
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtecting microRNAs from RNase degradation with steric DNA nanostructures†
H.
Qian
 ab,
C. Y.
Tay
acd,
M. I.
Setyawati
a,
S. L.
Chia
a,
D. S.
Lee
a and
D. T.
Leong
*a
ab,
C. Y.
Tay
acd,
M. I.
Setyawati
a,
S. L.
Chia
a,
D. S.
Lee
a and
D. T.
Leong
*a
aDepartment of Chemical and Biomolecular Engineering, National University of Singapore, 4 Engineering Drive 4, Singapore 117585, Singapore. E-mail: cheltwd@nus.edu.sg; Fax: +65 6779 1936; Tel: +65 6516 7262
bInstitute of Respiratory Diseases and Critical Care, Xinqiao Hospital of Third Military Medical University, 183 Xinqiao Street, Chongqing 400037, China
cSchool of Materials Science and Engineering, Nanyang Technological University, N4.1, Nanyang Avenue, Singapore 639798, Singapore
dSchool of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore
First published on 14th September 2016
Abstract
Tumor suppressive microRNAs are potent molecules that might cure cancer, one day. Despite the many advanced strategies for delivery of these microRNAs to the cell, there are few therapeutic microRNAs in clinical use. Progress in microRNA bioapplications is hindered by a high vulnerability of exogeneous microRNA molecules to RNase degradation that occurs in extra- and intracellular physiological conditions. In this proof-of-concept study, we use a programmable self-assembled DNA nanostructure bearing a “shuriken” shape to not only deliver but more importantly protect a tumor suppressive microRNA-145 for a sufficiently long time to exert its therapeutic effect in human colorectal cancer cells. Our DNA nanostructure harbored complementary sequences that can hybridize with the microRNA cargo. This brings the microRNA–DNA duplex very close to the core structure such that the microRNA cargo becomes sterically shielded from RNase's degradative activity. Our novel DNA nanostructure based protector concept removes the degradative bottleneck that may plague other nucleic acid delivery strategies and presents a new paradigm towards exploiting these microRNAs for anti-cancer therapy.
Introduction
Self-assembled DNA nanostructures hold great potential in the fields of photonics, bioimaging, nanomedicine and biosensing due to their programmable capability from one to three dimensions, their biocompatibility, the readiness of their uptake by cells and their relative stability inside cells. These capabilities have spawned many interesting strategies for designing and constructing structures out of DNA; however, there has not been a deluge of applications arising from these interesting structures. There are pockets of bioapplications of these fascinating structures as nanosized carriers of drugs,1–5 siRNA,6,7 peptides8,9 and in- and out-of-cell sensors.10–14 This window clearly presents an emerging area for the applications of DNA nanostructures in such fields.MicroRNA (miRNA) is a class of short RNAs that governs multiple intracellular and extracellular biofunctional processes.15,16 Amongst this very large family of miRNA, miRNA-145 (miR-145) stands out as a very important therapeutic molecule due to its inherent powerful tumor suppressive effect on cancer. It was reported to be down-regulated in most cancer cell lines and tissues17 such that reinstating miR-145 could almost immediately suppress tumor growth, and block cell invasion and metastasis.18 However, while the biological effect is strong, pragmatic issues of delivery, target specificity and successful protection against endogenous nucleases prevent any real progress towards its intended use as a clinically usable therapeutic, as with most miRNA driven anti-cancer therapeutics, which face similar issues. Commercially available specially designed transfection reagents for miRNAs and siRNAs may work well on cell cultured cells but these reagents are typically liposomes of micrometer sizes with a wide size distribution and earlier versions do exhibit some toxicity.19 Much effort has gone disproportionately into developing better and more advanced delivery strategies20–24 but this suffers from the law of diminishing returns. In reality, delivery is only the beginning of the journey that each exogeneous miRNA undergoes. Once in the intracellular environment, the exogeneous miRNAs are tagged as foreign and shuttled to the lysosomes25 where degradation takes place. This degradative process greatly diminishes the actual working concentration of the therapeutic miRNAs and immediately curtails the overall efficacy of the miRNAs, even those delivered at high efficiency. The irony is that ever-increasing delivery efficiency cannot solve the core stability problem. With high delivery efficiency, a high dose with non-specificity will negatively affect the non-targeted non-cancer cells. This lack of tools to protect exogenous miR-145 has prompted the imperfect solution of using plasmid or viral vectors to overexpress miR-145 in cancer cells; this strategy has limited real efficacy in clinical settings due to collateral damage to non-cancer cells, lower than 100% uptake by the target cancer cells and an overly complicated multi-stage process. Non-viral DNA nanostructures have instead shown some degree of success in the delivery of other molecular cargo like siRNA,6,7 aptamers,26–28 and proteins,29,30 into cells. While the mechanisms are unclear, DNA nanostructures showed the potential to protect their cargo11 in harsh degradative conditions. Coupled with their natural biocompatible characteristics, DNA nanostructures are well poised to be multi-scenario, programmable and powerful tools for miRNA delivery. We further reasoned that if we can protect the introduced miRNA for a sufficiently long time for its effect to be exerted, we might be able to remove this bottleneck holding back the broad clinical use of therapeutic miRNAs. We designed and assembled a DNA nanostructure bearing the shape of a “Shuriken”. We further reasoned that the shuriken's multi-pronged configuration would increase the residence time for robust uptake by cells without any transfection agents. More importantly, the hybridization of miRNA to the complementary DNA segment brings the miRNA to within less than 5 nm of the DNA crossover segment and the triad arms of the shuriken (Fig. 1a). The close proximity provided steric shielding from degradative RNase enzymes. In this paper, we not only showed this steric hindrance DNA-nanostructure conferred protection but also the rest of the expected chain of events from successful delivery to protection and finally effective inhibition of cancer growth of otherwise highly proliferative colorectal cancer cells in vitro and in three dimensional (3D) cell culture systems.
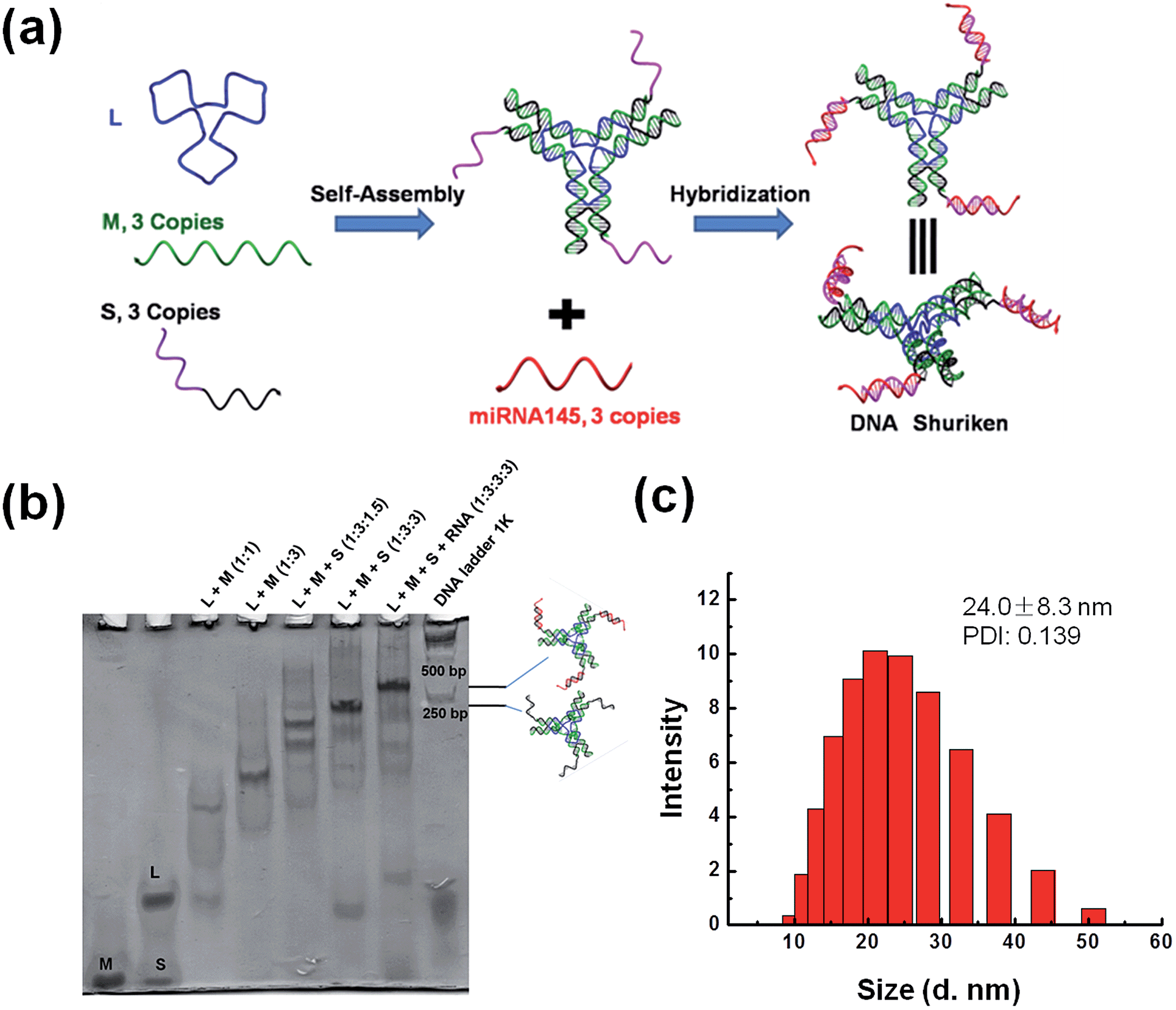 | ||
| Fig. 1 Design and characterization of the DNA shuriken. (a) A schematic illustration of the DNA shuriken design. The DNA shuriken is a DNA star motif carrying 3 miR-145 strands. (b) Native PAGE analysis of the DNA shuriken. (c) DLS characterization of the DNA shuriken. | ||
Results and discussion
The DNA shuriken was designed based on a DNA star motif (Fig. 1a).31,32 Briefly, the DNA star motif consisted of one long central strand L (blue), three copies of edge strands M (green) and three copies of peripheral strands S (black). Strand S contained a piece of a DNA sequence (17 bases) that could hybridize to miR-145 5p (23 bases) and protrude out as an overhang. The DNA star motifs then hybridized with three copies of miR-145 to form a DNA shuriken. A toehold 6 bases long is left on the end of the DNA and RNA hybrid duplex to prevent strand displacement. Each arm of the DNA star motif is approximately 10 nm long, while the length of the hybrid duplex overhang is approximately 6 nm. Note that the three arms of the DNA shuriken are not necessarily always in the same plane. The three arms and overhangs are deliberately designed to be very flexible to maximize the contact area and minimize the interaction enthalpy with the cell membrane to further drive its cellular uptake. The outcome of this design is exactly similar to the concept of a “ninja shuriken” rotating, striking a surface and embedding itself on the surface. This embedding theoretically increases the residence time for successful uptake.Native PAGE was utilized to characterize the formation of the DNA shuriken. As shown in Fig. 1b, both the DNA star motif and the DNA shuriken formed a sharp, dominant band with corresponding expected mobility differences in the gel. The band position of the DNA shuriken is higher than that of the DNA star motif because of the three identical miR-145 cargoes, and the size of the DNA shuriken (205 bp) is comparable to that of the DNA ladder. Dynamic light scattering results (Fig. 1c) further confirmed the formation of the DNA shuriken. The DNA shuriken has an average hydrodynamic diameter of 24.0 ± 8.3 nm, which is consistent with the theoretical value of the design (Fig. S1†).
DNA nanostructures have been proven to be readily taken up by cells33 without any transfection agents. In this study, we thoroughly investigated the cellular uptake of the DNA shuriken and the effectiveness of this delivery system. Fig. 2a(i and ii) shows the confocal fluorescence image and the cross-sectional view of the cells respectively treated with DNA shuriken for 2 hours. Internalization of the DNA shuriken particles tagged with Cy3 by DLD-1 cells was observed. We also observed that substantial naked miR-145 was internalized by the cells (Fig. 2a(iii and iv)), similar to another report.33 The time needed for cells to internalize sufficient amounts of DNA shuriken is very important when it comes to its therapeutic efficacy. It was reported that the cellular uptake of DNA origami structures with a size of dozens of nanometers requires more than 12 hours,3,34 while smaller DNA structures (<20 nm) need 2 to 6 hours.7,33 In our study, the uptake process is therefore comparatively very fast. Our microplate quantification shows that the relative fluorescence intensities reached a maximum of around three fold higher compared to that of the control within a brief 30 min incubation (Fig. S2†). The observed punctate staining of DNA shuriken–miR-145 (Fig. 2a(i and ii)) suggested endosomal localization of our constructs instead of random dispersion in the cytosol. Thus, we did a colocalization study of DNA shuriken and lysosomes with an incubation time of 2 hours. Most of the internalized DNA shuriken resided in the lysosomes, as indicated by the fluorescent images in Fig. 2b. Statistical analysis also showed that about 80% of the DNA shuriken particles were trapped in endosomes or lysosomes. To gauge the DNA shuriken performance as a miRNA delivery vehicle, we conducted a comparative study against the common transfection agent, Lipofectamine 2000. Our comparative study shows that the DNA shuriken significantly improved the miRNA-145 delivery as well. We detected close to 2-fold improvement in the DNA shuriken performance when compared to utilizing the Lipofectamine 2000 (Fig. 2c and S3†). To test the versatility of our DNA shuriken platform, we expanded our comparative study to other cell lines. Breast cancer cells, MCF-7, readily took up significant amounts of DNA shuriken particles while in contrast, HMVEC endothelial cells did not (Fig. S3†). In order to further examine the effectiveness of the DNA shuriken platform, we directly quantified the intracellular delivered miR-145 levels with quantitative reverse transcription PCR (RT-qPCR). It was previously reported that different DNA nanostructures can resist enzymatic DNA degradation and stay intact for 4 to 24 hours.33,34
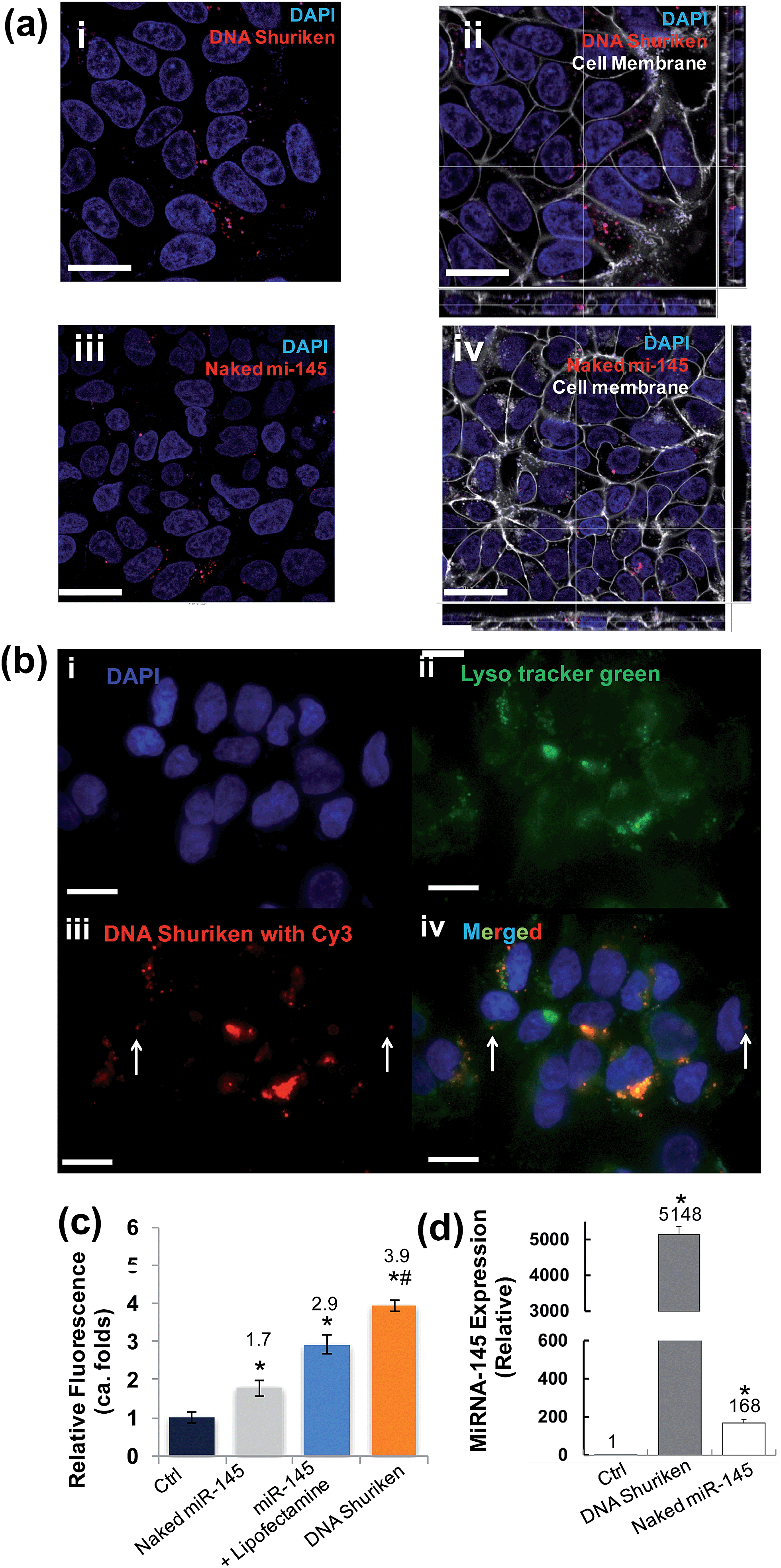 | ||
| Fig. 2 Internalization of DNA shuriken into cells. (a) Cells were incubated with DNA shuriken–miR-145 or naked miR-145 for two hours before imaging. Cell nuclei were stained with DAPI; miR-145 was tagged with Cy3. (a(i)) Cellular uptake of DNA shuriken–miR145. (a(ii)) The sectioning analysis of DNA shuriken–miR-145 treated cells showed intracellular presence but it was outside of the nucleus. (a(iii)) Comparatively lower cellular uptake of naked miR-145. (a(iv)) The sectional analysis of intracellular localization. (b) Colocalization study of DNA shuriken and lysosomes. Cells were stained with DAPI (b(i)) and Lyso tracker green (b(ii)). Most of the DNA shuriken particles (b(iii), tagged with Cy3) colocalized with lysosomes, and only a few DNA shuriken existed in the cytoplasm, indicated with arrows (b(iv)). (c) Quantification analysis of naked miR-145, miR-145 lipoplex (miR-145 + Lipofectamine) and DNA shuriken (with miR-145) using a microplate reader. The fluorescence signal is normalized to cell numbers and the untreated control. Data represent the mean ± SD, n = 3. Student's t-test, p < 0.05. *Significantly different from the control group (control). #Significantly different from the Lipofectamine treatment group. A complete data set can be found in Fig. S3.† (d) RT-qPCR quantification of the miR-145 level of DLD-1 cells after 24 hours' treatment. Let-7a was used as an internal control. The concentrations of DNA shuriken and naked miR-145 were kept at 200 nM (each DNA shuriken bears three copies of miR-145) and 600 nM respectively in all experiments. Data represent the mean ± SD, n = 3. Student's t-test, p < 0.05. *Significantly different from the control. Cell membranes were stained and are indicated in white. Scale bar: 20 μm. | ||
Along the same lines, it is therefore believed that this relative stability of DNA nanostructures in physiological conditions can help keep the cargo, single stranded DNA or RNA, safe from enzymatic digestion. Herein, RT-qPCR quantification showed that the miR-145 level is more than 5000 times higher than that of the untreated control even after a long 24 hours in a “foreign-nucleic-acid” unfriendly environment like the cell (Fig. 2d). In contrast, the intracellular level of naked miR-145 treated cells is approximately 30 fold lower than the DNA shuriken counterpart over the same time period of incubation. Our findings clearly show that the designed DNA shuriken can effectively deliver the miR-145 into cells, and stabilize and subsequently release the cargo.
We further investigated the anti-cancer effects of DNA shuriken on the highly proliferative colorectal cancer cell line DLD-1 which does not normally express significant amounts of miR-145. Since uncontrolled cell proliferation is a hallmark of cancer, we therefore employed a flow cytometry-based Ki67 cell proliferation assay to evaluate the therapeutic effect of miR-145. Ki67 is a potent cellular marker for proliferation.35 As can be seen in Fig. 3a, the percentage of proliferating cells (Ki67 positive) is 77.8%, 85.9% and 81.2% for negative control, the naked miR-145 strand and DNA star motif treated cells, respectively. Strikingly, a significant decrease in the percentage of proliferating cells (i.e. 46.3%) was observed for the cells that were treated with the DNA shuriken. It is noteworthy that the anti-proliferative effects of the DNA shuriken are even higher (approximately 1.4 folds) than those of the cells that are cultured in serum free medium devoid of any growth factors (without miR-145 or DNA shuriken treatment). The excellent cell proliferation suppression effect of miR-145 seen here is consistent with another study which directly overexpressed miR-145 from within the cell itself.36 Our results demonstrated that DNA nanostructures are suitable and effective for delivering external synthetic miR-145 and maintaining efficacy similar to endogenous expressed miR-145. To further the therapeutic effect of the DNA nanostructure platform on a more tumor-like format, we set up a 3D spheroid tumor model based on DLD-1 cells using a micropatterned hydrogel platform.37,38 The tumors were formed and treated with or without DNA shuriken for 5 days. Then we did a size change with time course analysis on the spheroids (Fig. 3b). After only two days there was a pronounced size difference, and the DNA shuriken treated tumor was 31% smaller (apparent size) than the untreated tumor. Fig. 3c shows the gross morphological evaluation of representative tumor spheroids. For the untreated group, the spheroids grew very fast and had a smooth surface of robust and uniform cell growth, while the DNA shuriken treated tumors were significantly smaller and had many cells “flaking off” from the outer surface. The reduced tumor size and morphology change paralleled the DNA shuriken-mediated anti-proliferative effects on 2D culture plastic growth and further demonstrated the efficacy to curb tumor growth in 3 dimensions. These results clearly showed that DNA shuriken can be an effective and powerful platform for miRNA therapeutics.
 | ||
| Fig. 3 The therapeutic effect and the stability of DNA shuriken in cell proliferation and 3D tumor growth. (a) Cell proliferation analysis of the DNA shuriken treated cells. (b) Impact of DNA shuriken treatment on tumor size over different time periods. The growth of tumors was assessed by apparent size change. Tumors were treated with DNA shuriken at a final concentration of 200 nM. (c) The effect of DNA shuriken on the tumor spheroid morphology. (d) Extracellular stability of DNA shuriken and naked miR-145. (e) RNase A degradative enzyme assay was employed to represent the efficient miRNA degradative process found in the intracellular environment. Scale bar: 200 μm. Data represent the mean ± SD, n = 3. Student's t-test compared to control, *p < 0.05, Bonferroni test compared to control, #p < 0.001. | ||
To the best of our knowledge, our current work is the first report that uses miRNA loaded DNA nanostructures for miRNA targeted cancer therapy. Although several reports showed that DNA nanostructures carrying siRNA6,7 or peptides8,9 had been successfully delivered into cells and taken effect, the details, such as the extracellular and intracellular stability of the DNA nanostructures, cellular uptake mechanisms, and the endosomal escape and cargo release, still remain largely unclear. Thus, elucidating such issues based on current DNA shuriken design is crucial for the future design of more effective, targeted and sophisticated therapeutic DNA nanostructure platforms. Firstly, we investigated the stability of DNA shuriken both in extracellular and intracellular environments (Fig. 3d and e). PAGE gel showed that the DNA shuriken structure was intact in the cell medium for up to 4 hours, whereas naked miR-145 was degraded within one hour. Secondly, to test the protective effect of DNA shuriken inside cells, we subjected the construct to a realistic estimated concentration of intracellular RNase A (1 μg mL−1). Fig. 3e shows that the DNA shuriken was stable for at least 8 hours, and naked miR-145 (unprotected) was quickly broken into two (or more) small fragments in a comparatively short time of 30 min. This reinforces our design strategy of steric hindrance conferred protection of the miRNA cargo. This result is also supported by the much higher measurable intracellular miR-145 level even after 24 hours in Fig. 2d.
This stability is important considering that the majority of our DNA shuriken was entrapped in lysosomes (Fig. 2b). While the mechanism of the lysosomal escape of the miR-145 was not delved into in this study, it did not contradict our findings that the miR-145 complexed in DNA shuriken was able to still exert its anti-cancer bioactivity (Fig. 3a–c). Even in the worst case scenario of being in the lysosomes for 24 hours (Fig. 2b), the all important sequence specificity of miR-145 was not compromised (the qPCR assay is extremely sensitive to sequence specificity changes (Fig. 2d)). Thus, this demonstrated the protective anti-degradative power of DNA shuriken in ensuring the safe passage of miR-145 from cell entry to successfully exerting its bioactivity (Fig. 3).
The non-toxic DNA nanostructures with their powerful structural control and precise spatial functionalization may launch the new era of delivery tools. Future design of DNA nanostructure based drugs emphasizing avoidance of endosome entrapment, customizable intracellular transportation,25 targeted delivery,39,40 and tracking of these DNA nanostructures in the cell would further improve the efficacy of this novel DNA nanotechnology.
Conclusions
In summary, we have designed and synthesized a DNA shuriken structure that contains miR-145 as a therapeutic agent against cancer. Its multi-pronged configuration helped with increasing the residence time for robust uptake by cells without any transfection agents. After entering the cell, the nanostructure continues to protect the miR-145 cargo to greatly suppress cancer cell proliferation and tumor growth. The significant therapeutic effect against cancer was attributed to the protection of the DNA nanostructure carrier as shown in both simulated extracellular and real intracellular environments. Our work showed that DNA nanostructures can be excellent customizable platforms for miRNA based cancer therapies, and have great potential to be expanded to other bioapplications, such as in vivo biosensing and bioimaging with specific targeting capability if necessary.41–44Acknowledgements
This work is supported by the Faculty of Engineering-Cross Faculty Grant (R-279-000-414-112).Notes and references
- V. Bagalkot, O. C. Farokhzad, R. Langer and S. Jon, Angew. Chem., Int. Ed., 2006, 45, 8149–8152 CrossRef CAS PubMed.
- Q. Zhang, Q. Jiang, N. Li, L. Dai, Q. Liu, L. Song, J. Wang, Y. Li, J. Tian, B. Q. Ding and Y. Du, ACS Nano, 2014, 8, 6633–6643 CrossRef CAS PubMed.
- Q. Jiang, C. Song, J. Nangreave, X. W. Liu, L. Lin, D. L. Qiu, Z. G. Wang, G. Z. Zou, X. J. Liang, H. Yan and B. Q. Ding, J. Am. Chem. Soc., 2012, 134, 13396–13403 CrossRef CAS PubMed.
- Y. X. Zhao, A. Shaw, X. H. Zeng, E. Benson, A. M. Nystrom and B. Hogberg, ACS Nano, 2012, 6, 8684–8691 CrossRef CAS PubMed.
- J. Li, C. Fan, H. Pei, J. Shi and Q. Huang, Adv. Mater., 2013, 25, 4386–4396 CrossRef CAS PubMed.
- H. Lee, A. K. R. Lytton-Jean, Y. Chen, K. T. Love, A. I. Park, E. D. Karagiannis, A. Sehgal, W. Querbes, C. S. Zurenko, M. Jayaraman, C. G. Peng, K. Charisse, A. Borodovsky, M. Manoharan, J. S. Donahoe, J. Truelove, M. Nahrendorf, R. Langer and D. G. Anderson, Nat. Nanotechnol., 2012, 7, 389–393 CrossRef CAS PubMed.
- Z. You, H. Qian, C. Z. Wang, B. F. He, J. W. Yan, C. Mao and G. S. Wang, Biomaterials, 2015, 67, 137–150 CrossRef CAS PubMed.
- J. Li, H. Pei, B. Zhu, L. Liang, M. Wei, Y. He, N. Chen, D. Li, Q. Huang and C. Fan, ACS Nano, 2011, 5, 8783–8789 CrossRef CAS PubMed.
- V. J. Schuller, S. Heidegger, N. Sandholzer, P. C. Nickels, N. A. Suhartha, S. Endres, C. Bourquin and T. Liedl, ACS Nano, 2011, 5, 9696–9702 CrossRef PubMed.
- S. Surana, J. M. Bhat, S. P. Koushika and Y. Krishnan, Nat. Commun., 2011, 2, 340 CrossRef PubMed.
- C. Y. Tay, L. Yuan and D. T. Leong, ACS Nano, 2015, 9, 5609–5617 CrossRef CAS PubMed.
- M. I. Setyawati, R. V. Kutty, C. Y. Tay, X. Yuan, J. Xie and D. T. Leong, ACS Appl. Mater. Interfaces, 2014, 6, 21822–21831 CAS.
- M. Giovanni, M. I. Setyawati, C. Y. Tay, W. S. Kuan and D. T. Leong, Adv. Funct. Mater., 2015, 25, 3840–3846 CrossRef CAS.
- D. S. Lee, H. Qian, C. Y. Tay and D. T. Leong, Chem. Soc. Rev., 2016, 45, 4199–4225 RSC.
- V. Ambros, Nature, 2004, 431, 350–355 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2009, 136, 215–233 CrossRef CAS PubMed.
- M. Sachdeva and Y. Y. Mo, Am. J. Transl. Res., 2010, 2, 170–180 CAS.
- M. Fuse, N. Nohata, S. Kojima, S. Sakamoto, T. Chiyomaru, K. Kawakami, H. Enokida, M. Nakagawa, Y. Naya, T. Ichikawa and N. Seki, Int. J. Oncol., 2011, 38, 1093–1101 CAS.
- B. Dalby, S. Cates, A. Harris, E. C. Ohki, M. L. Tilkins, P. J. Price and V. C. Ciccarone, Methods, 2004, 33, 95–103 CrossRef CAS PubMed.
- A. Sharei, J. Zoldan, A. Adamo, W. Y. Sim, N. Cho, E. Jackson, S. Mao, S. Schneider, M. J. Han, A. Lytton-Jean, P. A. Basto, S. Jhunjhunwala, J. Lee, D. A. Heller, J. W. Kang, G. C. Hartoularos, K. S. Kim, D. G. Anderson, R. Langer and K. F. Jensen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2082–2087 CrossRef CAS PubMed.
- A. K. Chen, M. A. Behlke and A. Tsourkas, Nucleic Acids Res., 2008, 36, 69 CrossRef PubMed.
- P. J. Santangelo, B. Nix, A. Tsourkas and G. Bao, Nucleic Acids Res., 2004, 32, 57 CrossRef PubMed.
- C. D. Medley, T. J. Drake, J. M. Tomasini, R. J. Rogers and W. Tan, Anal. Chem., 2005, 77, 4713–4718 CrossRef CAS PubMed.
- Q. Wang, H. Cheng, H. Peng, H. Zhou, P. Y. Li and R. Langer, Adv. Drug Delivery Rev., 2015, 91, 125–140 CrossRef CAS PubMed.
- L. Liang, J. Li, Q. Huang, J. Shi, H. Yan and C. H. Fan, Angew. Chem., Int. Ed., 2014, 53, 7745–7750 CrossRef CAS PubMed.
- C. Wu, D. Han, T. Chen, L. Peng, G. Zhu, M. You, L. Qiu, K. Sefah, X. Zhang and W. Tan, J. Am. Chem. Soc., 2013, 135, 18644–18650 CrossRef CAS PubMed.
- P. Charoenphol and H. Bermudez, Mol. Pharmaceutics, 2014, 11, 1721–1725 CrossRef CAS PubMed.
- R. Mo, T. Jiang, W. Sun and Z. Gu, Biomaterials, 2015, 50, 67–74 CrossRef CAS PubMed.
- X. Liu, Y. Xu, T. Yu, C. Clifford, Y. Liu, H. Yan and Y. Chang, Nano Lett., 2012, 12, 4254–4259 CrossRef CAS PubMed.
- S. M. Douglas, I. Bachelet and G. M. Church, Science, 2012, 335, 831–834 CrossRef CAS PubMed.
- Y. He, T. Ye, M. Su, C. Zhang, A. E. Ribbe, W. Jiang and C. Mao, Nature, 2008, 452, 198–201 CrossRef CAS PubMed.
- H. Qian, C. Tian, J. Yu, F. Guo, M. S. Zheng, W. Jiang, Q. F. Dong and C. Mao, Small, 2014, 10, 855–858 CrossRef CAS PubMed.
- A. S. Walsh, H. Yin, C. M. Erben, M. J. Wood and A. J. Turberfield, ACS Nano, 2011, 5, 5427–5432 CrossRef CAS PubMed.
- C. E. Castro, F. Kilchherr, D. Kim, E. L. Shiao, T. Wauer, P. Wortmann, M. Bathe and H. Dietz, Nat. Methods, 2011, 8, 221–229 CrossRef CAS PubMed.
- T. Scholzen and J. Gerdes, J. Cell. Physiol., 2000, 182, 311–322 CrossRef CAS PubMed.
- H. Zhu, U. Dougherty, V. Robinson, R. Mustafi, J. Pekow, S. Kupfer, Y. C. Li, J. Hart, K. Goss, A. Fichera, L. Joseph and M. Bissonnette, Mol. Cancer Res., 2011, 9, 960–975 CrossRef CAS PubMed.
- S. L. Chia, C. Y. Tay, M. I. Setyawati and D. T. Leong, Small, 2015, 11, 702–712 CrossRef CAS PubMed.
- C. Y. Tay, M. S. Muthu, S. L. Chia, K. T. Nguyen, S. S. Feng and D. T. Leong, Adv. Funct. Mater., 2016, 26, 4046–4065 CrossRef CAS.
- K. Ma, D. D. Wang, Y. Lin, J. Wang, V. Petrenko and C. Mao, Adv. Funct. Mater., 2013, 23, 1172–1181 CrossRef CAS PubMed.
- N. K. Niu, J. J. Yin, Y. X. Yang, Z. L. Wang, Z. W. Zhou, Z. X. He, X. W. Chen, X. J. Zhang, W. Duan, T. X. Yang and S. F. Zhou, Drug Des., Dev. Ther., 2015, 9, 4441 Search PubMed.
- C. Li, F. Li, Y. Zhang, W. Zhang, X. E. Zhang and Q. Wang, ACS Nano, 2015, 9, 12255–12263 CrossRef CAS PubMed.
- C. Li, L. Cao, Y. Zhang, P. Yi, M. Wang, B. Tan, Z. Deng, D. Wu and Q. Wang, Small, 2015, 11, 4517–4525 CrossRef CAS PubMed.
- J. Li, P. Cai, A. Shalviri, J. T. Henderson, C. S. He, W. D. Foltz, P. Prasad, P. M. Brodersen, Y. H. Chen, R. DaCosta, A. M. Rauth and X. Y. Wu, ACS Nano, 2014, 8, 9925–9940 CrossRef CAS PubMed.
- M. I. Setyawati, R. V. Kutty and D. T. Leong, Small, 2016 DOI:10.1002/smll.201601669.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6sc01829g |
| This journal is © The Royal Society of Chemistry 2017 |