DOI:
10.1039/C6SC01389A
(Edge Article)
Chem. Sci., 2016,
7, 5219-5223
New insights into the biosynthesis of fosfazinomycin†
Received
29th March 2016
, Accepted 22nd April 2016
First published on 6th May 2016
Abstract
The biosynthetic origin of a unique hydrazide moiety in the phosphonate natural product fosfazinomycin is unknown. This study presents the activities of five proteins encoded in its gene cluster. The flavin-dependent oxygenase FzmM catalyses the oxidation of L-Asp to N-hydroxy-Asp. When FzmL is added, fumarate is produced in addition to nitrous acid. The adenylosuccinate lyase homolog FzmR eliminates acetylhydrazine from N-acetyl-hydrazinosuccinate, which in turn is the product of FzmQ-catalysed acetylation of hydrazinosuccinate. Collectively, these findings suggest a path to N-acetylhydrazine from L-Asp. The incorporation of nitrogen from L-Asp into fosfazinomycin was confirmed by isotope labelling studies. Installation of the N-terminal Val of fosfazinomycin is catalysed by FzmI in a Val-tRNA dependent process.
Introduction
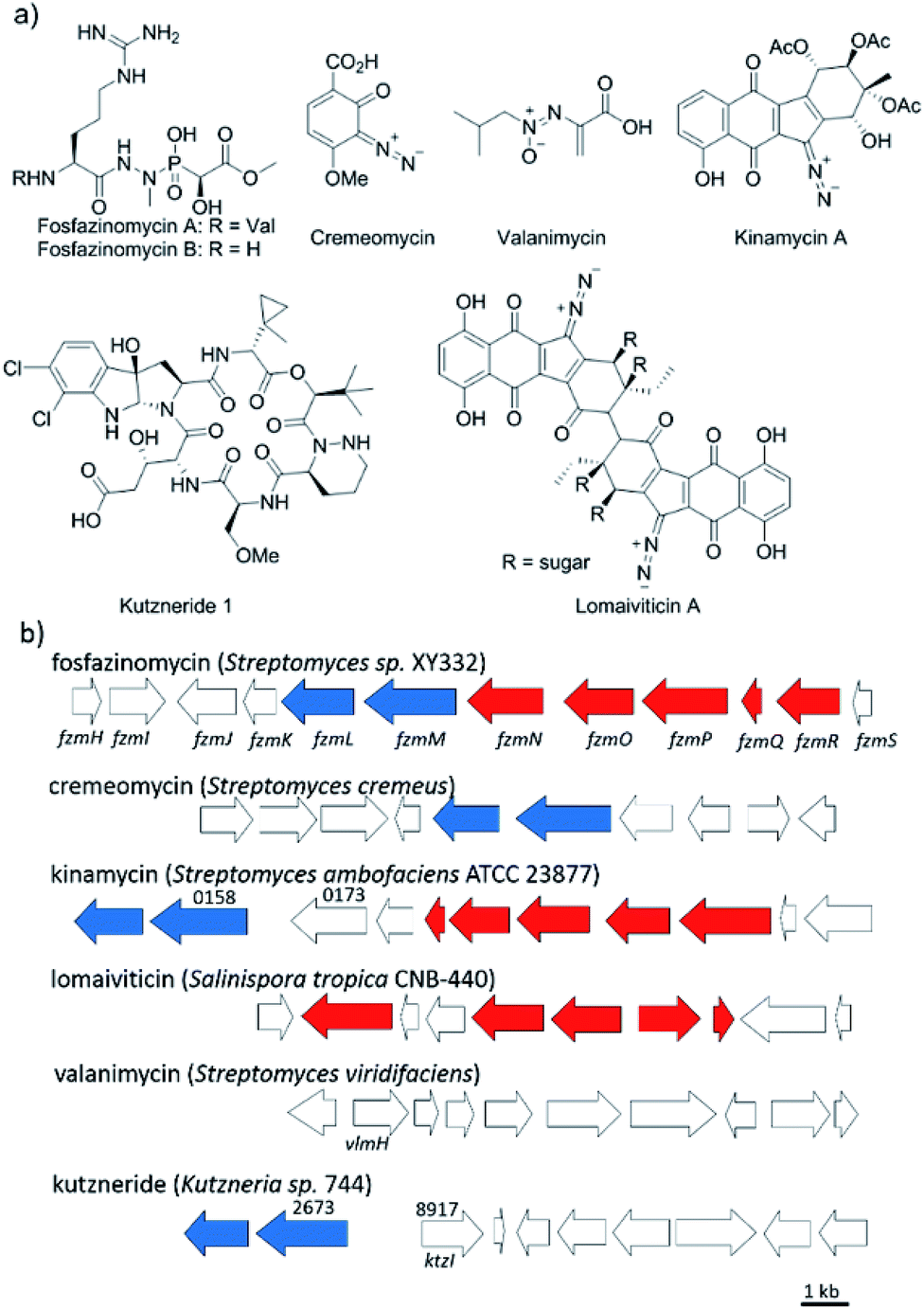
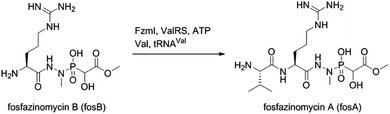
Nitrogen–nitrogen bonds are found in a number of natural products with antimicrobial, antifungal, and anti-tumor activities.1,2 At present, the mechanism of N–N bond formation is not understood.3,4 In recent years, the biosynthetic gene clusters have been reported for several such compounds, including valanimycin,3,5 kutzneride,4,6 and fosfazinomycin,7 as well as molecules containing diazo groups such as kinamycin,8,9 lomaiviticin,10,11 and cremeomycin12,13 (Fig. 1a). Comparison of these gene clusters does not identify genes that are present in all, but a subset of genes is found in more than one cluster (Fig. 1b). For instance, the kinamycin, cremeomycin, and fosfazinomycin clusters contain a pair of genes encoding a flavin-dependent oxidoreductase as well as a homolog of 3-carboxymuconate cycloisomerase (for fosfazinomycin, these are fzmM and fzmL, respectively). For kutzneride, a similar pair of genes is located at a remote locus (Fig. 1b), but they are absent from the cluster of valanimycin and the genome of the lomaiviticin producer. Conversely, a set of five other genes is conserved in the clusters of fosfazinomycin, the kinamycins, and lomaiviticin (Fig. 1b). These genes encode homologs of glutamine synthetase, amidase, adenylosuccinate lyase, and acetyl transferase enzymes, as well as a protein of unknown function. These five genes are not found in the clusters of cremeomycin and the kutznerides. Interestingly, the biosynthetic cluster for fosfazinomycin contains all seven genes. Prompted by the recent disclosure of activities of the 3-carboxymuconate cycloisomerase and oxidoreductase homologs from cremeomycin biosynthesis,13 we report here activities of these two enzymes from the fosfazinomycin cluster (FzmM and FzmL) as well as activities for the adenylosuccinate lyase (FzmR), acetyl transferase (FzmQ), and a protein involved in incorporation of the N-terminal Val (FzmI).
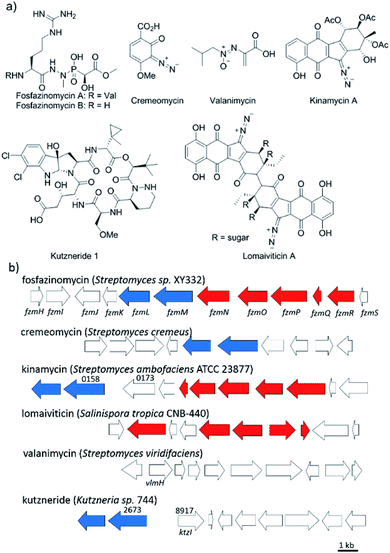 |
| | Fig. 1 (a) Structures of a selection of compounds containing N–N bonds for which the biosynthetic gene clusters have been reported. (b) Partial biosynthetic gene clusters of the compounds shown in panel (a). Genes conserved in fosfazinomycin and lomaiviticin clusters are shown in red, whereas genes conserved in fosfazinomycin and cremeomycin clusters are shown in blue. The locus numbers of the genes are labelled above the genes. | |
Results and discussion
Fosfazinomycins are unique in that both nitrogens are engaged in hydrazide bonds. Hence, unlike the other molecules in Fig. 1, which are thought to be assembled from the stepwise addition of a nitrogen to a pre-installed amino group,3–6,12–17 fosfazinomycin biosynthesis most likely involves a separately biosynthesised hydrazine derivative. To investigate this hypothesis, we conducted in vitro studies with a subset of N-terminally hexa-histidine tagged biosynthetic enzymes expressed in E. coli (Fig. S1–S5 in the ESI†). We first investigated the activity of the set of five conserved enzymes. Adenylosuccinate lyase delivers a nitrogen from Asp in purine biosynthesis (Fig. 2a), suggesting that its homolog FzmR might deliver a nitrogen from Asp to the hydrazine moiety of fosfazinomycin. FzmR did not exhibit any activity when incubated with the canonical substrate adenylosuccinate. We next investigated the hydrazine analog of Asp (hydrazinosuccinate), which was also not a substrate. However, FzmR converted N-acetylhydrazinosuccinate into AcNHNH2 and fumarate (eqn (1); Fig. S6 and S7†).| | 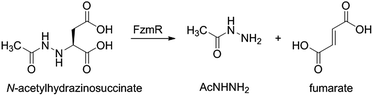 | (1) |
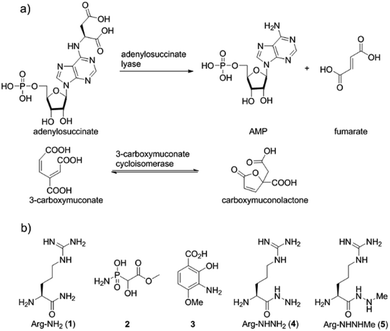 |
| | Fig. 2 (a) Canonical reactions catalysed by homologs of FzmR and FzmL. (b) Structures of compounds discussed in the text. | |
We then investigated whether hydrazinosuccinate could be acetylated by the acetyltransferase FzmQ that is one of the aforementioned set of five conserved enzymes. Incubation of FzmQ with Ac-CoA and hydrazinosuccinate indeed generated the acetylated product as confirmed by LC-MS (Fig. S9†) and NMR analysis (Fig. 3a and b). Acetylation was not observed for L-Asp, L-Arg, and L-Glu. The kinetics of hydrazinosuccinate acetylation were determined using a spectrophotometric assay that monitored the formation of CoA.18 The apparent kcat and Km values (for hydrazinosuccinate) were 2.1 ± 0.1 s−1 and 4.2 ± 0.8 μM, respectively, giving a value for kcat/Km of 5 × 105 M−1 s−1 (Fig. S10†). This high catalytic efficiency suggests hydrazinosuccinate is likely the physiological substrate of FzmQ (eqn (2)).
| | 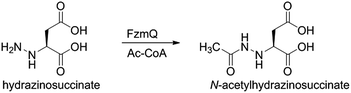 | (2) |
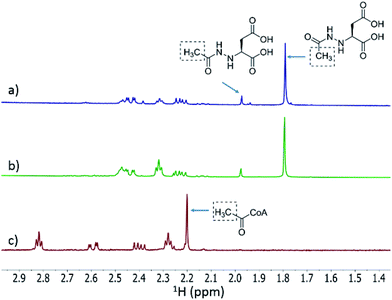 |
| | Fig. 3
1H NMR spectra of the assay of His6-FzmQ with hydrazinosuccinate and Ac-CoA. (a) 1H NMR spectrum of the assay in panel (b) spiked with an authentic standard of N-acetylhydrazinosuccinate. (b) NMR spectrum of the reaction mixture containing hydrazinosuccinate, Ac-CoA, and His6-FzmQ. (c) NMR spectrum of the reaction mixture in the absence of His6-FzmQ. Note: both enzymatically prepared and synthetic N-acetylhydrazinosuccinate exist as a pair of rotational isomers (see ESI†). | |
Because the flavin-dependent oxidoreductase FzmM was predicted based on its sequence to be an N-hydroxylase, we next used a Hansatech O2 electrode to investigate its potential substrates. O2 was rapidly consumed in the presence of L-Asp, FzmM, FAD, and NADPH. Other amino acids that were tested such as L-Glu, L-Orn, L-Lys, and D-Asp, and cofactors like NADH and FMN resulted in negligible O2 consumption. We also tested other potential substrates such as compounds 1 and 2 (Fig. 2b),19 but observed no activity. We anticipated that N-hydroxyaspartic acid (N-hydroxy-Asp) would be the product of the FzmM reaction with L-Asp. Because of its reported instability,20 we derivatised the enzymatic product with Fmoc-Cl followed by LC-MS analysis (Fig. S11–S14†), which showed that Fmoc-N-hydroxy-Asp was indeed produced (Scheme 1).
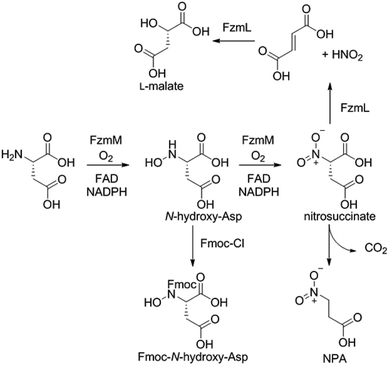 |
| | Scheme 1 | |
When L-3-13C-Asp was used as substrate for FzmM, two new peaks A and B were observed in the 13C NMR spectrum (Fig. 4). The disappearance of peak A at longer incubation times suggested that peak A was likely the unstable product N-hydroxy-Asp that was identified by LC-MS. To identify the structure of product B, a phosphite dehydrogenase NADPH regeneration system was used that boosted the reaction conversion (Fig. S15†).21 Spectroscopic analysis (Fig. S15–S19†) revealed that product B is 3-nitropropionic acid22 (NPA, Scheme 1). Addition of catalase and superoxide dismutase did not alter the reaction outcome. The apparent kcat and Km with L-Asp as substrate were 2.48 ± 0.06 s−1 and 0.79 ± 0.07 mM, respectively, giving an overall efficiency of 3.1 × 103 M−1 s−1 (Fig. S20†). This value is comparable to that of other flavin-dependent N-hydroxylases, including CreE from the cremeomycin and KtzI from the kutzneride pathways,4,13 suggesting L-Asp is the physiological substrate of FzmM.
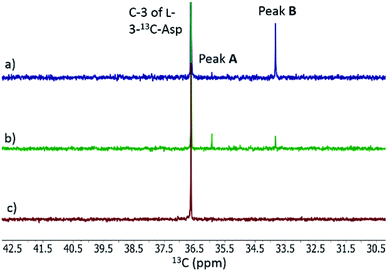 |
| | Fig. 4
13C NMR analysis of the activity of His6-FzmM on L-3-13C-Asp. (a) Same as panel (b) except the spectrum was recorded after incubation for 5 h. (b) NMR spectrum of the reaction mixture containing L-3-13C-Asp, FAD, NADPH, O2, and His6-FzmM after incubation for 10 min. (c) NMR spectrum of the reaction mixture in the absence of His6-FzmM after incubation for 23 h. | |
We also investigated whether the initial N-hydroxy-Asp product could potentially be acetylated by FzmQ by co-incubation of FzmM and FzmQ with L-3-13C-Asp, O2, NADPH, FAD, and Ac-CoA. In addition to N-hydroxy-Asp and NPA, no other product peaks were observed in the 13C NMR spectra, suggesting FzmQ is unable to acetylate N-hydroxy-Asp.
We next investigated potential alternative fates of the FzmM product(s). Coincubation of L-3-13C-Asp, FzmM, and the 3-carboxymuconate cycloisomerase (FzmL) afforded fumarate and L-malate (Scheme 1 and Fig. S21†). The latter was demonstrated to be the product of FzmL-catalysed hydration of fumarate. In addition, nitrous acid was detected in the coincubation assay using Saltzman's reagent (Fig. S22†).13 Similar results were very recently reported for the combination of the two homologous enzymes from the cremeomycin pathway (sequence identities 54 and 60%, respectively).13 Nitrous acid was not observed upon coincubation of FzmR with L-Asp and FzmM. Instead, NPA was formed with or without the addition of FzmR (Fig. S21†). This observation shows once again that FzmR has high preference for N-acetylhydrazinosuccinate.
The findings reported thus far in combination with the recent report on cremeomycin biosynthesis13 allow formulation of a putative pathway for hydrazine formation (Scheme 2). Our data show that FzmM together with FzmL produces nitrous acid from Asp. In cremeomycin biosynthesis, it was suggested that this nitrous acid converts the aniline precursor 3 (Fig. 2b) to the final diazo product.13 Similarly, nitrite has been shown to be incorporated into the terminal nitrogen of the diazo moiety of SF2415A3.14 Here we propose that a similar reaction with Asp as the substrate would result in formation of the N–N bond (Scheme 2). This step would probably be catalysed by a currently unknown enzyme in the cluster, in which several proteins remain functionally unassigned. Because of the highly reactive nature of aliphatic diazo compounds, enzymatic reduction of the proposed diazo intermediate would need to immediately occur to generate hydrazinosuccinate. The latter would be acetylated by FzmQ, and the hydrazine unit would be liberated by FzmR in the form of acetylhydrazine, which then sets the stage for forming the hydrazide bonds in fosfazinomycin. This process requires removal of the acetyl group (possibly by the amidase FzmO) and condensation with Arg and methyl 2-hydroxy-2-phosphono-acetate. We previously suggested that the cluster contains several genes that could form the two hydrazide bonds and the amide bond between Val and Arg: an ATP-GRASP enzyme (FzmF), a Gln and an Asn synthase (FzmN and A), and an aminoacyl-tRNA peptidyl transferase (FzmI).19 At present we have not been able to produce soluble amidase, and we have not observed condensation of Arg with hydrazine or acetylhydrazine in the presence of any of these four proteins, which we expressed in E. coli and purified. However, incubation of FzmI with fosfazinomycin B (Fig. 1a), L-Val, E. coli total tRNAs, Val-tRNA synthetase, and ATP resulted in the production of fosfazinomycin A (eqn (3); Fig. S23–S25†). A brief substrate scope study indicated that in addition to fosfazinomycin B, FzmI was able to ligate Arg, Arg-NHNH2, and Arg-NHNHMe19 to Val under the same reaction conditions to afford the corresponding dipeptides (Fig. S26–S28†). Thus, the list of potential enzymes that can form the hydrazide linkages is reduced to three, which will be the focus of future work.
| |  | (3) |
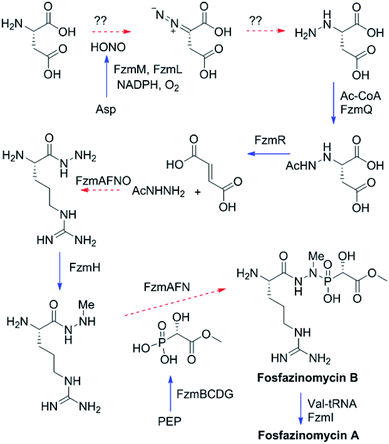 |
| | Scheme 2 Proposed biosynthetic pathway of fosfazinomycin A. Solid blue arrows indicate steps that have been reconstituted in this or a previous study;19 dashed red arrows are proposed and the potential proteins catalyzing these steps are indicated. PEP, phosphoenolpyruvate. | |
The proposed pathway in Scheme 2 predicts that both nitrogens in fosfazinomycin may derive from Asp. At present, no studies have provided direct evidence that in vivo Asp is the donor of any of the nitrogens involved in N–N bonds in natural products. In kutzneride and valanimycin, both nitrogens have been shown to originate from other amino acids (Orn, and Ser and Val, respectively).4,23,24 We therefore grew the fosfazinomycin producing organism Streptomyces sp. NRRL S-149 on minimal medium containing 15N-Asp and/or 15NH4Cl. As shown in Fig. 5a, when both sources of labelled nitrogen were present, a doublet signal was observed for fosfazinomycin by 31P NMR spectroscopy (J = 10.2 Hz). When only 15N-Asp was labelled, the doublet was replaced by two overlapping signals (Fig. 5b). MS analysis showed that the majority of fosfazinomycin was unlabelled and a minor fraction contained one 15N. These findings suggest the NMR peaks are made up of a singlet from unlabelled fosfazinomycin A and a smaller doublet from material that contains a 15N adjacent to the phosphorus atom. When unlabelled Asp was used along with 15NH4Cl (Fig. 5c) again two signals were observed by 31P NMR spectroscopy. MS analysis indicated that the majority of the sample contained seven 15N atoms and that in about 40% of the samples one of the 15N atoms was replaced by an unlabelled nitrogen that must come from Asp. This observation suggests that the incorporation is specific and not the result of scrambling. Collectively, these findings indicate that nitrogen from exogenously supplied Asp is indeed incorporated into fosfazinomycin at the position adjacent to the phosphorus atom.
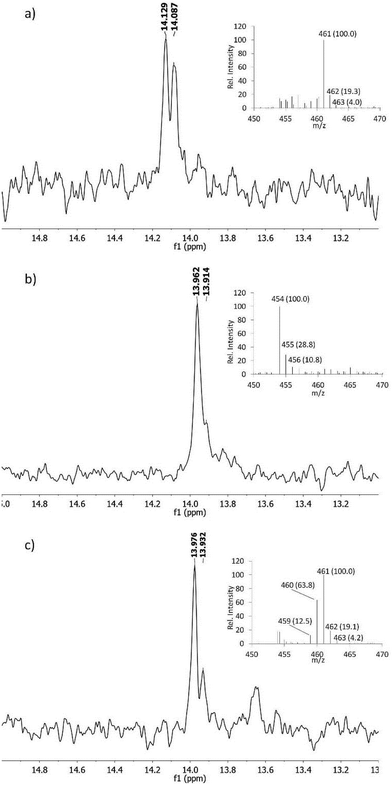 |
| | Fig. 5
31P NMR spectra of fosfazinomycin produced by Streptomyces sp. NRRL S-149. The strain was grown on 15NH4Cl and 15N-Asp (a), NH4Cl and 15N-Asp (b), and 15NH4Cl and unlabelled Asp (c). MS data for each spectrum are shown as insets. 31P NMR chemical shifts for phosphonates are sensitive to pH because their pKa,2 is close to the pH of the producing medium. Hence, the three samples show slightly different chemical shift values. | |
In all experiments, the incorporation pathway using exogenously supplied Asp competes with endogenous biosynthesis of Asp from NH4Cl and the efficiency of incorporation varies somewhat but is <40%. We tried to ascertain whether both nitrogens in fosfazinomycin derive from Asp. However, the dilution caused by endogenous Asp biosynthesis prevented us to unambiguously confirm or refute this hypothesis (see ESI†). Use of an Asp auxotrophic strain will be needed to answer this question, but because heterologous production of fosfazinomycin has been challenging,7 this approach has not yet been possible.
Conclusions
In summary, we report in vitro activities for five enzymes in the fosfazinomycin biosynthetic pathway. As in cremeomycin biosynthesis, two of these make nitrite from Asp. However, the pathways differ in that the N–N bond of fosfazinomycin appears to be made on Asp rather than on a biosynthetic intermediate that already has one of the nitrogens installed. In addition, through metabolic labelling studies, we provide the first experimental evidence that at least one, and possibly both, of the nitrogens in the hydrazide moiety of this natural product derive from Asp. These findings set the stage for the investigation of the remaining steps of the biosynthetic pathway.
Methods
For details regarding molecular biology procedures, protein expression and purifications, and enzyme assays, see the ESI.†
Acknowledgements
The authors thank Dr Despina Bougioukou (UIUC) for help with cloning of valine tRNA synthetase, Dr Jaeheon Lee (UIUC) for collecting FTICR-MS data, Xiling Zhao (UIUC) for help with the preparation of phosphite dehydrogenase, and Emily Ulrich (UIUC) for help with generating the Michaelis–Menten graphs. This work was supported by the National Institutes of Health (GM PO1 GM077596 to W. A. V.) NMR spectra were recorded on a 600 MHz instrument purchased with support from NIH Grant S10 RR028833.
Notes and references
- L. M. Blair and J. Sperry, J. Nat. Prod., 2013, 76, 794–812 CrossRef CAS PubMed.
- G. Le Goff and J. Ouazzani, Bioorg. Med. Chem., 2014, 22, 6529–6544 CrossRef CAS PubMed.
- R. P. Garg, L. B. Alemany, S. Moran and R. J. Parry, J. Am. Chem. Soc., 2009, 131, 9608–9609 CrossRef CAS PubMed.
- C. S. Neumann, W. Jiang, J. R. Heemstra Jr, E. A. Gontang, R. Kolter and C. T. Walsh, ChemBioChem, 2012, 13, 972–976 CrossRef CAS PubMed.
- R. P. Garg, Y. Q. Ma, J. C. Hoyt and R. J. Parry, Mol. Microbiol., 2002, 46, 505–517 CrossRef CAS PubMed.
- D. G. Fujimori, S. Hrvatin, C. S. Neumann, M. Strieker, M. A. Marahiel and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16498–16503 CrossRef PubMed.
- J. T. Gao, K. S. Ju, X. M. Yu, J. E. Vélasquez, S. Mukherjee, J. Lee, C. M. Zhao, B. S. Evans, J. R. Doroghazi, W. W. Metcalf and W. A. van der Donk, Angew. Chem., Int. Ed., 2014, 53, 1334–1337 CrossRef CAS PubMed.
- S. J. Gould, S. T. Hong and J. R. Carney, J. Antibiot., 1998, 51, 50–57 CrossRef CAS PubMed.
- R. Bunet, L. Song, M. V. Mendes, C. Corre, L. Hotel, N. Rouhier, X. Framboisier, P. Leblond, G. L. Challis and B. Aigle, J. Bacteriol., 2011, 193, 1142–1153 CrossRef CAS PubMed.
- R. D. Kersten, A. L. Lane, M. Nett, T. K. Richter, B. M. Duggan, P. C. Dorrestein and B. S. Moore, ChemBioChem, 2013, 14, 955–962 CrossRef CAS PubMed.
- J. E. Janso, B. A. Haltli, A. S. Eustaquio, K. Kulowski, A. J. Waldman, L. Zha, H. Nakamura, V. S. Bernan, H. Y. He, G. T. Carter, F. E. Koehn and E. P. Balskus, Tetrahedron, 2014, 70, 4156–4164 CrossRef CAS PubMed.
- A. J. Waldman, Y. Pechersky, P. Wang, J. X. Wang and E. P. Balskus, ChemBioChem, 2015, 16, 2172–2175 CrossRef CAS PubMed.
- Y. Sugai, Y. Katsuyama and Y. Ohnishi, Nat. Chem. Biol., 2015, 12, 73–75 CrossRef PubMed.
- J. M. Winter, A. L. Jansma, T. M. Handel and B. S. Moore, Angew. Chem., Int. Ed., 2009, 48, 767–770 CrossRef CAS PubMed.
- S. J. Gould, C. R. Melville, M. C. Cone, J. Chen and J. R. Carney, J. Org. Chem., 1997, 62, 320–324 CrossRef CAS PubMed.
- S. J. Gould and C. R. Melville, Bioorg. Med. Chem. Lett., 1995, 5, 51–54 CrossRef CAS.
- P. J. Seaton and S. J. Gould, J. Am. Chem. Soc., 1988, 110, 5912–5914 CrossRef CAS.
- A. L. Sikora, B. A. Frankel and J. S. Blanchard, Biochemistry, 2008, 47, 10781–10789 CrossRef CAS PubMed.
- Z. D. Huang, K. K. A. Wang, J. Lee and W. A. van der Donk, Chem. Sci., 2015, 6, 1282–1287 RSC.
- B. Weiner, G. J. Poelarends, D. B. Janssen and B. L. Feringa, Chem.–Eur. J., 2008, 14, 10094–10100 CrossRef CAS PubMed.
- J. E. Hung, E. J. Fogle, H. D. Christman, T. W. Johannes, H. M. Zhao, W. W. Metcalf and W. A. van der Donk, Biochemistry, 2012, 51, 4254–4262 CrossRef CAS PubMed.
- R. L. Baxter, S. L. Smith, J. R. Martin and A. B. Hanley, J. Chem. Soc., Perkin Trans. 1, 1994, 2297–2299 RSC.
- M. Yamato, T. Takeuchi, H. Umezawa, N. Sakata, H. Hayashi and M. Hori, J. Antibiot., 1986, 39, 1263–1269 CrossRef CAS PubMed.
- R. J. Parry, Y. Li and F. L. Lii, J. Am. Chem. Soc., 1992, 114, 10062–10064 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures, molecular biology processes, enzyme purifications, and synthetic procedures for the preparation of substrates and standards and their spectroscopic characterization. See DOI: 10.1039/c6sc01389a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*