
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative investigation of human cell surface N-glycoprotein dynamics†
Haopeng
Xiao
and
Ronghu
Wu
*
School of Chemistry and Biochemistry, The Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: Ronghu.Wu@chemistry.gatech.edu; Fax: +1-404-894-7452; Tel: +1-404-385-1515
First published on 15th August 2016
Abstract
Surface glycoproteins regulate nearly every extracellular event and they are dynamic for cells to adapt to the ever-changing extracellular environment. These glycoproteins contain a wealth of information on cellular development and disease states, and have significant biomedical implications. Systematic investigation of surface glycoproteins will result in a better understanding of surface protein functions, cellular activities and the molecular mechanisms of disease. However, it is extraordinarily challenging to specifically and globally analyze surface glycoproteins. Here we designed the first method to systematically analyze surface glycoprotein dynamics and measure their half-lives by integrating pulse-chase labeling, selective enrichment of surface glycoproteins, and multiplexed proteomics. The current results clearly demonstrated that surface glycoproteins with catalytic activities were more stable than those with binding and receptor activities. Glycosylation sites located outside of any domain had a notably longer median half-life than those within domains, which strongly suggests that glycans within domains regulate protein interactions with other molecules while those outside of domains mainly play a role in protecting the protein from degradation. This method can be extensively applied to biological and biomedical research.
Introduction
Nearly all proteins on the cell surface are glycosylated, and surface glycoproteins are essential for cell survival.1 Protein glycosylation plays crucial roles in a wide variety of extracellular activities, including antibody recognition, cell adhesion, microorganism binding, facilitating ligand binding, and affecting receptor multimerization.2–8 Aberrant surface protein glycosylation impacts on cellular properties, such as cell solubility and mobility, which are related to human disease,9–11 including cancer,12,13 congenital disorders and infectious diseases.14,15 It has long been understood that the covalent attachment between glycans and proteins is extremely complicated because of the heterogeneity of glycan structures, which makes the comprehensive analysis of protein glycosylation challenging.16–24 It is even more difficult to analyze glycoproteins located only on the cell surface. The elegant and pioneering work of using sugar analogs to engineer cell surface glycans and glycoproteins has opened a new avenue in the study of cell surface glycoproteins.25,26Surface glycoproteins are dynamic for cells to adapt to the ever-changing extracellular environment. The presence of glycans on proteins not only facilitates protein folding and trafficking, but also protects proteins from degradation.27–30 Glycans create a steric hindrance around the peptide backbone, which mechanically prevents proteases from properly binding to proteins. In addition, protein glycosylation also protects the protein backbone from being damaged or degraded through oxidation, chemical crosslinking, precipitation, and denaturation.31,32 However, the systematic study of glycoprotein dynamics and half-lives has yet to be reported, including the dynamics of crucial cell surface glycoproteins, due to the lack of effective methods. In recent years, MS-based proteomics has enabled the global analysis of proteins and protein modifications, including glycosylation.33–44 Due to the complexity of biological samples, effective separation and enrichment are required to comprehensively analyze every type of protein modification.13,45,46 In order to analyze glycoproteins located only on the cell surface, it is essential to selectively separate and enrich them from high abundance intracellular proteins prior to MS analysis.
In this work, we have designed a method to target surface N-glycoproteins and quantify their half-lives by combining pulse-chase metabolic labelling, click chemistry, and multiplexed proteomics. A sugar analog, N-azidoacetylgalactosamine (GalNAz), was employed to label cells to generate a chemical handle for further surface glycoprotein tagging via copper-free click chemistry under mild physiological conditions. Pulse-chase labelling allowed us to track the abundance changes of cell surface glycoproteins while avoiding any contribution from newly synthesized glycoproteins during cell growth because they were not labelled with the functional azido group. After enrichment of tagged glycopeptides, six-plexed Tandem Mass Tag (TMT) reagents47 were used to label enriched glycopeptides at six different time points for quantification with MS-based proteomics. Eventually the glycoprotein abundance changes as a function of time were measured, and their half-lives were globally determined. This integrated method, specifically targeting surface glycoproteins, can be extensively applied to biological and biomedical research.
Results
The principle of surface glycoprotein enrichment and identification
The incorporation of bio-orthogonal groups into proteins or modified proteins has recently been demonstrated to be very effective for the study of proteins in complex biological systems.25,48–56 In this work glycoproteins were labelled with a sugar analog containing a biologically inert but chemically functional azido group, and the labelled surface glycoproteins in living cells were specifically tagged with biotin via copper-free click chemistry (Fig. 1). Here we performed the click reaction prior to switching the media, which can eliminate potential negative effects from cells using stored GalNAz and protein internalization on protein half-life quantification. After cell lysis and protein digestion, only biotin-tagged glycopeptides were selectively enriched with NeutrAvidin beads through specific biotin–avidin interactions (the detailed procedure is given in the Experimental section). Enriched and purified samples were analyzed by an online LC-MS system, and both full MS and MS2 were recorded in the Orbitrap cell with high resolution and high mass accuracy.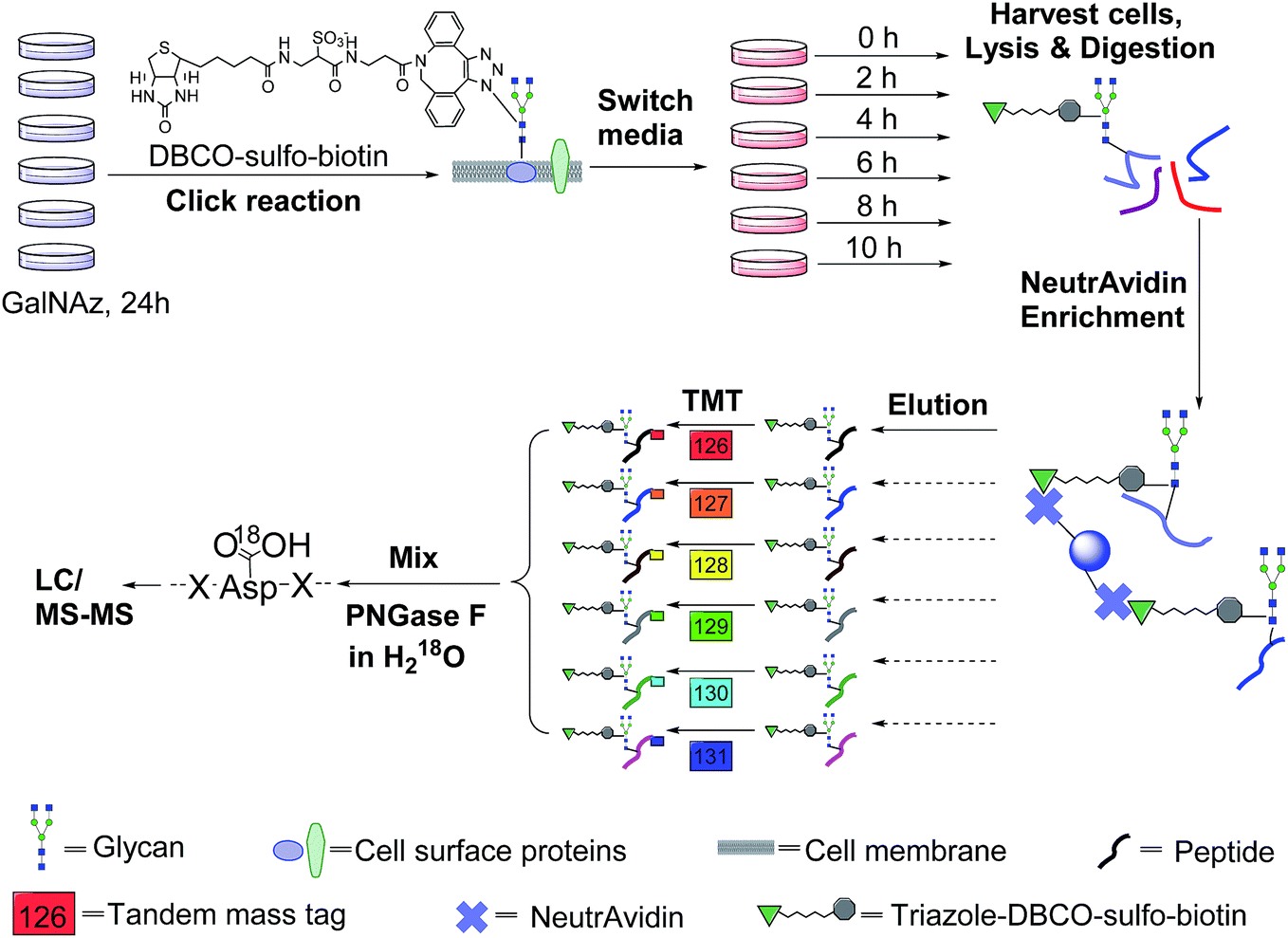 | ||
| Fig. 1 Experimental procedure for studying surface glycoprotein dynamics and measuring their half-lives. All six flasks of cells were labelled in DMEM containing 100 μM GalNAz for 24 h, then cell surface glycoproteins were tagged with click chemistry under physiological conditions. After tagging, media were switched to the normal chase media, and each flask of cells was harvested at a different time point (0, 2, 4, 6, 8 or 10 h). Enriched glycopeptides were labelled with TMT reagents for quantification. | ||
The TMT method enables the identification and quantitation of glycopeptides and glycoproteins in different samples in combination with tandem mass spectrometry. The tags contain four regions, namely, a mass reporter region, a cleavable linker region, a mass normalization region, and a protein/peptide reactive group. In this case, the reactive group of N-hydroxysuccinimide (NHS) can react quickly with the amine group at the N-terminus and/or the side chain of the lysine residue for every peptide. Each of six samples was tagged with one channel of TMT reagent, then mixed. For the same peptide in six samples, they all carry isobaric tags, and have the same elution time and m/z in the full mass spectrum. When the peptides are fragmented, the reporter ions generated from the tagged peptides have intensities proportional to the amount of peptide in each sample. Eventually, peptide backbone fragments allow us to identify peptides and the reporter ion intensities enable us to quantify the peptide abundance changes in the six samples.
An example of peptide identification and quantification is displayed in Fig. 2(a). The peptide N#VSVAEGK (# denotes the glycosylation site) was confidently identified with an XCorr of 3.2. The XCorr value is the cross-correlation value from SEQUEST search, which reflects how good the match is between theoretical and experimental tandem mass spectra.57 XCorr values are usually higher for well-matched, larger peptides, and lower for smaller peptides. Considering the short length of this peptide, this XCorr value can allow us to confidently identify this glycopeptide, and as shown in Fig. 2(a), nearly all y and b ions were detected and matched. The ModScore for the glycosylation site (N286) is 1000 because there is only one possible site localization. This peptide is from PTGFRN, which is a well-known receptor regulator located on the cell surface.58 The reporter ion intensities enabled us to accurately quantify the glycopeptide abundance changes as a function of time (Fig. 2(a), left insert). Correspondingly we were able to calculate the half-life of 15.5 h based on the abundance changes (Fig. 2(a), right insert).
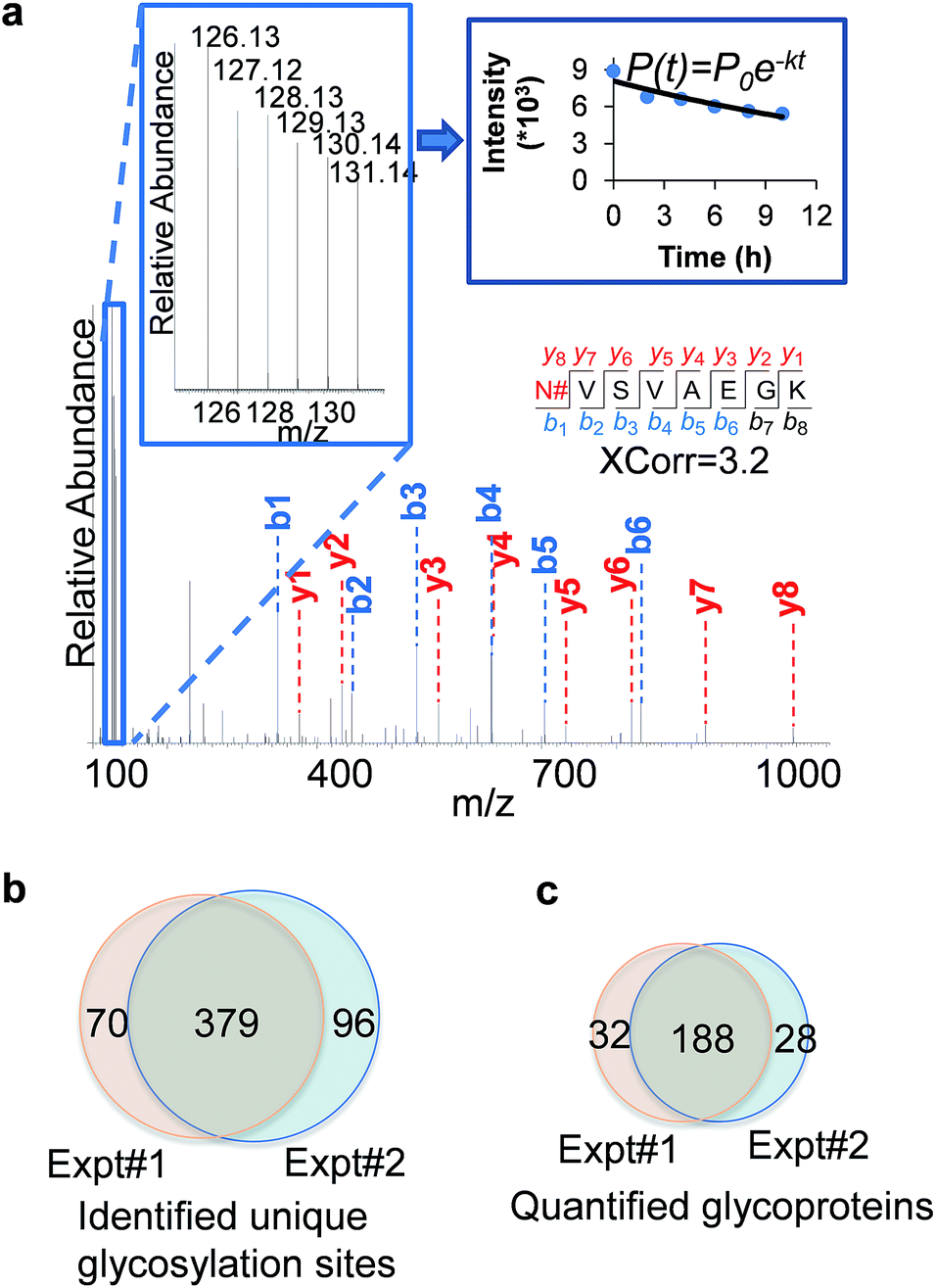 | ||
| Fig. 2 An example of glycopeptide identification and quantification and the comparison of identified unique glycosylation sites and quantified surface glycoproteins. (a) Example MS showing peptide identification and quantification. Based on the fragments, we were able to confidently identify the glycopeptide N#VSVAEGK (# denotes the glycosylation site) from the protein PTGFRN, and based on the reporter ion intensities, the half-life of this glycopeptide was 15.5 hours. (b) Comparison of the unique surface protein glycosylation sites identified in two parallel experiments. (c) Comparison of the quantified surface glycoproteins in duplicate experiments. | ||
In the current duplicate experiments, we identified a total of 545 unique glycosylation sites on 265 glycoproteins (ESI Tables 1, 2, and 7†), and most of them (480 sites) were well localized with a ModScore > 13. The overlap of unique glycosylation sites identified between two replicates is around 80% across all identification and quantification results (Fig. 2(b) and (c)), which demonstrated that the current method is highly reproducible. The majority of unique glycopeptides contained a single glycosylation site, and there was a small group of proteins bearing more than five sites, including IGF1R, ECE1, LAMP1, CELSR2, PLXNB2, CEACAM5, ITGB1, and PTPRJ. For example, for IGF1R, a receptor tyrosine kinase which mediates the actions of insulin-like growth factor 1 (IGF1) located on the plasma membrane, we identified eleven glycosylation sites: N244, N314, N607, N622, N638, N640, N747, N756, N764, N900, and N913. All these sites exist in the extracellular space, which is further discussed below.
We clustered the identified glycoproteins according to cellular compartment and pathway using the Database for Annotation, Visualization, and Integrated Discovery 6.7 (DAVID 6.7) (Fig. S1†).59 For cellular compartments, membrane-related categories were highly enriched, including intrinsic to membrane (P = 1.80 × 10−81), plasma membrane (8.50 × 10−49), cell surface (6.20 × 10−20), external side of the plasma membrane (5.50 × 10−14), and receptor complex (6.20 × 10−11). Among the pathways, the ECM–receptor interactions (5.60 × 10−11) and cell adhesion molecules (CAMs) (1.30 × 10−10) pathways were prominently enriched. CAMs are cell-surface proteins involved in binding with the extracellular matrix (ECM) or with other cells during cell adhesion. These enriched categories are consistent with the expected functions of cell-surface glycoproteins.
Site location of type I and II glycoproteins based on the transmembrane domain
The site-specific virtue of our method allowed us to localize each glycosylation site in this experiment. In Fig. 3, we illustrated the site localization on type I and II transmembrane glycoproteins identified in this experiment. Type I transmembrane proteins have their N-termini located in the extracellular space while type II transmembrane proteins have their C-termini located in the extracellular space. As shown in Fig. 3, the x-axis represents the transmembrane (TM) domain of any protein, and the y-axis denotes the number of amino acid residues away from the transmembrane domain. The space above the x-axis is the extracellular space and below is the intracellular space. Each line depicts a protein, and the yellow dots represent the glycosylation sites. All glycosylation sites are clearly located in the extracellular space, which is in agreement with the experimental design and the common belief that glycans on surface proteins are located outside of the cell. We identified many more type I transmembrane proteins than type II, which corresponds well to the ratio of type I to II transmembrane proteins in UniProt (http://www.uniprot.org).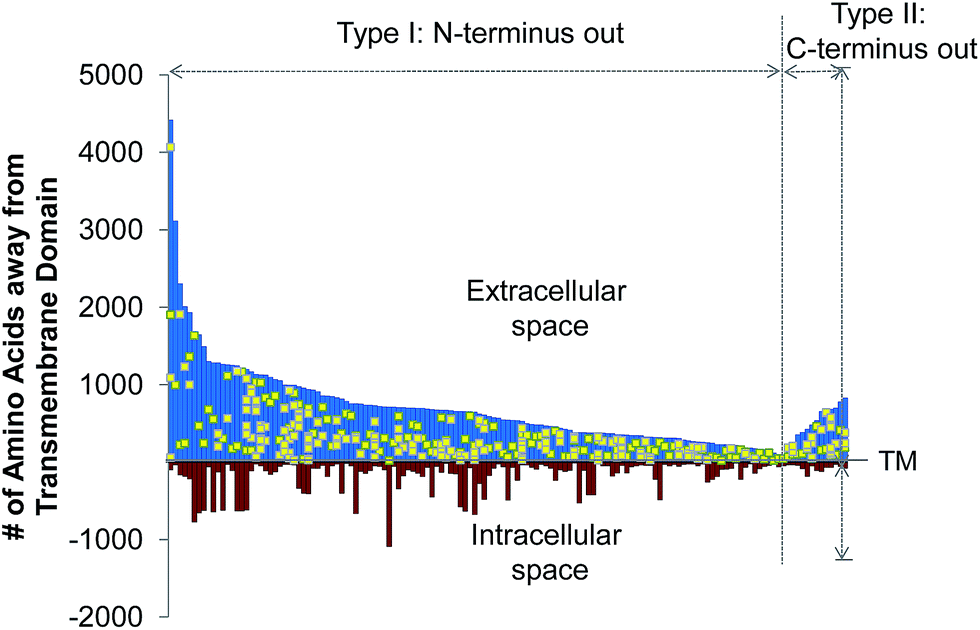 | ||
| Fig. 3 Site location of the type I and II N-glycoproteins based on the transmembrane domain (TM). We aligned each glycoprotein according to their transmembrane domain, which is known to be anchored in the plasma membrane, and each yellow dot refers to one glycosylation site. | ||
Quantification of surface glycoprotein abundance changes
Enriched peptides in each sample were labelled with one of six TMT reagents. TMT labelling allowed us to quantify multiple samples at once. Here we measured six samples from six time points simultaneously. This can dramatically increase the experimental throughput and reduce potential quantification errors. The starting amount of labelled surface glycoproteins was similar for each sample before the medium was switched. Based on this, we then quantified these surface glycoprotein abundance changes as a function of time. The six groups of TMT-labelled glycopeptides were mixed and subjected to PNGase F cleavage in heavy-oxygen water (H218O) to generate a common tag (+2.9883 Da) for MS analysis.18,60,61 This enabled us to distinguish authentic N-glycosylation sites from those caused by the naturally occurring deamidation of Asn. Finally, the peptide mixture was purified and loaded into an online LC-MS system for further analysis.The TMT reporter ion intensities in the MS2 provided us an opportunity to accurately measure the abundance changes of glycopeptides from different time points. Potential interferences from TMT labelling were likely avoided in this experiment because these samples were much simpler than whole cell lysates since surface glycoproteins only represent a very small portion of the whole proteome, and we also further fractionated the mixed sample into three fractions. Furthermore, long LC gradients were used to separate each fraction. Because the abundances of the same glycopeptide from six samples can be measured in one MS2 spectrum, this dramatically lowered the measurement error. In some cases, for instance, if one of the six TMT channels had abnormal signal intensity, then it was dropped and the half-life was calculated based on the signal intensities from the other five channels.
Measurement of surface glycoprotein half-lives
Based on the abundance changes of glycopeptides at six time points, their half-lives were simulated by the following exponential decay equation, as performed previously:62,63P(t) = P0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−kt) exp(−kt) |
In the duplicate experiments, we quantified 386 glycosylation sites (ESI Tables 3, 4, and 8†) based on two criteria: glycopeptides were singly glycosylated and the ModScore was larger than 13. If a glycoprotein contained two or more unique glycosylation sites, the half-life refers to the median half-life of the mixed different glycoforms. The half-life values for the 248 glycoproteins were determined, and are listed in ESI Tables 5, 6, and 9.† We plotted the half-lives of glycosylation sites in replicate 1 against those in replicate 2 (Fig. 4(a)), and a good linear simulation and a high R2 value were obtained. The reproducibility is much better when the half-lives are relatively short, which is discussed below.
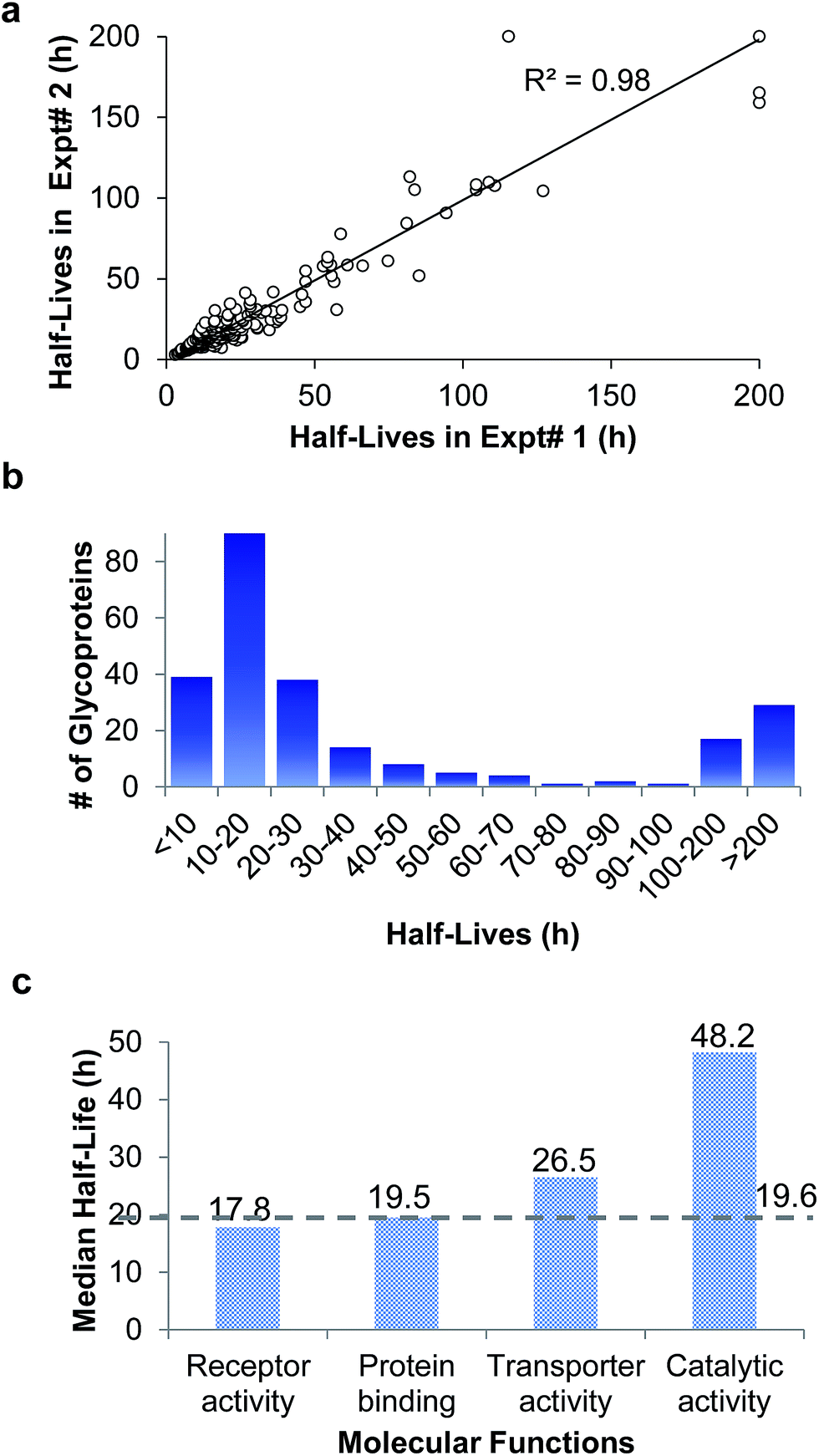 | ||
| Fig. 4 (a) Comparison of the half-lives of surface protein glycosylation sites measured in the duplicate experiments. (b) Distribution of the half-lives of surface glycoproteins. (c) The median half-lives of glycoproteins with different molecular functions. Proteins with receptor activity have the shortest median half-life (17.8 h), while proteins with catalytic activity have a longer median half-life (48.2 h). | ||
The distribution of the half-lives of surface glycoproteins is shown in Fig. 4(b). Most proteins have a half-life between 10 and 30 h. A total of 39 glycoproteins have a half-life of less than 10 h, while about one fifth of the glycoproteins (46) have a half-life of longer than 100 h. The median half-life of all glycoproteins quantified in our experiment was 19.6 h, which is much longer than the half-life of 8.7 h for over 800 newly synthesized proteins in our previous work,63 and also longer than a half-life of 8.2 h for 100 proteins measured with a MS-independent method.64 This is consistent with the assumption that glycans can stabilize proteins by preventing them from being degraded.
The functions associated with relatively long- or short-lived proteins were also investigated. Proteins with a half-life longer than 100 h or shorter than 10 h were clustered according to biological processes (Fig. S2†). While cell adhesion is enriched in both categories, notably, positive regulation of catalytic activity is enriched among long-lived proteins.
The median half-lives of proteins with various molecular functions were examined. As shown in Fig. 4(c), the median half-lives of proteins with receptor activity (17.8 h) and binding activity (19.5 h) are very similar to the overall protein median half-life (19.6 h), while proteins related to catalytic activity (receptor-type tyrosine kinases and phosphatases are not included because they only have intracellular catalytic activities) have a longer median half-life of 48.2 h, which suggests that glycan may protect enzymes on the cell surface more effectively.
Half-lives of glycosylation sites within or outside of domains
Among 386 quantified glycosylation sites, nearly half of them (170 sites) were located in different domains based on the domain information on UniProt, while 216 sites were not located in any protein domains. The median half-life for the 216 sites located outside of any domain is 21.5 h, which is 21% longer than the median half-life of 170 sites located within a specific domain (17.7 h), as shown in Fig. 5(a). The domains containing the greatest number of quantified glycosylation sites are Ig-like, fibronectin type-III, cadherin, and sema domains, which are shown in Fig. 5(b), along with their median half-lives. These domains are frequently contained in cell surface proteins, and play crucial roles in regulating cell–matrix interactions and cell surface receptor protein–ligand interactions. 68 sites are located in the Ig-like domain with a median half-life of 19.0 h. The median half-life of sites located in the cadherin domain is only 11.0 h, which is drastically shorter than the median half-life of 21.5 h for sites located outside of any domains. These results suggest that glycans located within a domain may play a major role in regulating protein interactions with other molecules, while glycans located outside of any domain are mostly involved in protecting proteins from degradation.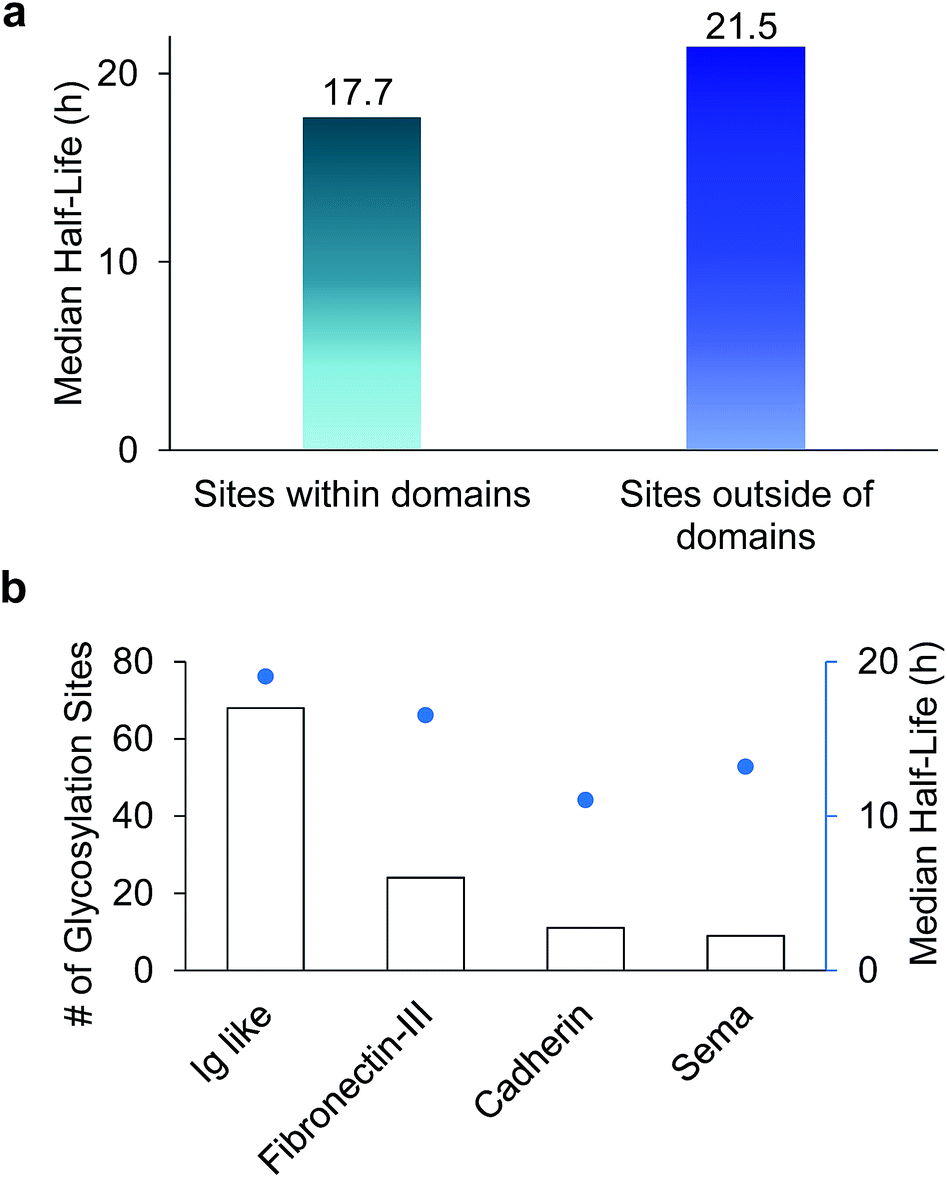 | ||
| Fig. 5 (a) Comparison of median half-lives for sites located outside domains and within domains. (b) The number of glycosylation sites located in different domains and their median half-lives. | ||
Half-lives of CD proteins and receptors
Cluster of differentiation (CD) molecules are of great biomedical significance because they serve as cell markers in immunophenotyping to distinguish and classify cells.65 CDs refer not only to proteins but can also be assigned to lipids and glycans on the cell surface. Among all of the glycoproteins quantified here, 62 are CD proteins (Table 1 and ESI Table 10†). The site-specific nature of this method provides an avenue to quantify the real glycosylated form of proteins. For example, we identified the glycosylation sites N365, N381, and N424 on CD98, which is involved in sodium-independent, high-affinity transport of large neutral amino acids. The half-life of the glycosylated form of CD98 is 27.2 h, which is much longer than the half-life (10.1 h) in the literature.63 Furthermore, the half-life of CD71 (transferrin receptor protein 1) is 18.3 h in this work for its glycoform on the cell surface, while the half-life of this protein was previously reported to be 4.4 h.63 Glycosylated and non-glycosylated forms of a protein normally coexist at any given time. Traditional gel-based or MS-based methods measure the half-life of the mixed glycosylated and non-glycosylated forms of a protein, but here we were able to measure the half-lives of only the glycosylated form of each protein because only surface glycoproteins were separated and analyzed.| UniProt ID | Gene symbol | CD name | Protein half-life | Annotation | |
|---|---|---|---|---|---|
| This work (h) | Previous worka (h) | ||||
| a Half-lives of corresponding proteins reported in the literature. | |||||
| P02786 | TFRC | CD71 | 18.3 | 4.4 (ref. 63) | Transferrin receptor protein 1 |
| P05556 | ITGB1 | CD29 | 24.2 | Integrin beta-1 | |
| P08069 | IGF1R | CD221 | 12.6 | Insulin-like growth factor 1 receptor | |
| P08195 | SLC3A2 | CD98 | 27.2 | 10.1 (ref. 63) | 4F2 cell-surface antigen heavy chain |
| P08962 | CD63 | CD63 | 24.2 | CD63 antigen | |
| P25445 | FAS | CD95 | 39.1 | Tumor necrosis factor receptor superfamily member 6 | |
| P26006 | ITGA3 | CD49c | 37.3 | Integrin alpha-3 | |
| P48960 | CD97 | CD97 | 13.2 | CD97 antigen | |
| P54709 | ATP1B3 | CD298 | 61.1 | Sodium/potassium-transporting ATPase subunit beta-3 | |
| P78536 | ADAM17 | CD156b | 112.7 | Disintegrin and metalloproteinase domain-containing protein 17 | |
Discussion
The mammalian cell surface is typically covered with sugars, and these sugars may be bound to lipids or proteins. Glycoproteins located on the cell surface regulate nearly every extracellular activity. Systematic and quantitative analysis of surface glycoproteins can aid in a better understanding of protein structure, properties and functions and also cellular activities. Due to the heterogeneity of glycans and low abundance of many glycoproteins, it is extremely challenging to globally identify and quantify glycoproteins in complex biological samples.46 It is significantly more challenging to specifically analyze surface glycoproteins. Fluorescence experiments have obtained very valuable information about cell surface glycans.66 However, it is hard to identify which proteins are bound to glycans and the exact glycosylation sites. MS-based proteomics provides the possibility to identify and quantify glycoproteins, but in order to analyze surface glycoproteins, selective enrichment of surface glycoproteins is required prior to MS analysis. It has remained a daunting task to systematically investigate the dynamics of cell surface glycoproteins. Integrating pulse-chase metabolic labelling, selective enrichment of surface glycoproteins, and multiplexed proteomics, for the first time, we site-specifically and systematically quantified changes in abundance of surface glycoproteins as a function of time, and measured their half-lives.Besides protein degradation, other contributions to cell surface glycoprotein dynamics include protein internalization, recycling, and deglycosylation. By tagging surface glycoproteins immediately before the medium switch, the effect of protein internalization on the measurement of surface glycoprotein half-lives could be avoided because, even if a protein was internalized, the biotin tag could ensure that it was eventually analyzed. Only tagged glycoproteins on the cell surface were investigated. Therefore, newly synthesized glycoproteins and recycled surface glycoproteins did not have any effect because they were not tagged. In other cases, when a deglycosylation event happens, the protein turns into a non-glycoform, which does not fit into our experimental subject and thus is not enriched and analyzed.
The trafficking behavior of surface proteins has always been of great interest to the science community.67–69 For relatively short-lived proteins quantified in our experiment, such as ErbB 3 (4.8 h) and ErbB 4 (11.9 h), they were reported to be slowly internalized, but quickly recycled.69 The constitutive degradation rate of EGFR (ErbB1) is slower than other ErbB family members, and the internalization rate of ErbB2 is similar to that of EGFR. ErbB family proteins, especially EGFR, may couple with G protein-coupled receptors and induce cell proliferation and migration.70 In this work, we quantified the half-lives of two G-protein-coupled receptors: CELSR1 (8.0 h) and CELSR2 (9.3 h), which are similar to the two ErbB proteins.
One limitation of this method is that proteins with very long half-lives might not be accurately determined because the full length of the time course may only cover the very beginning of the simulation curve, thus a minor variation could result in a large error. We applied a 200 h cut-off value to those long-lived proteins, namely, any protein with a half-life longer than 200 h was included in the >200 h category. Although this category did not present actual half-life values, it still indicates that these glycoproteins are very stable. Since the majority of the glycoproteins have half-lives shorter than 200 h, this category did not affect the calculation of the median half-life nonetheless.
The current experimental results have clearly demonstrated that glycans can more effectively protect enzymes than receptors and binding proteins located on the cell surface from being degraded, because proteins related to catalytic activity have a long median half-life of 48.2 h (quantified surface enzymes are listed in ESI Table 11†). It is well-known that there are many proteases in the extracellular space, but these quantified surface enzymes were still relatively more stable. Proteins in the mitochondria or nuclei typically have a longer half-life because proteins located in these compartments may avoid being accessed by many proteases. Cell surface proteins are exposed to different environments, but glycans on surface proteins may provide one layer of protection, especially for proteins with catalytic activity.
Experimental
Cell culture, metabolic labeling, and copper-free click reaction
MCF-7 cells (from American type culture collection (ATCC)) were equally seeded into twelve T175 cell culture flasks (Thermo) with Dulbecco's Modified Eagle's Medium (DMEM) (Sigma-Aldrich) containing 10% fetal bovine serum (FBS) (Thermo). Cells were grown in a humidified incubator with 5.0% CO2 at 37 °C. When cells reached 50% confluency, 100 μM GalNAz (Click Chemistry Tools) was added to the media and cells were cultured for another 24 h. The click reaction was then performed for all flasks. Briefly, cells were gently washed twice with phosphate buffered saline (PBS), then 100 μM dibenzocyclooctyne (DBCO)-sulfo-biotin in DMEM was added into the cell culture flasks. Cells were incubated for 1 h at 37 °C, and then washed twice using PBS. The media were then switched to normal DMEM with 10% FBS and different flasks were further cultured for 0, 2, 4, 6, 8, and 10 hours. Cells were pelleted by centrifugation at 300 g for 5 minutes, and washed twice with cold PBS. Cells were then incubated in a buffer containing 150 mM NaCl, 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH = 7.6, 25 μg mL−1 digitonin, and 1 tablet per 10 mL protease inhibitor (complete mini, EDTA-free, Roche) on ice for 10 minutes. Cytosolic proteins were removed by centrifuging the samples at 2500 g for 10 minutes and discarding the supernatant. Cell pellets were lysed through end-over-end rotation at 4 °C for 45 minutes in lysis buffer (50 mM HEPES pH = 7.6, 150 mM NaCl, 0.5% sodium deoxycholate (SDC), 10 units per mL benzonase and 1 tablet per 10 mL protease inhibitor). Lysates were centrifuged, and the resulting supernatant was transferred to new tubes. Proteins were subjected to disulfide reduction with 5 mM dithiothreitol (DTT) (56 °C, 25 minutes) and alkylation with 14 mM iodoacetamide (RT, 20 minutes in the dark). Detergent was removed by methanol–chloroform protein precipitation. The purified proteins were digested with 10 ng μL−1 Lys-C (Wako) in 50 mM HEPES pH = 8.6, 1.6 M urea, 5% ACN at 31 °C for 16 hours, followed by further digestion with 8 ng μL−1 trypsin (Promega) at 37 °C for 4 hours.Glycopeptide separation and enrichment
Digestion mixtures were acidified by addition of trifluoroacetic acid (TFA) to a final concentration of 0.1%, clarified by centrifugation and desalted using a tC18 Sep-Pak cartridge (Waters). Purified peptides were dried and then enriched with NeutrAvidin beads (Thermo) at 37 °C for 30 minutes. The samples were transferred to spin columns, followed by thoroughly washing according to the manufacturer's protocol. Peptides were then eluted from the beads three times by 2 min incubations with 200 μL of 8 M guanidine–HCl (pH = 1.5) at 56 °C. Eluates were combined, desalted using tC18 Sep-Pak cartridge, and lyophilized.TMT labelling and PNGase F cleavage
Purified peptides from each of the six time points were labelled with one of the sixplex TMT reagents (Thermo) following the manufacturer's protocol. Briefly, purified and lyophilized peptides were dissolved in 100 μL of 100 mM triethylammonium bicarbonate (TEAB) buffer, pH = 8.5. Each tube of TMT reagents was dissolved in 41 μL of anhydrous DMSO and transferred into the peptide tube. The reaction lasted for 1 h at room temperature, and then was quenched by adding 8 μL of 5% hydroxylamine. Peptides from all six tubes were then mixed, desalted again using a tC18 Sep-Pak cartridge, and lyophilized overnight. Completely dried peptides were deglycosylated with two units of peptide-N-glycosidase F (PNGase F, Sigma-Aldrich) in 40 μL buffer containing 50 mM NH4HCO3 (pH = 9) in heavy-oxygen water (H218O) for 3 h at 37 °C. The reaction was quenched by adding formic acid (FA) to a final concentration of 1%. Peptides were further purified via stage tip and separated into three fractions using 20%, 50% and 80% ACN containing 1% HOAc, respectively.LC-MS/MS analysis
Purified and dried peptide samples were dissolved in 10 μL of solvent containing 5% ACN and 4% FA, and 4 μL of dissolved sample were loaded onto a microcapillary column packed with C18 beads (Magic C18AQ, 3 μm, 200 Å, 100 μm × 16 cm, Michrom Bioresources) by a Dionex WPS-3000TPLRS autosampler (UltiMate 3000 thermostatted Rapid Separation Pulled Loop Wellplate Sampler). Peptides were separated by reversed-phase chromatography using an UltiMate 3000 binary pump with a 112 minute gradient of 1–12%, 3–14%, or 3–24% ACN (with 0.125% FA) for the three fractions. Peptides were detected with a data-dependent Top15 method63 in a hybrid dual-cell quadrupole linear ion trap – Orbitrap mass spectrometer (LTQ Orbitrap Elite, ThermoFisher, with Xcalibur 3.0.63 software). For each cycle, one full MS scan (resolution: 60000) in the Orbitrap at 106 AGC target was followed by up to 15 MS/MS for the most intense ions. The selected ions were excluded from further analysis for 90 seconds. Ions with single or unassigned charge were discarded. MS2 scans were performed in the Orbitrap cell by activating with high energy collision dissociation (HCD) at 40% normalized collision energy with 1.2 m/z isolation width.
Database search and data filtering
All MS2 spectra were converted into an mzXML format, and then searched using the SEQUEST algorithm (version 28).57 Spectra were matched against a database containing sequences of all proteins in the UniProt Human (Homo sapiens) database (downloaded in February 2014). The following parameters were used during the search: 10 ppm mass tolerance; fully digested with trypsin; up to 2 missed cleavages; fixed modifications: carbamidomethylation of cysteine (+57.0214), TMT modification of lysine (+229.1629) and N-terminus (+229.1629); variable modifications: oxidation of methionine (+15.9949), 18O tag on asparagine (+2.9883). False discovery rates (FDR) of peptide and protein identifications were evaluated and controlled by the target-decoy method.71,72 Each protein sequence was listed in both forward and reversed orders. Linear discriminant analysis (LDA), which is similar to other methods in the literature,73 was used to control the quality of peptide identifications using parameters such as Xcorr, precursor mass error, and charge state.74 Peptides fewer than seven amino acid residues in length were deleted. Furthermore, peptide spectral matches were filtered to <1% FDR. The dataset was restricted to glycopeptides when determining FDRs for glycopeptide identification.75Glycosylation site localization, glycopeptide quantification, and bioinformatics analysis
The confidence associated with each glycosylation site localization was represented by their ModScore, which is calculated from a probabilistic algorithm.75 ModScore is similar to AScore,75 and it considers all possible modification sites in a modified peptide, and matches the fragments with theoretical peaks from the peptide with each of potential modification site. If the ModScore for a residue is relatively high, then the probability of that site being modified is also high. On the contrary, there may be a low score for potential sites, which means that there are not sufficient fragments to confidently locate the modification site. Sites with ModScore > 13 (P < 0.05) were considered as confidently localized. The TMT reporter ion intensities obtained in MS2 were recorded and calibrated prior to performing glycopeptide quantification. If the same glycopeptide was quantified several times, the median value was used as the glycopeptide abundance change. The protein ratio was calculated based on the median ratios of all unique glycopeptides. Protein annotations were extracted from the UniProt database (http://www.uniprot.org). The Database for Annotation, Visualization and Integrated discovery (DAVID) v6.7 (http://david.abcc.ncifcrf.gov/home.jsp)76 was employed to perform functional analysis.All raw files are accessible in the following public accessible website (http://www.peptideatlas.org/PASS/PASS00913, password: BE6745wv).
Conclusions
We designed the first method to target surface glycoproteins, site-specifically study their dynamics and measure their half-lives by incorporating metabolic labelling, click chemistry, and multiplexed proteomics. The current method has several advantages. Firstly, only surface glycoproteins were selectively tagged and enriched for MS analysis. Secondly, site-specific protein glycosylation information was obtained in this work, and only authentic glycosylated forms of proteins were analyzed. Thirdly, multiplexed proteomics enabled to quantify glycoproteins at several time points simultaneously, increasing the accuracy of measuring protein abundance changes and the corresponding half-lives. Furthermore, the high throughput MS-based experiment allowed us to systematically study surface glycoprotein dynamics.By using this new method, we quantified 248 surface glycoprotein dynamics with the median half-life as 19.6 h, which is over two times longer than that of newly synthesized proteins measured in our recent work (8.7 h).63 The median half-life of glycopeptides with sites located outside of any domain is longer than that of glycopeptides with sites within different domains. Surface glycoproteins corresponding to catalytic activities were more stable with a median half-life of 48.2 h. Although there are many proteases outside of the cells, glycans can effectively protect surface enzymes from being degraded. Investigation of surface glycoprotein dynamics can aid in better understanding their properties and functions. This method can be extensively applied to investigate surface glycoproteins and their dynamics in biological and biomedical research.
Acknowledgements
This work was supported by the National Science Foundation (CAREER Award, CHE-1454501). R. W. is also supported by the Blanchard Assistant Professorship.References
- A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 2nd edn, 2008 Search PubMed.
- H. Lis and N. Sharon, Eur. J. Biochem., 1993, 218, 1–27 CrossRef CAS PubMed.
- A. Pulsipher, M. E. Griffin, S. E. Stone and L. C. Hsieh-Wilson, Angew. Chem., Int. Ed., 2015, 54, 1466–1470 CrossRef CAS PubMed.
- P. M. Rudd, T. Elliott, P. Cresswell, I. A. Wilson and R. A. Dwek, Science, 2001, 291, 2370–2376 CrossRef CAS PubMed.
- H. Takeuchi and R. S. Haltiwanger, Semin. Cell Dev. Biol., 2010, 21, 638–645 CrossRef CAS PubMed.
- S. G. Bi and L. G. Baum, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 1599–1610 CrossRef CAS PubMed.
- H. J. Gabius, H. C. Siebert, S. Andre, J. Jimenez-Barbero and H. Rudiger, ChemBioChem, 2004, 5, 740–764 CrossRef CAS PubMed.
- J. Kuball, B. Hauptrock, V. Malina, E. Antunes, R. H. Voss, M. Wolfl, R. Strong, M. Theobald and P. D. Greenberg, J. Exp. Med., 2009, 206, 463–475 CrossRef CAS PubMed.
- J. W. Dennis, M. Granovsky and C. E. Warren, BioEssays, 1999, 21, 412–421 CrossRef CAS PubMed.
- E. Maverakis, K. Kim, M. Shimoda, M. E. Gershwin, F. Patel, R. Wilken, S. Raychaudhuri, L. R. Ruhaak and C. B. Lebrilla, J. Autoimmun., 2015, 57, 1–13 CrossRef CAS PubMed.
- L. Lehle, S. Strahl and W. Tanner, Angew. Chem., Int. Ed., 2006, 45, 6802–6818 CrossRef CAS PubMed.
- M. N. Christiansen, J. Chik, L. Lee, M. Anugraham, J. L. Abrahams and N. H. Packer, Proteomics, 2014, 14, 525–546 CrossRef CAS PubMed.
- S. Holst, A. J. M. Deuss, G. W. van Pelt, S. J. van Vliet, J. J. Garcia-Vallejo, C. A. M. Koeleman, A. M. Deelder, W. E. Mesker, R. A. Tollenaar, Y. Rombouts and M. Wuhrer, Mol. Cell. Proteomics, 2016, 15, 124–140 CAS.
- D. J. Vigerust, Cent. Eur. J. Biol., 2011, 6, 802–816 CAS.
- H. H. Freeze and M. Aebi, Curr. Opin. Struct. Biol., 2005, 15, 490–498 CrossRef CAS PubMed.
- B. Wollscheid, D. Bausch-Fluck, C. Henderson, R. O'Brien, M. Bibel, R. Schiess, R. Aebersold and J. D. Watts, Nat. Biotechnol., 2009, 27, 378–386 CrossRef CAS PubMed.
- H. Zhang, X. J. Li, D. B. Martin and R. Aebersold, Nat. Biotechnol., 2003, 21, 660–666 CrossRef CAS PubMed.
- H. Kaji, H. Saito, Y. Yamauchi, T. Shinkawa, M. Taoka, J. Hirabayashi, K. Kasai, N. Takahashi and T. Isobe, Nat. Biotechnol., 2003, 21, 667–672 CrossRef CAS PubMed.
- V. Dotz, R. Haselberg, A. Shubhakar, R. P. Kozak, D. Falck, Y. Rombouts, D. Reusch, G. W. Somsen, D. L. Fernandes and M. Wuhrer, TrAC, Trends Anal. Chem., 2015, 73, 1–9 CrossRef CAS.
- K. Marino, J. Bones, J. J. Kattla and P. M. Rudd, Nat. Chem. Biol., 2010, 6, 713–723 CrossRef CAS PubMed.
- C. Steentoft, S. Y. Vakhrushev, M. B. Vester-Christensen, K. Schjoldager, Y. Kong, E. P. Bennett, U. Mandel, H. Wandall, S. B. Levery and H. Clausen, Nat. Methods, 2011, 8, 977–982 CrossRef CAS PubMed.
- I. Hang, C. W. Lin, O. C. Grant, S. Fleurkens, T. K. Villiger, M. Soos, M. Morbidelli, R. J. Woods, R. Gauss and M. Aebi, Glycobiology, 2015, 25, 1335–1349 CrossRef CAS PubMed.
- M. Schubert, M. J. Walczak, M. Aebi and G. Wider, Angew. Chem., Int. Ed., 2015, 54, 7096–7100 CrossRef CAS PubMed.
- Z. J. Bie, Y. Chen, J. Ye, S. S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2015, 54, 10211–10215 CrossRef CAS PubMed.
- L. K. Mahal, K. J. Yarema and C. R. Bertozzi, Science, 1997, 276, 1125–1128 CrossRef CAS PubMed.
- S. C. Hubbard, M. Boyce, C. T. McVaugh, D. M. Peehl and C. R. Bertozzi, Bioorg. Med. Chem. Lett., 2011, 21, 4945–4950 CrossRef CAS PubMed.
- A. Helenius and M. Aebi, Science, 2001, 291, 2364–2369 CrossRef CAS PubMed.
- D. Shental-Bechor and Y. Levy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8256–8261 CrossRef CAS PubMed.
- M. R. Wormald and R. A. Dwek, Structure, 1999, 7, R155–R160 CrossRef CAS PubMed.
- S. R. Hanson, E. K. Culyba, T. L. Hsu, C. H. Wong, J. W. Kelly and E. T. Powers, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3131–3136 CrossRef CAS PubMed.
- R. J. Sola and K. Griebenow, J. Pharm. Sci., 2009, 98, 1223–1245 CrossRef CAS PubMed.
- C. Q. Wang, M. Eufemi, C. Turano and A. Giartosio, Biochemistry, 1996, 35, 7299–7307 CrossRef CAS PubMed.
- J. R. Yates, C. I. Ruse and A. Nakorchevsky, Annual Review of Biomedical Engineering, Annual Reviews, Palo Alto, 2009, vol. 11, pp. 49–79 Search PubMed.
- J. E. Rexach, C. J. Rogers, S. H. Yu, J. F. Tao, Y. E. Sun and L. C. Hsieh-Wilson, Nat. Chem. Biol., 2010, 6, 645–651 CrossRef CAS PubMed.
- D. F. Zielinska, F. Gnad, K. Schropp, J. R. Wisniewski and M. Mann, Mol. Cell, 2012, 46, 542–548 CrossRef CAS PubMed.
- J. Munoz and A. J. R. Heck, Angew. Chem., Int. Ed., 2014, 53, 10864–10866 CrossRef CAS PubMed.
- Y. Li, F. R. Cross and B. T. Chait, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11323–11328 CrossRef CAS PubMed.
- Z. J. Tan, H. D. Yin, S. Nie, Z. X. Lin, J. H. Zhu, M. T. Ruffin, M. A. Anderson, D. M. Simone and D. M. Lubman, J. Proteome Res., 2015, 14, 1968–1978 CrossRef CAS PubMed.
- L. Hwang, S. Ayaz-Guner, Z. R. Gregorich, W. X. Cai, S. G. Valeja, S. Jin and Y. Ge, J. Am. Chem. Soc., 2015, 137, 2432–2435 CrossRef CAS PubMed.
- D. Bausch-Fluck, A. Hofmann, T. Bock, A. P. Frei, F. Cerciello, A. Jacobs, H. Moest, U. Omasit, R. L. Gundry, C. Yoon, R. Schiess, A. Schmidt, P. Mirkowska, A. Hartlova, J. E. Van Eyk, J. P. Bourquin, R. Aebersold, K. R. Boheler, P. Zandstra and B. Wollscheid, PLoS One, 2015, 10, e0121314 Search PubMed.
- W. X. Chen, J. M. Smeekens and R. H. Wu, Chem. Sci., 2015, 6, 4681–4689 RSC.
- R. H. Wu, W. Haas, N. Dephoure, E. L. Huttlin, B. Zhai, M. E. Sowa and S. P. Gygi, Nat. Methods, 2011, 8, 677–683 CrossRef CAS PubMed.
- T. N. C. Ramya, E. Weerapana, B. F. Cravatt and J. C. Paulson, Glycobiology, 2013, 23, 211–221 CrossRef CAS PubMed.
- X. Wang, Z. F. Yuan, J. Fan, K. R. Karch, L. E. Ball, J. M. Denu and B. A. Garcia, Mol. Cell. Proteomics, 2016, 15, 2462–2475 CAS.
- M. R. Larsen, S. S. Jensen, L. A. Jakobsen and N. H. H. Heegaard, Mol. Cell. Proteomics, 2007, 6, 1778–1787 CAS.
- S. S. Sun, P. Shah, S. T. Eshghi, W. M. Yang, N. Trikannad, S. Yang, L. J. Chen, P. Aiyetan, N. Hoti, Z. Zhang, D. W. Chan and H. Zhang, Nat. Biotechnol., 2015, 34, 84–88 CrossRef PubMed.
- A. Thompson, J. Schafer, K. Kuhn, S. Kienle, J. Schwarz, G. Schmidt, T. Neumann and C. Hamon, Anal. Chem., 2003, 75, 1895–1904 CrossRef CAS PubMed.
- K. L. Kiick, E. Saxon, D. A. Tirrell and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 19–24 CrossRef CAS PubMed.
- D. C. Dieterich, A. J. Link, J. Graumann, D. A. Tirrell and E. M. Schuman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9482–9487 CrossRef CAS PubMed.
- A. J. M. Howden, V. Geoghegan, K. Katsch, G. Efstathiou, B. Bhushan, O. Boutureira, B. Thomas, D. C. Trudgian, B. M. Kessler, D. C. Dieterich, B. G. Davis and O. Acuto, Nat. Methods, 2013, 10, 343–346 CrossRef CAS PubMed.
- S. T. Dieck, L. Kochen, C. Hanus, M. Heumueller, I. Bartnik, B. Nassim-Assir, K. Merk, T. Mosler, S. Garg, S. Bunse, D. A. Tirrell and E. M. Schuman, Nat. Methods, 2015, 12, 411–414 CrossRef PubMed.
- Y. H. Tsai, S. Essig, J. R. James, K. Lang and J. W. Chin, Nat. Chem., 2015, 7, 554–561 CrossRef CAS PubMed.
- V. Hong, N. F. Steinmetz, M. Manchester and M. G. Finn, Bioconjugate Chem., 2010, 21, 1912–1916 CrossRef CAS PubMed.
- B. Belardi, A. de la Zerda, D. R. Spiciarich, S. L. Maund, D. M. Peehl and C. R. Bertozzi, Angew. Chem., Int. Ed., 2013, 52, 14045–14049 CrossRef CAS PubMed.
- M. Grammel and H. C. Hang, Nat. Chem. Biol., 2013, 9, 475–484 CrossRef CAS PubMed.
- Y. Y. Yang, J. M. Ascano and H. C. Hang, J. Am. Chem. Soc., 2010, 132, 3640–3641 CrossRef CAS PubMed.
- J. K. Eng, A. L. McCormack and J. R. Yates, J. Am. Soc. Mass Spectrom., 1994, 5, 976–989 CrossRef CAS PubMed.
- A. Teckchandani, N. Toida, J. Goodchild, C. Henderson, J. Watts, B. Wollscheid and J. A. Cooper, J. Cell Biol., 2009, 186, 99–110 CrossRef CAS PubMed.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Nucleic Acids Res., 2009, 37, 1–13 CrossRef CAS PubMed.
- H. P. Xiao, G. X. Tang and R. H. Wu, Anal. Chem., 2016, 88, 3324–3332 CrossRef CAS PubMed.
- W. X. Chen, J. M. Smeekens and R. H. Wu, Mol. Cell. Proteomics, 2014, 13, 1563–1572 CAS.
- B. Schwanhausser, D. Busse, N. Li, G. Dittmar, J. Schuchhardt, J. Wolf, W. Chen and M. Selbach, Nature, 2011, 473, 337–342 CrossRef PubMed.
- W. X. Chen, J. M. Smeekens and R. H. Wu, Chem. Sci., 2016, 7, 1393–1400 RSC.
- E. Eden, N. Geva-Zatorsky, I. Issaeva, A. Cohen, E. Dekel, T. Danon, L. Cohen, A. Mayo and U. Alon, Science, 2011, 331, 764–768 CrossRef CAS PubMed.
- H. Zola, B. Swart, A. Banham, S. Barry, A. Beare, A. Bensussan, L. Boumsell, C. D. Buckley, H. J. Buhring, G. Clark, P. Engel, D. Fox, B. Q. Jin, P. J. Macardle, F. Malavasi, D. Mason, H. Stockinger and X. F. Yang, J. Immunol. Methods, 2007, 319, 1–5 CrossRef CAS PubMed.
- J. Seibel, S. Konig, A. Gohler, S. Doose, E. Memmel, N. Bertleff and M. Sauer, Expert Rev. Proteomics, 2013, 10, 25–31 CrossRef CAS PubMed.
- I. Chen, M. Howarth, W. Y. Lin and A. Y. Ting, Nat. Methods, 2005, 2, 99–104 CrossRef CAS PubMed.
- N. George, H. Pick, H. Vogel, N. Johnsson and K. Johnsson, J. Am. Chem. Soc., 2004, 126, 8896–8897 CrossRef CAS PubMed.
- H. S. Wiley, Exp. Cell Res., 2003, 284, 78–88 CrossRef CAS PubMed.
- A. Gschwind, S. Hart, O. M. Fischer and A. Ullrich, EMBO J., 2003, 22, 2411–2421 CrossRef CAS PubMed.
- J. M. Peng, J. E. Elias, C. C. Thoreen, L. J. Licklider and S. P. Gygi, J. Proteome Res., 2003, 2, 43–50 CrossRef CAS PubMed.
- J. E. Elias and S. P. Gygi, Nat. Methods, 2007, 4, 207–214 CrossRef CAS PubMed.
- L. Kall, J. D. Canterbury, J. Weston, W. S. Noble and M. J. MacCoss, Nat. Methods, 2007, 4, 923–925 CrossRef PubMed.
- E. L. Huttlin, M. P. Jedrychowski, J. E. Elias, T. Goswami, R. Rad, S. A. Beausolei, J. Villen, W. Haas, M. E. Sowa and S. P. Gygi, Cell, 2010, 143, 1174–1189 CrossRef CAS PubMed.
- S. A. Beausoleil, J. Villen, S. A. Gerber, J. Rush and S. P. Gygi, Nat. Biotechnol., 2006, 24, 1285–1292 CrossRef CAS PubMed.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Nat. Protoc., 2008, 4, 44–57 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Two supplementary figures, and eleven supplementary tables. See DOI: 10.1039/c6sc01814a |
| This journal is © The Royal Society of Chemistry 2017 |