
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd-catalyzed dehydrogenative annulation approach for the efficient synthesis of phenanthridinones†
Xinyao
Li
a,
Jun
Pan
a,
Song
Song
a and
Ning
Jiao
*ab
aState Key Laboratory of Natural and Biomimetic Drugs, School of Pharmaceutical Sciences, Peking University, Xue Yuan Rd. 38, Beijing 100191, China. E-mail: jiaoning@pku.edu.cn
bState Key Laboratory of Elemento-organic Chemistry, Nankai University, Weijin Rd. 94, Tianjin 300071, China
First published on 26th April 2016
Abstract
A novel Pd-catalyzed intermolecular dehydrogenative annulation of aryl iodides and aryl carbamic chlorides for the efficient synthesis of phenanthridinone derivatives was developed. Simple aryl iodides and carbamic chlorides readily made from various anilines, a broad substrate scope with hetero/polycycles, as well as high-value products, make this direct dehydrogenative annulation approach very practical and attractive.
Introduction
Phenanthridinones are ubiquitous fused heterocyclic motifs found in many natural alkaloids and pharmaceutically active compounds, exhibiting broad biological activities such as antitumor, antivirus, and DNA topoisomerase I inhibition.1 The rising use of these biologically significant fused phenanthridinones has stimulated considerable interest in developing their synthetic methods with enhanced generality, scope, and cost effectiveness. Accordingly, many synthetic approaches, including annulation of prefunctionalized substrates to construct phenanthridinones and their derivatives, were developed in the past decades.2 Despite their significance, most of reported methods suffer from the prefunctionalized substrates low efficiency or limited substrate scope. Therefore, direct strategies for the efficient synthesis of diverse phenanthridinones are still highly demanded.Transition-metal-catalyzed dehydrogenative annulation of simple substrates has proven to be a versatile and powerful synthetic strategy to construct polycyclic and heteroaromatic compounds.3 The retrosynthetic analysis of phenanthridinones through a dehydrogenative strategy could be orientated to typical C–C bond formation (I), amide directed C–N bond formation (II), or acylation (III) (Scheme 1a). Using a one chemical bond formation strategy, the intramolecular dehydrogenative cyclization of aryl amides via path I has been significantly developed by the groups of Åkermark,4a Fagnou,4b Dong,4c Cheng,4d and Curran4e (Scheme 1b).
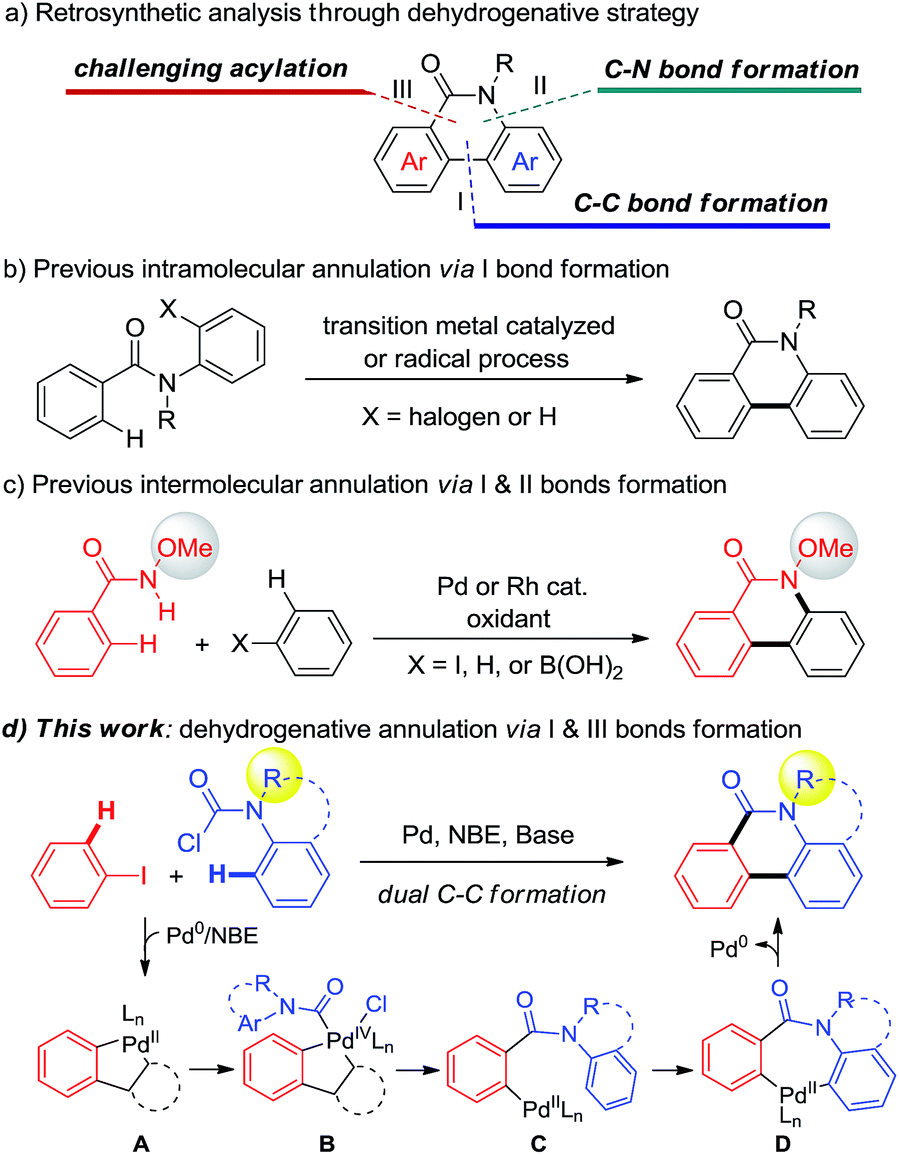 | ||
| Scheme 1 Dehydrogenative annulation strategies for phenanthridinone construction. | ||
In order to disclose new approaches from more simple substrates, a strategy using two chemical bond formation is desired. Recently, Wang5 and Cheng6 independently developed elegant Pd- or Rh-catalyzed intermolecular dehydrogenative cyclization via C–C and C–N bond formation (pathways I and II) to prepare N-methoxy phenanthridinones (Scheme 1c). However, despite the significance, the requirement of stoichiometric oxidant and the CONHOMe directing group limit the substrate scope and their application in the construction of hetero and polycycles. Alternatively, a straightforward approach through two C–C bond formation (pathways I and III) (Scheme 1a) enables the use of various functional groups including some heterocyclic partners from corresponding readily available aniline precursors (Scheme 1d). However, to the best of our knowledge, this strategy is still unknown and poses a challenge due to the adjacent dual C–C bond construction via C–H bond cleavage.7
Recently, Pd/norbornene (NBE) chemistry has been demonstrated to be a significant protocol that allows activation of both the ipso and ortho-positions of arenes.8,9 In continuation of our interest in dehydrogenative annulation for the construction of heterocycles,10 we envisioned that if a proper carbonyl reagent could be employed to form PdIV intermediate B through the palladacycle A species generated in situ from aryl iodides in the presence of NBE, acylation at the ortho-position would be realized and produce the aryl-Pd intermediate C. Subsequently, if species C could undergo the CMD11 process, followed by reductive elimination to realize arylation,12 the desired phenanthridinone derivatives would be constructed (Scheme 1d). Very recently, Dong,13a Gu,13b and Liang13c independently reported unprecedented ortho C–H acylation of aryl iodides through Pd/NBE chemistry. These results encouraged us to test the above dehydrogenative design. Nevertheless, three challenges have to be solved: (1) the carbonyl reagent not only needs to provide the carbonyl group but also has to be a stronger oxidant than aryl halides, to avoid homo-Catellani coupling,14 for oxidizing A. (2) The subsequent dehydrogenative annulation that implicates the intermediacy of seven-membered palladacycle D should be favoured over other quenched processes. (3) Multiple steps including acylation and annulation have to take place successively under mild conditions. Herein, we report the discovery of a novel Pd-catalyzed dehydrogenative annulation of aryl iodides and aryl carbamic chlorides for the efficient synthesis of phenanthridinones (Scheme 1d).
Results and discussion
Initially, we endeavoured to realize the Pd-catalyzed dehydrogenative annulation reaction using iodobenzenes and anilines with norbornene under CO, however, only the ipso position of the iodobenzenes was carbonylated, providing liner amides without cyclization. We hypothesized that properly pre-functionalized carbonyl anilines may inhibit the carbonylation at the ipso position in order to preclude the by-products. As we expected, when 1-iodo-2-methylbenzene (1a) was reacted with methyl(phenyl)carbamic chloride (2a), the intermolecular annulation performed well affording the desired phenanthridinone 3a (Table S1, see ESI†). The desired product, 3a, could be obtained in up to 98% yield when using Pd(OAc)2/PPh3 as the metal/ligand combination, NBE, and Cs2CO3 as the base in DCE at 95 °C (3a, Table 1). A series of control experiments indicated that the palladium, phosphine ligand, NBE, and base were all essential for this transformation (entries 2–5, Table S1, see ESI†).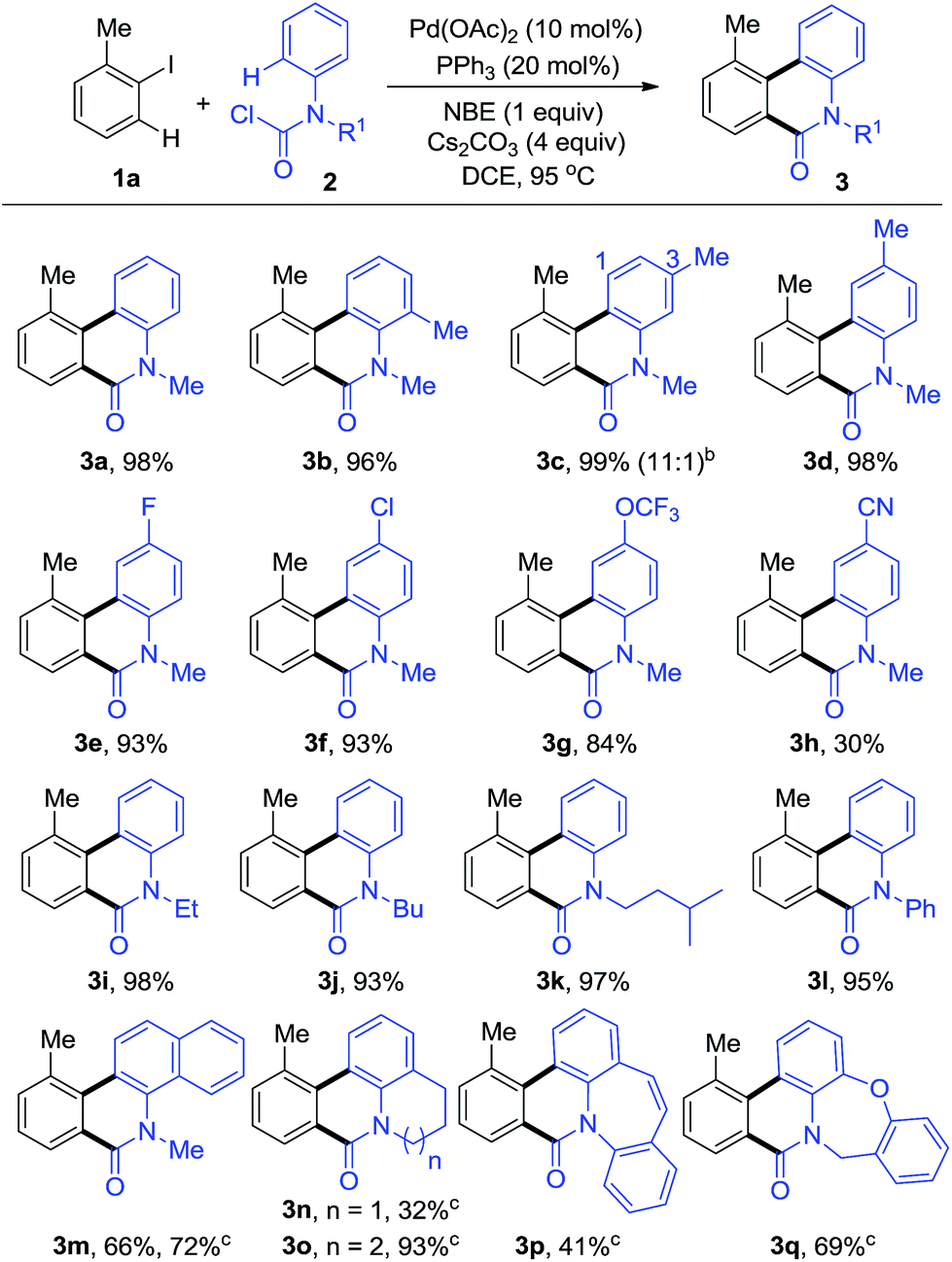
With the optimized reaction conditions in hand, we next explored the scope of aryl carbamic chlorides 2 (Table 1). o-, m-, p-Methyl substituents on the phenyl ring of 2 were well tolerated and the desired phenanthridinones 3b–d were isolated in excellent yields. Among them, 3c was synthesized with good regioselectivity. A series of 2, bearing electron-donating groups or weak electron-withdrawing groups such as F and Cl, and OCF3, were tested and showed excellent performance in this reaction, giving the corresponding products in high yields (3d–g). It is worthy to note that N-alkyl and aryl groups were compatible in this transformation with almost equivalent transformation into corresponding products (3i–l). Furthermore, methyl(naphthalen-1-yl)carbamic chloride (2m) and polycyclic carbamic chlorides (2n–q) were smoothly converted into polycyclic phenanthridinones (3m–q) in moderate to good yields, which are the common core structural motifs in some natural products and bioactive compounds. For these substrates, the reaction proceeded better in toluene than in DCE.
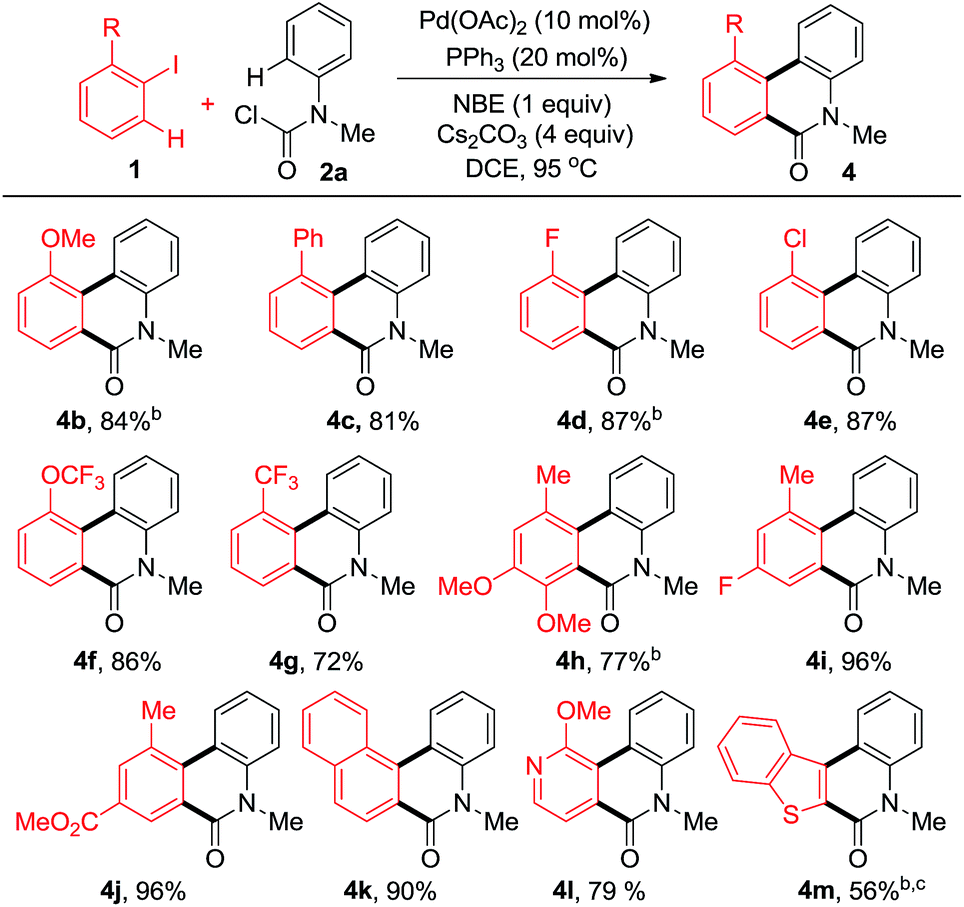
In order to explore the effect of substituents on iodobenzenes, a variety of aromatic and heteroaromatic iodides were employed (Table 2). Different kinds of electron-rich or electron-deficient aryl iodides were compatible under these reaction conditions. Multiple substituted aryl iodides with versatile groups reacted smoothly to give the corresponding products in moderate to good yields (4h–j). Notably, even poly and heterocyclic fused phenanthridinones (4k–m) could be obtained in good yields.
In addition, other kinds of aryl carbamic chlorides were also employed in the annulation reaction (Scheme 2). Although the N–H carbamic chloride did not work, N-PMB and N-benzyl substituted carbamic chlorides (2r–s) could smoothly undergo the annulation to afford the phenanthridinones 3r–s in excellent yields (eqn (1)). Furthermore, the removal of the PMB group proceeded under acidic conditions to give N–H phenanthridinone 5 in 91% yield. When allyl(phenyl)carbamic chloride (2t) was employed, 6a and 6b, instead of the expected N-allyl phenanthridinone, were obtained in 80% and 15% yields, respectively, which actually proceeded through acylation followed by a Heck-reaction (eqn (2)). When vinyl(phenyl)carbamic chloride (2u) took part in the reaction, to our surprise, two spiro-isoindolines 7a and 7b were formed (eqn (3)). Simple iodobenzene without an ortho substituent gave an unseparated mixture containing mono- and di-acylation annulation products, and unknown byproducts.
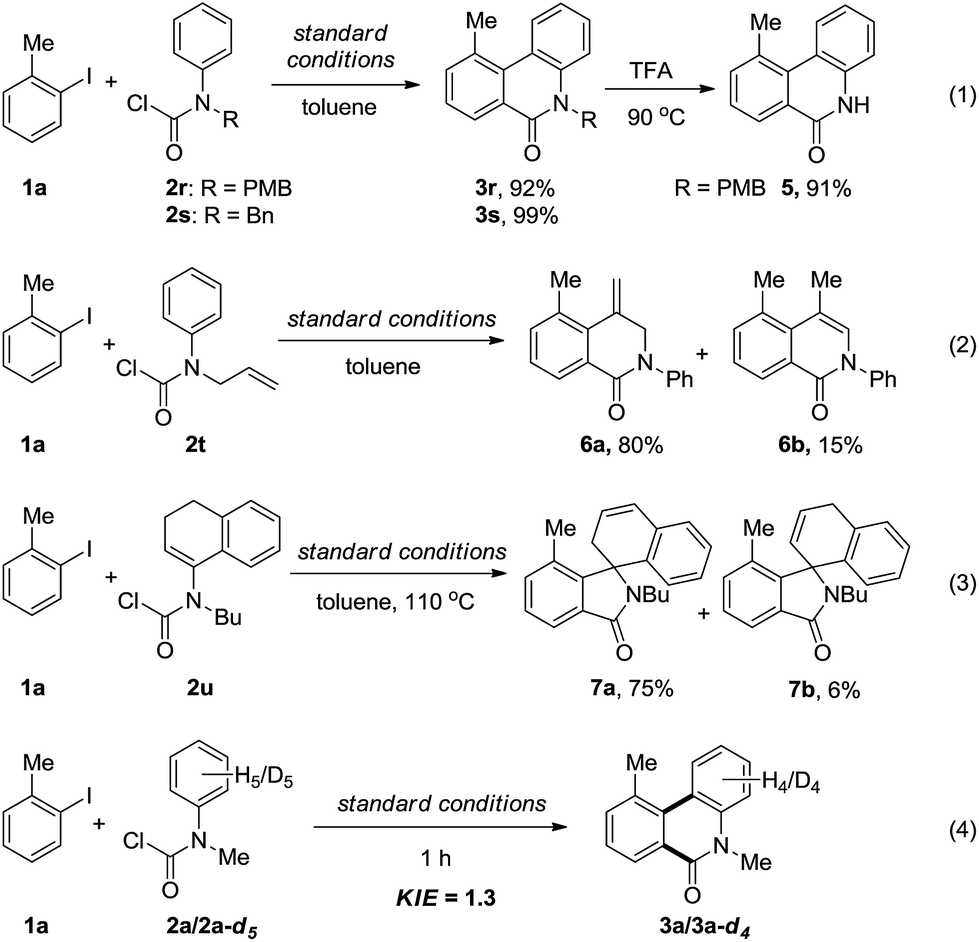 | ||
| Scheme 2 Other substrates and isotope-labeling experiment. | ||
To gain insight into the mechanism, the intermolecular KIE for the annulation reaction was determined to be kH/kD = 1.3, indicating that the C–H bond cleavage should not be involved in the rate-determining step of the catalytic cycle (eqn (4)). Moreover, a linear relationship between log(KR/KH) and substitute constant σ is established for substituted methyl(phenyl)carbamic chloride (R = p-Me, p-H, p-F, p-Cl, p-OCF3, and p-CN) (Fig. 1).15 The resulting Hammett parameter ρ is −0.46, suggesting the reaction is substituent-sensitive and the influence of the electronic effects correspond to the oxidative addition step. Electron withdrawing substituents can decrease the negative charge in carbamic chloride to attenuate its coordination ability and thus destabilize the transition state of oxidant addition.
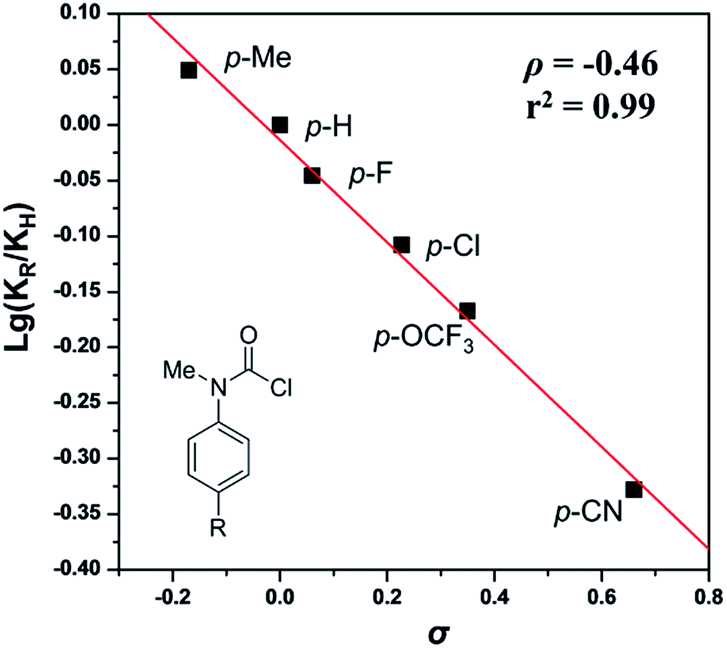 | ||
| Fig. 1 Hammett plot of the annulation of 2. | ||
Based on our preliminary mechanistic studies, and assuming a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry between all reaction components in the catalyst turnover to simplify the discussion, we conducted a density functional theory (DFT) calculation investigation into the direct dehydrogenative annulation of 1a and 2a to better understand the mechanism of this transformation (Fig. 2).16,17 The phenyl groups in the ligand are replaced by methyl groups to save computational time but without sacrificing credibility. Initially, the oxidative addition of 1a with Pd0 could occur on the bisphosphine or monophosphine complex.18 The calculations show that the oxidative addition on the monophosphine complex via transition state TS1 is slightly favoured over that on the bisphosphine complex viaTS1′, indicating that the monophosphine complex is the dominant active species, while the bisphosphine complex is the minor one (see ESI† for details). However, both pathways are facile, requiring an activation free energy of only 16.2 and 16.7 kcal mol−1 to form PdII species INTa exergonicly. The next step, corresponding to the insertion of NBE, is reversible to afford INTb. After base exchange, the formed INTb′ undergoes a CMD process to provide the stable intermediate palladacycle INTc irreversibly. Then, oxidative addition takes place through transition state TS4 with an activation free energy of 29.7 kcal mol−1 to deliver PdIV intermediate INTd. The following reductive elimination of PdIV is facile and exergonic. After acylation, β-C elimination of NBE occurs endergonicly. The subsequent CMD step readily proceeds to furnish cyclic intermediate INTg. Finally, the reductive elimination of INTg is also facile to give the final phenanthridinone 3a, whilst regenerating the Pd0 catalyst. Reviewing the whole energy profile, we find that the direct oxidative addition of the in situ formed palladacycle with aryl carbamic chloride is the rate-determining step, in good agreement with the experimental observation of the Hammett plot.
:1 stoichiometry between all reaction components in the catalyst turnover to simplify the discussion, we conducted a density functional theory (DFT) calculation investigation into the direct dehydrogenative annulation of 1a and 2a to better understand the mechanism of this transformation (Fig. 2).16,17 The phenyl groups in the ligand are replaced by methyl groups to save computational time but without sacrificing credibility. Initially, the oxidative addition of 1a with Pd0 could occur on the bisphosphine or monophosphine complex.18 The calculations show that the oxidative addition on the monophosphine complex via transition state TS1 is slightly favoured over that on the bisphosphine complex viaTS1′, indicating that the monophosphine complex is the dominant active species, while the bisphosphine complex is the minor one (see ESI† for details). However, both pathways are facile, requiring an activation free energy of only 16.2 and 16.7 kcal mol−1 to form PdII species INTa exergonicly. The next step, corresponding to the insertion of NBE, is reversible to afford INTb. After base exchange, the formed INTb′ undergoes a CMD process to provide the stable intermediate palladacycle INTc irreversibly. Then, oxidative addition takes place through transition state TS4 with an activation free energy of 29.7 kcal mol−1 to deliver PdIV intermediate INTd. The following reductive elimination of PdIV is facile and exergonic. After acylation, β-C elimination of NBE occurs endergonicly. The subsequent CMD step readily proceeds to furnish cyclic intermediate INTg. Finally, the reductive elimination of INTg is also facile to give the final phenanthridinone 3a, whilst regenerating the Pd0 catalyst. Reviewing the whole energy profile, we find that the direct oxidative addition of the in situ formed palladacycle with aryl carbamic chloride is the rate-determining step, in good agreement with the experimental observation of the Hammett plot.
 | ||
| Fig. 2 DFT-computed energy profiles for Pd-catalyzed dehydrogenative acylation and annulation of 1a and 2a. | ||
Conclusions
In conclusion, we have demonstrated the first Pd-catalyzed intermolecular direct dehydrogenative annulation of aryl iodides and aryl carbamic chlorides through dual C–H bond activation for the efficient synthesis of phenanthridinone derivatives. This protocol is easy to handle with simple aryl iodide and aryl carbamic chloride substrates readily prepared from various anilines. The use of various functional groups, including some heterocyclic partners, makes this approach very attractive for the construction of high-value and biologically significant phenanthridinone heterocycles. Preliminary experiments and DFT calculations suggest that the in situ formed palladacycle INTc could undergo oxidative addition of aryl carbamic chloride to generate a PdIV intermediate in the rate-determining step. Efforts toward expanding the reaction scope of other fused heterocycles are underway.Acknowledgements
Financial support from the National Basic Research Program of China (973 Program) (grant No. 2015CB856600), National Natural Science Foundation of China (No. 21325206), and National Young Top-notch Talent Support Program are greatly appreciated. We thank Xiaojin Wen in this group for reproducing the results of 3l and 4e.Notes and references
-
(a) D. Weltin, V. Picard, K. Aupeix, M. Varin, D. Oth, J. Marchal, P. Dufour and P. Bischoff, Int. J. Immunopharmacol., 1995, 17, 265 CrossRef CAS PubMed
; (b) S. Patil, S. Kamath, T. Sanchez, N. Neamati, R. F. Schinazi and J. K. Buolamwini, Bioorg. Med. Chem., 2007, 15, 1212 CrossRef CAS PubMed
-
(a) T. N. Le, S. G. Gang and W.-J. Cho, J. Org. Chem., 2004, 69, 2768 CrossRef CAS PubMed
- For reviews on the dehydrogenative annulations, see:
(a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed
-
(a) B. Åkermark, L. Eberson, E. Jonsson and E. Pettersson, J. Org. Chem., 1975, 40, 1365 CrossRef
- G.-W. Wang, T.-T. Yuan and D.-D. Li, Angew. Chem., Int. Ed., 2011, 50, 1380 CrossRef CAS PubMed
-
(a) J. Karthikeyan and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 9880 CrossRef CAS PubMed
-
(a) U. Umeda, H. Tsurugi, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2008, 47, 4019 CrossRef PubMed
-
(a) M. Catellani, F. Frignani and A. Rangoni, Angew. Chem., Int. Ed. Engl., 1997, 36, 119 CrossRef CAS
- For some recent examples, see:
(a) X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334 CrossRef CAS PubMed
-
(a) Z. Shi, C. Zhang, S. Li, D. Pan, S. Ding, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 4572 CrossRef CAS PubMed
-
(a) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754 CrossRef CAS PubMed
-
(a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed
-
(a) Z. Dong, J. Wang, Z. Ren and G. Dong, Angew. Chem., Int. Ed., 2015, 54, 12664 CrossRef CAS PubMed
-
(a) E. Motti, N. Della Ca', S. Deledda, E. Fava, F. Panciroli and M. Catellani, Chem.
Commun., 2010, 46, 4291 CAS
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS
- All of the DFT calculations were performed with the Gaussian 09 program package at the M06 level of theory with the LACVP(d) basis set. All of the energies discussed in the paper are Gibbs free energies in the gas-phase (ΔG). Gibbs free energies with the CPCM solvation correction in DCE (ΔGDCE) based on the structures in the gas phase with the same basis set are also given in parenthesis. Computational details and references are given in the ESI.†.
- G. Maestri, E. Motti, N. Della Ca', M. Malacria, E. Derat and M. Catellani, J. Am. Chem. Soc., 2011, 133, 8574 CrossRef CAS PubMed
-
(a) K. Vikse, T. Naka, J. S. McIndoe, M. Besora and F. Maseras, ChemCatChem, 2013, 5, 3604 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and spectral data. See DOI: 10.1039/c6sc01148a |
| This journal is © The Royal Society of Chemistry 2016 |