro-7-bromoquinoline which reacts at C7 (87 → 88)170 (Scheme 22).

 | ||
| Fig. 8 Coupling outcomes for isoquinolines. | ||
That SMC reactions are favoured in the annelated benzo-ring over the pyridyl ring holds also for 4,7-dibromo-1-chloroisoquinoline (89 → 90, Scheme 23).173b
 | ||
| Scheme 23 4,7-Dibromo-1-chloroisoquinoline undergoes SMC at C7 (89 → 90).173b | ||
 | ||
| Fig. 9 Coupling outcomes for benzodiazines. | ||
Symmetrical 1,4-dichlorophthalazines can undergo mono-SMC reactions176 but unsymmetrical derivatives do not appear to have been investigated.
 | ||
| Scheme 24 6-Bromo-2,4-dichloroquinazoline undergoes SMC at C4 (91 → 92).181 | ||
4d. Indoles, benzofurans, benzothiophenes, benzodiazoles, benz(is)oxazoles & benz(is)othiazoles
 | ||
| Fig. 10 Coupling outcomes for indoles, benzoxazoles, benzothiazoles and benzodiazoles. | ||
 | ||
| Scheme 25 A 3,5,7-trichlorobenzothiophene undergoes SMC at C3 (93 → 94).194 | ||
 | ||
| Scheme 26 6-Bromo-3-iodo-1-H-indazole undergoes SMC at C3 (95 → 96).195 | ||
4e. Aza(iso)quinolones (naphthyridines)
 | ||
| Fig. 11 Coupling outcomes for azaquinolines and azaisoquinolines. | ||
Our data show that 2,5-dichloro-6-azaquinoline ([1,6]-naphthyridine) undergoes SMC reactions at C2204 and 4,7-dichloro-6-azaquinoline reacts at C4. 7-Carbethoxy- and 7-carboxamido-2,8-dichloro-5-azaquinolines ([1,5]-naphthyridines) undergo SMC reactions at C2.205 3,7-Dibromo-5-azaquinoline ([1,5]-naphthyridine, 97) is a symmetrical molecule and has been reported to undergo mono-SMC reactions with a range of aryl pinnacolato boronates (→ 98, Scheme 27).69
 | ||
| Scheme 27 A SMC reaction of 3,7-dibromo-5-azaquinoline with pinacolatoborane (97 → 98).69 | ||
5. Conclusions
Given the complexity of the catalytic cycle involved in SMC reactions and the myriad of disparate Pd pre-catalysts, ligands, solvents and bases employed in these processes, it is not surprising that no absolutely rigid site-selectivity rules can be provided to predict the outcome of these reactions on heteroaryl polyhalides. As we have seen, experimental parameters can critically impact on the dominant catalytic species in solution and its ability to undergo the site-selectivity-determining OA step. However notwithstanding this, it is clear from the analysis presented in this review that for the majority of SMC reactions on this substrate class, particularly when using ‘standard’ conditions, the preferred site of reaction can be predicted with some confidence by paying attention to the nature of the halides present, the intrinsic relative electrophilicities of different ring-carbons (particularly for strongly polarised systems), and the electronic (and to a lesser extent steric) influence of substituents. We hope that by drawing together published data pertaining to this and summarising additional data mined from the Pfizer global chemistry RKB and the CAS Scifinder® databases, we have contributed to making the design of synthetic strategies for the construction of molecules containing polysubstituted heteroaryl motifs, for whatever purpose but especially in the context of pharmaceutical drug discovery, easier and more reliable.Acknowledgements
We thank Pfizer Worldwide Medicinal Chemistry for generous support of this work.Notes and references
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed
.
-
A. F. Pozharskii, A. T. Soldatenkov and A. R. Katritsky, Heterocycles in Life and Society - An Introduction to Heterocyclic Chemistry and Biochemistry and the Role of Heterocycles in Science, Technology, Medicine and Agriculture, Wiley, Chichester, 1997 Search PubMed
-
(a)
A. Kantak and B. DeBoef, in Cross-Coupling and Heck-Type Reactions 3: Metal Catalyzed Heck-Type Reactions and C-C Cross Coupling via C-H Activation, ed. M. Larhed, Georg Thieme Verlag, Stuttgart-New York, 2012, vol. 3, pp. 585–641 Search PubMed
-
(a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS
- In this context, a ‘pseudo-halogen’ is a functional group capable of undergoing oxidative addition (OA) with Pd(0) (e.g. a triflate). In this article the term ‘halide’ implicitly encompasses pseudo-halides.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed
- S. Schröter, C. Stock and T. Bach, Tetrahedron, 2005, 61, 2245–2267 CrossRef
- M. Schnürch, R. Flasik, A. F. Khan, M. Spina, M. D. Mihovilovic and P. Stanetty, Eur. J. Org. Chem., 2006, 3283–3307 CrossRef
- S. T. Handy and Y. Zhang, Chem. Commun., 2006, 299–301 RSC
- I. J. S. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036–1045 RSC
- J.-R. Wang and K. Manabe, Synthesis, 2009, 1405–1427 Search PubMed
- R. Rossi, F. Bellina and M. Lessi, Adv. Synth. Catal., 2012, 354, 1181–1255 CrossRef CAS
- Z. Hassan, T. Patonay and P. Langer, Synlett, 2013, 412–423 CAS
- The Pfizer RKB accesses both individual reaction (CeN) and library (PMC) datasets (>2.8 million reactions, 1993 onwards) which are rich in reactions on heterocyclic systems of medicinal interest often attempted in the first instance using standard conditions: Pd(dppf)Cl2 or Pd(PPh3)4 with Na2CO3 or NaHCO3 or K2CO3 in DME-H2O or THF-H2O or 1,4-dioxane-H2O.
- http://www.cas.org/products/scifinder, >60 million reactions, 1840 onwards.
- Deploying different boron derivatives and/or controlling the speciation/ligation state of boronic esters and consequent transmetallation reaction rates can be used to effect regioselective sequential SMC reactions but this strategy lies outside the scope of our survey. For details, see e.g. the reviews of Lloyd-Jones (ref. 17a and d) and Watson (ref. 17b). Using this approach, Watson achieved an elegant one-pot sequential SMC reaction of 2,4-di-chloropyrimidine at C4 then C2 using MIDA and Pin aryl boronates, see: ref. 17c.
-
(a) A. J. J. Lennox and G. C. Lloyd-Jones, Angew. Chem., Int. Ed., 2013, 52, 7362–7370 CrossRef CAS PubMed
- S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4544–4568 CrossRef CAS
-
B. W. Glasspoole, E. C. Keske and C. M. Crudden, in New Trends in Cross-Coupling New Trends in Cross-Coupling: Theory and Applications, ed. T. Colacot, The Royal Society of Chemistry, 2014, pp. 521–550 Search PubMed
- F. S. Han, Chem. Soc. Rev., 2013, 42, 5270–5298 RSC
- C. Amatore, G. Le Duc and A. Jutand, Chem.–Eur. J., 2013, 19, 10082–10093 CrossRef CAS PubMed
-
(a) L.-C. Liang, P.-S. Chien and M.-H. Huang, Organometallics, 2005, 24, 353–357 CrossRef CAS
-
(a) A. H. Roy and J. F. Hartwig, J. Am. Chem. Soc., 2001, 123, 1232–1233 CrossRef CAS PubMed
- J.-F. Fauvarque, F. Pflüger and M. Troupel, J. Organomet. Chem., 1981, 208, 419–427 CrossRef CAS
- N. A. Strotman, H. R. Chobanian, J. He, Y. Guo, P. G. Dormer, C. M. Jones and J. E. Steves, J. Org. Chem., 2010, 75, 1733–1739 CrossRef CAS PubMed
- I. J. S. Fairlamb, C. T. O'Brien, Z. Lin and K. C. Lam, Org. Biomol. Chem., 2006, 4, 1213–1216 CAS
-
(a) C. Y. Legault, Y. Garcia, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 12664–12665 CrossRef CAS PubMed
- Y. Zhang and S. T. Handy, Open Org. Chem. J., 2008, 2, 58–64 CrossRef CAS
- Y. Garcia, F. Schoenebeck, C. Y. Legault, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2009, 131, 6632–6639 CrossRef CAS PubMed
- Y. Feng, L. Liu, J.-T. Wang, H. Huang and Q.-X. Guo, J. Chem. Inf. Comput. Sci., 2003, 43, 2005–2013 CrossRef CAS PubMed
- K. C. Lam, T. B. Marder and Z. Lin, Organometallics, 2007, 26, 758–760 CrossRef CAS
- The lower rates of OA of alkyl cf. aryl halides has been attributed to the absence of the requisite π* orbital in the former; see refs. 27a and 31.
- W. A. Herrebout, N. Nagels, S. Verbeeck, B. J. van der Veken and B. U. W. Maes, Eur. J. Org. Chem., 2010, 3152–3158 CrossRef CAS
-
(a) E. Blaise, A. E. Kümmerle, H. Hammoud, J. X. de Araújo-Júnior, F. Bihel, J.-J. Bourguignon and M. Schmitt, J. Org. Chem., 2014, 79, 10311–10322 CrossRef CAS PubMed
-
(a) W. Yang, Y. Wang and J. R. Corte, Org. Lett., 2003, 5, 3131–3134 CrossRef CAS PubMed
- G. Y. Li, J. Org. Chem., 2002, 67, 3643–3650 CrossRef CAS PubMed
- M. Nawaz, M. F. Ibad, O.-U.-R. Abid, R. A. Khera, A. Villinger and P. Langer, Synlett, 2010, 150–152 CAS
- O.-u.-R. Abid, M. F. Ibad, M. Nawaz, A. Ali, M. Sher, N. H. Rama, A. Villinger and P. Langer, Tetrahedron Lett., 2010, 51, 1541–1544 CrossRef CAS
- X. Dai, Y. Chen, S. Garrell, H. Liu, L.-K. Zhang, A. Palani, G. Hughes and R. Nargund, J. Org. Chem., 2013, 78, 7758–7763 CrossRef CAS PubMed
- T. Kamikawa and T. Hayashi, Tetrahedron Lett., 1997, 38, 7087–7090 CrossRef CAS
- The divergent outcome of SMC reactions relative to Stille, Kumada and Negishi reactions for triflate-containing substrates has been attributed to the involvement of neutral vs. anionic Pd complexes respectively, see discussion of triflate reactions in the ESI†.
- A. F. Littke, C. Dai and G. C. Fu, J. Am. Chem. Soc., 2000, 122, 4020–4028 CrossRef CAS
- F. Schoenebeck and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 2496–2497 CrossRef CAS PubMed
- F. Proutiere and F. Schoenebeck, Angew. Chem., Int. Ed., 2011, 50, 8192–8195 CrossRef CAS PubMed
- U. Christmann and R. Vilar, Angew. Chem., Int. Ed., 2005, 44, 366–374 CrossRef CAS PubMed
- A comprehensive account of the evolution of our understanding of the various Pd(0) species that can participate in OA is beyond the scope of this article. The following key papers and references therein are should be consulted for orientation: PdL2: ref. 46a, [PdL2X]-: ref. 46b, PdL: ref. 44 and 46c–e; [PdLX]-: ref. 46f. In general, whether the active OA species is PdL or PdL2 is dependent on the naure of the ligand, the solvent polarity etc.: as a rule of thumb, bulky monodentate alkyl phosphines are likely to promote OA via PdL, whereas less bulky and bidentate phosphines favour OA via PdL2 (see: ref. 46g).
-
(a) C. Amatore and F. Pfluger, Organometallics, 1990, 9, 2276–2282 CrossRef CAS
-
(a) E. Lyngvi, I. A. Sanhueza and F. Schoenebeck, Organometallics, 2015, 34, 805–812 CrossRef CAS
- F. Proutiere and F. Schoenebeck, Synlett, 2012, 645–648 CAS
- F. Proutiere, M. Aufiero and F. Schoenebeck, J. Am. Chem. Soc., 2012, 134, 606–612 CrossRef CAS PubMed
-
(a) K. J. Bonney, F. Proutiere and F. Schoenebeck, Chem. Sci., 2013, 4, 4434–4439 RSC
- M. Aufiero, T. Scattolin, F. Proutière and F. Schoenebeck, Organometallics, 2015, 34, 5191–5195 CrossRef CAS
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS
- F. Proutiere, E. Lyngvi, M. Aufiero, I. A. Sanhueza and F. Schoenebeck, Organometallics, 2014, 33, 6879–6884 CrossRef CAS
- Z. L. Niemeyer, A. Milo, D. P. Hickey and M. S. Sigman, Nat. Chem., 2016, 8, 610–617 CrossRef CAS PubMed
- P. J. Ellis, I. J. S. Fairlamb, S. F. J. Hackett, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2010, 49, 1820–1824 CrossRef CAS PubMed
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS
- R. K. Arvela, N. E. Leadbeater, M. S. Sangi, V. A. Williams, P. Granados and R. D. Singer, J. Org. Chem., 2005, 70, 161–168 CrossRef CAS PubMed
- P. Fitton and E. A. Rick, J. Organomet. Chem., 1971, 28, 287–291 CrossRef CAS
-
(a) Z. Chamas, E. Marchi, A. Modelli, Y. Fort, P. Ceroni and V. Mamane, Eur. J. Org. Chem., 2013, 2316–2324 CrossRef
- T. D. Tran, F. Wakenhut, C. Pickford, S. Shaw, M. Westby, C. Smith-Burchnell, L. Watson, M. Paradowski, J. Milbank, R. A. Brimage, R. Halstead, R. Glen, C. P. Wilson, F. Adam, D. Hay, J.-Y. Chiva, C. Nichols, D. C. Blakemore, I. Gardner, S. Dayal, A. Pike, R. Webster and D. C. Pryde, ChemMedChem, 2014, 9, 1378–1386 CrossRef CAS PubMed
-
N. Bifulco, N. Brooijmans, B. L. Hodous, J. L. Kim and C. V. Miduturu, WO2014011900 A2, 2014
-
M. Kitade, S. Ohkubo and S. Yamashita, WO2012093708 A1, 2012
-
M. Weiss, E. F. Dimauro, T. Dineen, R. Graceffa, A. Guzman-Perez, H. Huang, C. Kreiman, I. E. Marx, H. N. Nguyen and E. Peterson, WO2014201173 A1, 2014
-
J. de Vicente Fidalgo, J. Li, R. C. Schoenfeld, F. X. Talamas and J. P. G. Taygerly, US20100311760 A1, 2010
- C. G. Barber, R. P. Dickinson and P. V. Fish, Bioorg. Med. Chem. Lett., 2004, 14, 3227–3230 CrossRef CAS PubMed
-
N. D. Patel, C. H. Senanayake, W. Tang, X. Wei and N. K. Yee, WO 2010129451 A1, 2010
-
Y. L. Qiu, C. Wang, X. Peng, H. Cao, L. Ying, X. Gao, B. Wang and Y. S. Or, WO2013052369 A1, 2013
-
J. B. J. Milbank, D. C. Pryde and T. D. Tran, WO2011004276 A1, 2011
- E. Saugues, A.-L. Debaud, F. Anizon, N. Bonnefoy and P. Moreau, Eur. J. Med. Chem., 2012, 57, 112–125 CrossRef CAS PubMed
- C. P. Ashcroft, S. J. Fussell and K. Wilford, Tetrahedron Lett., 2013, 54, 4529–4532 CrossRef CAS
-
(a) G. Audran, P. Brémond, S. R. A. Marque, D. Siri and M. Santelli, Tetrahedron, 2014, 70, 2272–2279 CrossRef CAS
- Generally, in these introductions, patents are only cited if there are no journal articles relating to that ring system.
- The reviews of Bach (ref. 7) and Rossi (ref. 12) should be consulted for discussion of other SMC reactions involving substituted substrates which correspond to some of the reactions in our numerical data and which conform to the intrinsic selectivity trends indicated in our figures.
-
(a) F. F. Wagner and D. L. Comins, Org. Lett., 2006, 8, 3549–3552 CrossRef CAS PubMed
-
(a) H. Benmansour, R. D. Chambers, P. R. Hoskin and G. Sandford, J. Fluorine Chem., 2001, 112, 133–137 CrossRef CAS
-
(a) S. Vice, T. Bara, A. Bauer, C. A. Evans, J. Ford, H. Josien, S. McCombie, M. Miller, D. Nazareno, A. Palani and J. Tagat, J. Org. Chem., 2001, 66, 2487–2492 CrossRef CAS PubMed
- H. Hu, C. Ge, A. Zhang and L. Ding, Molecules, 2009, 14, 3153–3160 CrossRef CAS PubMed
- S. Reimann, P. Ehlers, A. Petrosyan, S. Kohse, A. Spannenberg, A. E. Surkus, T. V. Ghochikyan, A. S. Saghyan, S. Lochbrunner, O. Kühn, R. Ludwig and P. Langer, Adv. Synth. Catal., 2014, 356, 1987–2008 CrossRef CAS
- P. Ehlers, S. Reimann, S. Erfle, A. Villinger and P. Langer, Synlett, 2010, 1528–1532 CAS
- S. Reimann, S. Parpart, P. Ehlers, M. Sharif, A. Spannenberg and P. Langer, Org. Biomol. Chem., 2015, 13, 6832–6838 CAS
- J. Liu, A. E. Fitzgerald and N. S. Mani, J. Org. Chem., 2008, 73, 2951–2954 CrossRef CAS PubMed
-
C. Ni, B. Shao, L. Tafesse, J. Yao, J. Yu and X. Zhou, WO2012035421 A3, 2012
- Y. Dai, K. Hartandi, N. B. Soni, L. J. Pease, D. R. Reuter, A. M. Olson, D. J. Osterling, S. Z. Doktor, D. H. Albert, J. J. Bouska, K. B. Glaser, P. A. Marcotte, K. D. Stewart, S. K. Davidsen and M. R. Michaelides, Bioorg. Med. Chem. Lett., 2008, 18, 386–390 CrossRef CAS PubMed
- D. Simoni, G. Grisolia, G. Giannini, M. Roberti, R. Rondanin, L. Piccagli, R. Baruchello, M. Rossi, R. Romagnoli, F. P. Invidiata, S. Grimaudo, M. K. Jung, E. Hamel, N. Gebbia, L. Crosta, V. Abbadessa, A. Di Cristina, L. Dusonchet, M. Meli and M. Tolomeo, J. Med. Chem., 2005, 48, 723–736 CrossRef CAS PubMed
- L. M. Daykin, J. S. Siddle, A. L. Ankers, A. S. Batsanov and M. R. Bryce, Tetrahedron, 2010, 66, 668–675 CrossRef CAS
-
(a) C. R. Woods, M. Benaglia, S. Toyota, K. Hardcastle and J. S. Siegel, Angew. Chem., Int. Ed., 2001, 40, 749–751 CrossRef CAS
- J. N. Cumming, E. M. Smith, L. Wang, J. Misiaszek, J. Durkin, J. Pan, U. Iserloh, Y. Wu, Z. Zhu, C. Strickland, J. Voigt, X. Chen, M. E. Kennedy, R. Kuvelkar, L. A. Hyde, K. Cox, L. Favreau, M. F. Czarniecki, W. J. Greenlee, B. A. McKittrick, E. M. Parker and A. W. Stamford, Bioorg. Med. Chem. Lett., 2012, 22, 2444–2449 CrossRef CAS PubMed
- S. Ahmed, M. Sharif, K. Shoaib, S. Reimann, J. Iqbal, T. Patonay, A. Spannenberg and P. Langer, Tetrahedron Lett., 2013, 54, 1669–1672 CrossRef CAS
- I. Ali, B. Siyo, Z. Hassan, I. Malik, I. Ullah, A. Ali, M. Nawaz, J. Iqbal, T. Patonay, A. Villinger and P. Langer, J. Fluorine Chem., 2013, 145, 18–34 CrossRef CAS
- M. J. Reyes, R. Castillo, M. L. Izquierdo and J. Alvarez-Builla, Tetrahedron Lett., 2006, 47, 6457–6460 CrossRef CAS
- S. Hilton, S. Naud, J. J. Caldwell, K. Boxall, S. Burns, V. E. Anderson, L. Antoni, C. E. Allen, L. H. Pearl, A. W. Oliver, G. Wynne Aherne, M. D. Garrett and I. Collins, Bioorg. Med. Chem., 2010, 18, 707–718 CrossRef CAS PubMed
- H. Hikawa and Y. Yokoyama, Tetrahedron, 2010, 66, 9552–9559 CrossRef CAS
-
(a)
L. Andrau, J. Atherall, L. C. Axford, M. Blair, I. Collins, L. Czaplewski, D. Davies, D. Haydon, C. J. Lunniss and D. Mitchell, WO2012045124 A1, 2012
-
A. Nardi, J. K. Christensen and D. Peters, WO2009112461 A1, 2009
-
D. A. Claremon, L. Zhuang, K. Leftheris, C. M. Tice, Z. Xu, Y. Ye, S. B. Singh, S. Cacatian, W. Zhao and F. Himmelsbach, WO2009134392 A1, 2009
- E. Sotelo and E. Raviña, Synlett, 2002, 0223–0226 CrossRef CAS
-
(a)
H. Strobel, P. Wohlfart, H. W. Kleemann, G. Zoller and D. W. Will, WO2008077507 A1, 2008
-
(a) S. Lin, Z. Liu and Y. Hu, J. Comb. Chem., 2007, 9, 742–744 CrossRef CAS PubMed
-
(a) S. Gronowitz, A.-B. Hornfeldt, V. Kristjansson and T. Musil, Chem. Scr., 1986, 26, 305–309 CAS
-
(a) J. M. Schomaker and T. J. Delia, J. Org. Chem., 2001, 66, 7125–7128 CrossRef CAS PubMed
- G. Hughes, C. Wang, A. S. Batsanov, M. Fern, S. Frank, M. R. Bryce, I. F. Perepichka, A. P. Monkman and B. P. Lyons, Org. Biomol. Chem., 2003, 1, 3069–3077 CAS
-
(a)
M. Pass, WO2006005918 A1, 2006
-
S. Asano, K. Kamimoto and Y. Isobe, WO2011152485 A1, 2011
-
L. J. Chen, P. Chen, H. Y. Hsu, Y. T. Huang, M. Y. Kuo, Y. S. Lee, Y. Y. Lu and P. K. Tsai, WO2011080568 A8, 2012
-
(a)
M. Burger and M. Lindvall, WO2010026121 A1, 2010
-
A. E. Fitzgerald, J. Liu and N. S. Mani, WO2009035668 A1, 2009
-
C. Akuche, L. D. Cantin, L. Chen, S. Choi, R. B. Clark, M. F. Hentemann, R. C. Lavoie, S. X. Liang, X. Ma and D. Majumdar, WO2004058174 A3, 2004
-
(a) N. Saygili, A. S. Batsanov and M. R. Bryce, Org. Biomol. Chem., 2004, 2, 852–857 RSC
- M. Hussain, N. T. Hung, R. A. Khera, I. Malik, D. S. Zinad and P. Langer, Adv. Synth. Catal., 2010, 352, 1429–1433 CrossRef CAS
- F. Turksoy, G. Hughes, A. S. Batsanov and M. R. Bryce, J. Mater. Chem., 2003, 13, 1554–1557 RSC
-
(a)
B. Corkey, E. Elzein, R. Jiang, R. Kalla, T. Kobayashi, D. Koltun, X. Li, G. Notte, E. Parkhill and T. Perry, US20110021521 A1, 2011
- C.-G. Yang, G. Liu and B. Jiang, J. Org. Chem., 2002, 67, 9392–9396 CrossRef CAS PubMed
-
(a) E. L. Kolychev, A. F. Asachenko, P. B. Dzhevakov, A. A. Bush, V. V. Shuntikov, V. N. Khrustalev and M. S. Nechaev, Dalton Trans., 2013, 42, 6859–6866 RSC
-
M. J. Arnost, J. Green, S. L. Harbeson and V. Savic, WO2003066629 A3, 2003
-
H. M. Armstrong, R. Beresis, J. L. Goulet, M. A. Holmes, X. Hong, S. G. Mills, W. H. Parsons, P. J. Sinclair, M. G. Steiner and F. Wong, WO2001000213 A1, 2001
-
R. Clark, B. A. Stearns, J. M. Scott, H. R. Coate, L. Zhao, T. J. Seiders, D. Volkots, J. M. Arruda, N. S. Stock and Y. P. Truong, WO2011041462 A3, 2011
- F. Buron, N. Plé, A. Turck and G. Queguiner, J. Org. Chem., 2005, 70, 2616–2621 CrossRef CAS PubMed
-
(a)
J. Hynes, L. G. V. De and H. Wu, WO2009155388 A1, 2009
- R. Castillo, M. J. Reyes, M. L. Izquierdo and J. Alvarez-Builla, Tetrahedron, 2008, 64, 1351–1370 CrossRef CAS
-
(a) I. Niculescu-Duvaz, E. Roman, S. R. Whittaker, F. Friedlos, R. Kirk, I. J. Scanlon, L. C. Davies, D. Niculescu-Duvaz, R. Marais and C. J. Springer, J. Med. Chem., 2008, 51, 3261–3274 CrossRef CAS PubMed
-
M. Nazaré, N. Halland, F. Schmidt, T. Weiss, U. Dietz and A. Hofmeister, WO2013041119 A1, 2013
- S.-M. T. Toguem, O. Fatunsin, A. Villinger and P. Langer, Tetrahedron Lett., 2011, 52, 3732–3735 CrossRef CAS
- T. T. Dang, R. Ahmad, T. T. Dang, H. Reinke and P. Langer, Tetrahedron Lett., 2008, 49, 1698–1700 CrossRef CAS
-
(a) T. Fukuda, E.-i. Sudo, K. Shimokawa and M. Iwao, Tetrahedron, 2008, 64, 328–338 CrossRef CAS
-
(a) F. Beaumard, P. Dauban and R. H. Dodd, Org. Lett., 2009, 11, 1801–1804 CrossRef CAS PubMed
-
M. Wennerstal, J. Lofstedt, X. Wu, L. Kruger and L. Hagberg, WO2011042477 A1, 2011
- S. T. Handy and Y. Zhang, Synthesis, 2006, 3883–3887 CrossRef CAS
-
R. W. Chau, C. A. Cullis, M. O. Duffey, K. E. Gipson, Y. Hu, G. Li, M. D. Sintchak, S. G. Stroud and T. J. Vos, WO2013096637 A1, 2013
-
(a) A. K. Takle, M. J. Bamford, S. Davies, R. P. Davis, D. K. Dean, A. Gaiba, E. A. Irving, F. D. King, A. Naylor, C. A. Parr, A. M. Ray, A. D. Reith, B. B. Smith, P. C. Staton, J. G. A. Steadman, T. O. Stean and D. M. Wilson, Bioorg. Med. Chem. Lett., 2008, 18, 4373–4376 CrossRef CAS PubMed
- M. Hussain, R. A. Khera, N. T. Hung and P. Langer, Org. Biomol. Chem., 2011, 9, 370–373 CAS
- S.-H. Son, Y. Abe, M. Yuasa, Y. Yamagishi, N. Sakai, T. Ayabe and K. Yamada, Chem. Lett., 2011, 40, 378–380 CrossRef CAS
-
C. A. Cullis, K. E. Granger, J. Guo, M. Hirose, G. Li, M. Mizutani and T. J. Vos, WO2012021615 A1, 2012
-
(a) R. Pereira, B. Iglesias and A. R. de Lera, Tetrahedron, 2001, 57, 7871–7881 CrossRef CAS
-
(a) M. Gallant, M. Belley, M.-C. Carrière, A. Chateauneuf, D. Denis, N. Lachance, S. Lamontagne, K. M. Metters, N. Sawyer, D. Slipetz, J. F. Truchon and M. Labelle, Bioorg. Med. Chem. Lett., 2003, 13, 3813–3816 CrossRef CAS PubMed
- S.-M. T. Toguem, A. Villinger and P. Langer, Synlett, 2010, 909–912 CAS
-
(a) S.-M. T. Toguem, A. Villinger and P. Langer, Synlett, 2009, 3311–3314 CAS
-
(a) P. A. Tempest and R. W. Armstrong, J. Am. Chem. Soc., 1997, 119, 7607–7608 CrossRef CAS
-
(a) M. Ankersen, B. Peschke, B. S. Hansen and T. K. Hansen, Bioorg. Med. Chem. Lett., 1997, 7, 1293–1298 CrossRef CAS
-
(a) A. Rahimi, J. C. Namyslo, M. H. H. Drafz, J. Halm, E. Hübner, M. Nieger, N. Rautzenberg and A. Schmidt, J. Org. Chem., 2011, 76, 7316–7325 CrossRef CAS PubMed
-
(a) T. T. Dang, N. Rasool, T. T. Dang, H. Reinke and P. Langer, Tetrahedron Lett., 2007, 48, 845–847 CrossRef CAS
-
(a)
J. P. Whitten, Y. Pei, J. Cao, Z. Wang and E. Rogers, US20110065724 A1, 2011
- P. Dallemagne, L. Pham Khanh, A. Alsaïdi, I. Varlet, V. Collot, M. Paillet, R. Bureau and S. Rault, Bioorg. Med. Chem., 2003, 11, 1161–1167 CrossRef CAS PubMed
- S. Jing, R. Zhang, H. Dai, C. Du and X. Cheng, Chin. J. Chem., 2012, 30, 577–584 CrossRef CAS
-
(a)
X. Sun, WO2011075478 A1, 2011
-
(a) I. Kawasaki, M. Yamashita and S. Ohta, Chem. Pharm. Bull., 1996, 44, 1831–1839 CrossRef
- J. Tan, Y. Chen, H. Li and N. Yasuda, J. Org. Chem., 2014, 79, 8871–8876 CrossRef CAS PubMed
-
Z. Huang, J. Jin, T. D. Machajewski, W. R. Antonios-Mccrea, M. Mckenna, D. Poon, P. A. Renhowe, M. Sendzik, C. M. Shafer and A. Smith, WO2009115572 A3, 2009
-
(a)
A. Abeywardane, B. Farmer, N. A. Farrow, D. A. Gao, A. Heim-Riether, L. L. S. Keenan, I. A. Mugge, S. J. Taylor, Z. Xiong and Y. Yu, WO2010045188 A1, 2010
-
(a) R. A. Khera, A. Ali, M. Hussain, J. Tatar, A. Villinger and P. Langer, Synlett, 2010, 1923–1926 CAS
- K. J. Hodgetts and M. T. Kershaw, Org. Lett., 2002, 4, 2905–2907 CrossRef CAS PubMed
- E. Ferrer Flegeau, M. E. Popkin and M. F. Greaney, J. Org. Chem., 2008, 73, 3303–3306 CrossRef PubMed
-
(a) P. Stanetty, M. Schnürch and M. D. Mihovilovic, J. Org. Chem., 2006, 71, 3754–3761 CrossRef CAS PubMed
-
(a) T. Tao, B.-B. Ma, Y.-X. Peng, X.-X. Wang, W. Huang and X.-Z. You, J. Org. Chem., 2013, 78, 8669–8679 CrossRef CAS PubMed
-
(a) H. Nakagawa, T. Nakashima and T. Kawai, Eur. J. Org. Chem., 2012, 4493–4500 CrossRef CAS
-
D. J. Pinto, J. R. Corte, P. J. Gilligan, T. Fang, I. L. M. Smith, Y. Wang, W. Yang and W. R. Ewing, WO2013022818 A1, 2013
-
(a) I. C. Christoforou, P. A. Koutentis and C. W. Rees, Org. Biomol. Chem., 2003, 1, 2900–2907 RSC
-
(a)
R. T. Backer, M. J. Fisher, S. P. Hollinshead, S. L. Kuklish, E. C. R. Smith and K. Takeuchi, WO2008103185 A3, 2008
- D. Csányi, G. Timári and G. Hajós, Synth. Commun., 1999, 29, 3959–3969 CrossRef
- T. Shiota and T. Yamamori, J. Org. Chem., 1999, 64, 453–457 CrossRef CAS
-
(a) A. Piala, D. Mayi and S. T. Handy, Tetrahedron, 2011, 67, 4147–4154 CrossRef CAS PubMed
- G. Zhou, D. Wu, B. Snyder, R. G. Ptak, H. Kaur and M. Gochin, J. Med. Chem., 2011, 54, 7220–7231 CrossRef CAS PubMed
-
(a) I. E. Nifant'ev, P. V. Ivchenko, V. V. Bagrov, S. M. Nagy, L. N. Winslow, J. A. Merrick-Mack, S. Mihan and A. V. Churakov, Dalton Trans., 2013, 42, 1501–1511 RSC
- F. X. Talamas, S. C. Abbot, S. Anand, K. A. Brameld, D. S. Carter, J. Chen, D. Davis, J. de Vicente, A. D. Fung, L. Gong, S. F. Harris, P. Inbar, S. S. Labadie, E. K. Lee, R. Lemoine, S. Le Pogam, V. Leveque, J. Li, J. McIntosh, I. Nájera, J. Park, A. Railkar, S. Rajyaguru, M. Sangi, R. C. Schoenfeld, L. R. Staben, Y. Tan, J. P. Taygerly, A. G. Villaseñor and P. E. Weller, J. Med. Chem., 2014, 57, 1914–1931 CrossRef CAS PubMed
-
S. Chang, D. P. Kang, J. K. Kim, S. W. Kim, S. Y. Ko, J. S. Lee, J. A. Park, S. B. Shim, H. C. Sung and Y. S. Yang, WO2007114672 A1, 2007
- I. P. Beletskaya, A. V. Tsvetkov, G. V. Latyshev and N. V. Lukashev, Russ. J. Org. Chem., 2003, 39, 1660–1667 CrossRef CAS
-
(a) D. Dubé, M. Blouin, C. Brideau, C.-C. Chan, S. Desmarais, D. Ethier, J.-P. Falgueyret, R. W. Friesen, M. Girard, Y. Girard, J. Guay, D. Riendeau, P. Tagari and R. N. Young, Bioorg. Med. Chem. Lett., 1998, 8, 1255–1260 CrossRef
-
(a) G. Srikanth and H. Machchhindra, J. Pharma Res., 2009, 2, 1448–1450 CAS
- D. L. Comins, J. M. Nolan and I. D. Bori, Tetrahedron Lett., 2005, 46, 6697–6699 CrossRef CAS
- R. Graham Robinett, A. J. Freemerman, M. A. Skinner, L. Shewchuk and K. Lackey, Bioorg. Med. Chem. Lett., 2007, 17, 5886–5893 CrossRef CAS PubMed
-
(a) J.-Y. Legros, G. Primault and J.-C. Fiaud, Tetrahedron, 2001, 57, 2507–2514 CrossRef CAS
- D.-W. Gao, Q. Gu and S.-L. You, ACS Catal., 2014, 4, 2741–2745 CrossRef CAS
-
(a) B. Herberich, G.-Q. Cao, P. P. Chakrabarti, J. R. Falsey, L. Pettus, R. M. Rzasa, A. B. Reed, A. Reichelt, K. Sham, M. Thaman, R. P. Wurz, S. Xu, D. Zhang, F. Hsieh, M. R. Lee, R. Syed, V. Li, D. Grosfeld, M. H. Plant, B. Henkle, L. Sherman, S. Middleton, L. M. Wong and A. S. Tasker, J. Med. Chem., 2008, 51, 6271–6279 CrossRef CAS PubMed
- C. Boezio, H. Bregman, J. R. Coats, E. F. Dimauro, T. Dineen, B. Du, R. Graceffa, C. Kreiman, D. La and I. E. Marx, WO2013086229 A1, 2013.
-
(a) L. H. Pettus, S. Xu, G.-Q. Cao, P. P. Chakrabarti, R. M. Rzasa, K. Sham, R. P. Wurz, D. Zhang, S. Middleton, B. Henkle, M. H. Plant, C. J. M. Saris, L. Sherman, L. M. Wong, D. A. Powers, Y. Tudor, V. Yu, M. R. Lee, R. Syed, F. Hsieh and A. S. Tasker, J. Med. Chem., 2008, 51, 6280–6292 CrossRef CAS PubMed
-
(a) M. A. J. Duncton, E. L. Piatnitski Chekler, R. Katoch-Rouse, D. Sherman, W. C. Wong, L. M. Smith Ii, J. K. Kawakami, A. S. Kiselyov, D. L. Milligan, C. Balagtas, Y. R. Hadari, Y. Wang, S. N. Patel, R. L. Rolster, J. R. Tonra, D. Surguladze, S. Mitelman, P. Kussie, P. Bohlen and J. F. Doody, Bioorg. Med. Chem., 2009, 17, 731–740 CrossRef CAS PubMed
- Q.-Y. Wang, S. J. Patel, E. Vangrevelinghe, H. Y. Xu, R. Rao, D. Jaber, W. Schul, F. Gu, O. Heudi, N. L. Ma, M. K. Poh, W. Y. Phong, T. H. Keller, E. Jacoby and S. G. Vasudevan, Antimicrob. Agents Chemother., 2009, 53, 1823–1831 CrossRef CAS PubMed
-
H. M. Eggenweiler, M. Wolf and H. P. Buchstaller, WO2006122631 A1, 2006
-
(a) P. Wipf and K. M. George, Synlett, 2010, 644–648 CrossRef CAS PubMed
-
J. M. Travins, R. C. Bernotas, J. E. Wrobel, D. H. Kaufman, B. Hu, J. W. Jetter, D. J. O'Neill and C. W. Mann, WO2010059627 A1, 2010
-
F. Stauffer and P. Furet, WO2008012326 A1, 2008
- I. Ali, B. Siyo, Y. Al-Soud, A. Villinger and P. Langer, Synthesis, 2012, 44, 1637–1646 CrossRef CAS
-
M. H. T. Bui, T. D. Cushing, L. De Turiso Felix Gonzalez, X. Hao and B. Lucas, WO2013152150 A1, 2013
-
Y. Chen, T. D. Cushing, J. A. Duquette, L. De Turiso Felix Gonzalez, X. Hao, X. He, B. Lucas, L. R. Mcgee, A. Reichelt and R. M. Rzasa, WO2008118468 A1, 2008
- L. Mao, H. Sakurai and T. Hirao, Synthesis, 2004, 2535–2539 CAS
-
(a) M. F. Ibad, M. Hussain, O.-U.-R. Abid, A. Ali, I. Ullah, D. S. Zinad and P. Langer, Synlett, 2010, 411–414 CAS
- Y. Liu and G. W. Gribble, Tetrahedron Lett., 2000, 41, 8717–8721 CrossRef CAS
- P. Li, Y. Ji, W. Chen, X. Zhang and L. Wang, RSC Adv., 2013, 3, 73–78 RSC
-
(a) N. T. Hung, M. Hussain, I. Malik, A. Villinger and P. Langer, Tetrahedron Lett., 2010, 51, 2420–2422 CrossRef
- A. L. Tsuhako, C. K. Marlowe, C. A. Zaharia, L. Kabigting, W. Bajjalieh, Z. Tesfai, P. Huang, K. Moon, N. Aay, A. Tambo-Ong, J. M. Nuss, W. Xu and P. Kearney, WO2012149528 A1, 2012.
- T. Bach and M. Bartels, Tetrahedron Lett., 2002, 43, 9125–9127 CrossRef CAS
-
(a) A. Heynderickx, A. Samat and R. Guglielmetti, Synthesis, 2002, 213–216 CrossRef CAS
- Y. Ji, P. Li, X. Zhang and L. Wang, Org. Biomol. Chem., 2013, 11, 4095–4101 CAS
- L. Li, M.-C. Mathieu, D. Denis, A. G. Therien and Z. Wang, Bioorg. Med. Chem. Lett., 2011, 21, 734–737 CrossRef CAS PubMed
-
(a) M. E. Fraley, J. T. Steen, E. J. Brnardic, K. L. Arrington, K. L. Spencer, B. A. Hanney, Y. Kim, G. D. Hartman, S. M. Stirdivant, B. A. Drakas, K. Rickert, E. S. Walsh, K. Hamilton, C. A. Buser, J. Hardwick, W. Tao, S. C. Beck, X. Mao, R. B. Lobell, L. Sepp-Lorenzino, Y. Yan, M. Ikuta, S. K. Munshi, L. C. Kuo and C. Kreatsoulas, Bioorg. Med. Chem. Lett., 2006, 16, 6049–6053 CrossRef CAS PubMed
-
(a) A. M. Hamdy, N. Eleya, H. H. Mohammed, T. Patonay, A. Spannenberg and P. Langer, Tetrahedron, 2013, 69, 2081–2086 CrossRef CAS
- A. G. Steinig, M. J. Mulvihill, J. Wang, D. S. Werner, Q. Weng, J. Kan, H. Coate and X. Chen, WO2009099982 A1, 2009.
- Y. Gravenfors, C. Jonasson, J. Malmstroem, G. Nordvall, D. Pyring, C. Slivo, D. Sohn, P. Stroem and D. Wensbo, WO2007086800 A1, 2007.
-
(a)
R. Narquizian, T. Woltering and W. Wostl, WO2012136603 A1, 2012
- B. Barlaam, P. Bernstein, C. Dantzman and P. Warwick, WO2002051821 A1, 2002.
-
(a)
J. L. Carr, M. D. Charles, C. H. Foxton, J. H. Gilliatt, T. R. Hammonds, N. S. Mistry, G. A. Pave and T. M. Raynham, WO2007125331 A3, 2008
-
C. Amendt, D. Dorsch, G. Hoelzemann, A. Jonczyk and F. Zenke, WO2012119690 A1, 2012
- I. Ali, Z. Hassan, M. Hein, A. Falodun, T. Patonay, A. Villinger and P. Langer, Synthesis, 2012, 44, 2255–2263 CrossRef CAS
-
S. An, WO2013185353 A1, 2013
- H. Cheng, C. Li, S. Bailey, S. M. Baxi, L. Goulet, L. Guo, J. Hoffman, Y. Jiang, T. O. Johnson, T. W. Johnson, D. R. Knighton, J. Li, K. K. C. Liu, Z. Liu, M. A. Marx, M. Walls, P. A. Wells, M.-J. Yin, J. Zhu and M. Zientek, ACS Med. Chem. Lett., 2013, 4, 91–97 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Notes on mixed halide–triflate heterocycles and details of the Pfizer RKB and CAS Scifinder® searches. See DOI: 10.1039/c6sc02118b |
| This journal is © The Royal Society of Chemistry 2017 |