
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural engineering of porphyrin-based small molecules as donors for efficient organic solar cells†
Hongda
Wang‡
ab,
Liangang
Xiao‡
c,
Lei
Yan‡
d,
Song
Chen‡
a,
Xunjin
Zhu
*ab,
Xiaobin
Peng
*c,
Xingzhu
Wang
*d,
Wai-Kwok
Wong
*a and
Wai-Yeung
Wong
*ab
aInstitute of Molecular Functional Materials, Department of Chemistry and Institute of Advanced Materials, Hong Kong Baptist University, Waterloo Road, Kowloon Tong, Hong Kong, P. R. China. E-mail: xjzhu@hkbu.edu.hk; wkwong@hkbu.edu.hk; rwywong@hkbu.edu.hk
bHKBU Institute of Research and Continuing Education, Shenzhen Virtual University Park, Shenzhen, 518057, P. R. China
cInstitute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, 381 Wushan Road, Guangzhou 510640, P. R. China. E-mail: chxbpeng@scut.edu.cn
dKey Laboratory of Advanced Functional Polymer Materials of Colleges and Universities of Hunan Province and Key Lab of Environment-friendly Chemistry and Application in Ministry of Education, College of Chemistry, Xiangtan University, Xiangtan 411105, Hunan Province, China. E-mail: xzwang@xtu.edu.cn
First published on 15th March 2016
Abstract
Porphyrin-based small molecules as donors have long been ignored in bulky heterojunction organic solar cells due to their unfavorable aggregation and the low charge mobility. With the aim of striking a delicate balance between molecular design, morphology, interfacial layer and device fabrication to maximize the power conversion efficiency (PCE) of organic solar cells, three comparable porphyrin-based small molecules with an acceptor–donor–acceptor configuration have been developed for use as donor materials in solution processed small molecule bulk heterojunction organic solar cells. In these molecules, electron-deficient 3-ethylrhodanine is introduced into the electron-rich porphyrin core through 5,15-bis(phenylethynyl) linkers. Structural engineering with 10,20-bis(2-hexylnonyl) aliphatic peripheral substituent on the porphyrin core, instead of the aromatic substituents such as 10,20-bis[3,5-di(dodecyloxyl)phenyl], and 10,20-bis(4-dodecyloxylphenyl), can simultaneously facilitate stronger intermolecular π–π stacking and higher charge transfer mobility in the film, leading to a maximum PCE of 7.70% in a conventional device. The inverted devices have also been demonstrated to have long-term ambient stability and a comparable PCE of 7.55%.
1. Introduction
Solution processed small molecule (SM) bulk heterojunction organic solar cells (BHJ OSCs) have shown great prospect in the realization of low-cost and large-scale devices due to their characteristics of well-defined structure and molecular weight, and easy synthesis and purification of small molecules.1,2 Generally, a small molecule as a donor in a solution processed BHJ OSC needs to possess the following features: (i) strong photo-absorption in the visible and near-infrared (NIR) region; (ii) sufficient solubility in solvents; (iii) strong intermolecular interaction in the solid state for efficient charge transfer; and (iv) excellent film formation ability. In the search for appropriate small molecule donors, various frameworks have been examined, including benzo[1,2-b:4,5-b′]dithiophene (BDT),3,4 boron dipyrromethene,5,6 diketopyrrolopyrrole,7–9 phthalocyanine,10 merocyanine,11 and squaraine.12–14 Over the past decade, the power conversion efficiencies (PCEs) of the solution processed SM BHJ OSCs have steadily improved. Since Bazan and Heeger et al. first reported a PCE of 6.7% based on a small molecule donor with 3,3′-di-2-ethylhexylsilylene as the central building block,15 the PCE was steadily enhanced to 9.02% by the structural optimization of both small molecules and devices in BHJ OSCs.16,17 Chen et al. also reported a series of small molecules with an oligothiophene unit as the core unit, and the efficiency was also over 9% after the optimization of the molecular structure.18,19 On the other hand, porphyrins and their analogues exhibit some intrinsic features that are attractive for their application in photovoltaics.20 They have strong absorption with high extinction coefficients (ε) in both the blue (Soret band) and red (Q-band) regions, and their photo- and electrochemistry can readily be adjusted through peripheral functionalization and by varying of the metal center. It is known that “push–pull” porphyrin derivatives have been successfully applied in dye-sensitized solar cells.21–23 However, discouraging PCEs were obtained when employing porphyrins and their analogues as donor materials in BHJ devices. Very recently, Peng and co-workers used {5,15-bis[3,5-di(dodecyloxyl)phenyl] porphyrinato}zinc(II) and 2,1,3-benzothiadiazole (BTD) to construct donor–acceptor–donor (A–D–A) small molecules as donors in BHJ OSCs. Since the two bulky 3,5-di(dodecyloxyl)phenyl substituents that are almost perpendicular to the porphyrin core suppress the intermolecular π–π stacking, a relatively poor photovoltaic performance was achieved.24Subsequently, they employed less bulky meso-substituents such as 4-octyloxy-phenyl and 5-(2-ethylhexyl)-thienyl in the new A–D–A porphyrin-cored molecules, which can exhibit stronger intermolecular π–π stacking in the solid state, leading to a higher hole mobility and better photovoltaic performance.25,26 It is obvious that the meso-phenyl substitutions with an almost orthogonal orientation relative to the porphyrin plane significantly prevent the intermolecular π–π stacking, as well as intermolecular charge transport.27 Our recent work further demonstrated that the direct peripheral meso-alkyl substitutions on the porphyrin ring could not only improve the solubility of small molecules in most organic solvents, but also control the film morphology and crystallinity, leading to an enhancement of charge transport.28
To rigorously evaluate the effect of peripheral aromatic or aliphatic meso-substituents of the porphyrin ring on the light harvesting, solubility, morphology, exciton diffusion and dissociation, charge transport and collection, and ultimate PCE, three A–D–A structural type porphyrin-based small molecules were carefully designed and prepared. Typically, 3-ethylrhodanine as the terminal unit bridged by a phenylethynyl moiety with porphyrins {5,15-bis[3,5-di(dodecyloxyl)phenyl]-porphyrinato}zinc(II), [5,15-bis(4-dodecyloxylphenyl)-porphyrinato]zinc(II), and [5,15-bis(2-hexylnonyl)-porphyrinato]zinc(II), afforded 4a, 4b and 4c, respectively (Scheme 1). Subsequently, their photovoltaic performances as donors were investigated systematically in conventional devices with a configuration of ITO/PEDOT:PSS/SM:PC71BM/PFN/Al (where PFN is poly[(9,9-bis(3′-(N,N-dimethylamino)-propyl)-2,7-fluorene)-alt-2,7-(9,9-dioctylfluorene)]). Inverted devices of ITO/Ca/SM:PC71BM/MoO3/Ag were also fabricated and tested in the following studies.
 | ||
| Scheme 1 Synthetic route for 4a–c. Reaction conditions: (a) NBS, CH2Cl2, pyridine, 0 °C; (b) Zn(AcO)2, CHCl3, reflux, 4 h; (c) Pd(PPh3)4, CuI, Et3N, THF, 50 °C, overnight; (d) 3-ethylrhodanine, CHCl3, piperidine, reflux, overnight. | ||
2. Results and discussion
2.1 Synthesis and characterization
The synthetic route for 4a–c is shown in Scheme 1. It should be noted that the use of the zinc porphyrin complex instead of a porphyrin free base is necessary, due to the higher stability of the metal complex and it also helps to improve the photovoltaic properties of the OSC devices.26 The Sonogashira coupling of 2a–c with 4-ethynyl-2,5-bis(hexyloxy)benzaldehyde produced the porphyrin intermediates 3a–c in good yields. The target compounds 4a–c were prepared by the Knoevenagel condensation of 3-ethylrhodanine with 3a–c, respectively. Although the solubility of 4a and 4b is not as good as 4c in organic solvents such as chloroform, THF, and toluene, all three π-conjugated small molecules could be readily processed in solution to form smooth and pinhole-free films using spin-coating technology.2.2 Optical and electrochemical properties
The normalized UV-vis absorption spectra of 4a–c in CH2Cl2 solutions and spin-casted thin films are depicted in Fig. 1, and the relevant optical data including the wavelengths of the absorption maxima (λmax), molar extinction coefficients, absorption edge wavelengths (λonset), and optical band gaps (Eoptg) are summarized in Table 1. As shown in Fig. 1, 4a in dilute CH2Cl2 solution shows a Soret band at 504 nm with a molar extinction coefficient (ε) of 1.55 × 105 M−1 cm−1 and a Q band at 682 nm with a molar extinction of 1.39 × 105 M−1 cm−1. Additionally, 4b in CH2Cl2 solution presents a similar Soret band at 505 nm with a ε of 1.61 × 105 M−1 cm−1 and a bathochromically shifted Q band at 720 nm with a ε of 1.53 × 105 M−1 cm−1. As for meso-2-hexylnonyl substituted 4c, a Soret band at 502 nm with a ε of 1.95 × 105 M−1 cm−1 and a Q band at 696 nm with a ε of 1.87 × 105 M−1 cm−1 were observed. Apparently, 4c exhibits much stronger absorption at both Soret and Q bands than 4a and 4b. | ||
| Fig. 1 UV-visible-NIR absorption spectra of 4a–c in CH2Cl2 solution (a) and films (b). | ||
| Comp. | λ max/nm (CH2Cl2) (ε/105 M−1 cm−1) | λ max/nm (film) | λ onset/nm (film) | E ox [V] | E HOMO [eV] | E LUMO [eV] | E optg [eV] |
|---|---|---|---|---|---|---|---|
| a HOMO levels were measured in DCM with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF) calibrated with ferrocene/ferrocenium (Fc/Fc+) as an external reference. b The HOMO and LUMO levels were estimated by the following equation: HOMO = −(4.70 + Eox), LUMO = HOMO − EOptg. Optical band gap estimated from the formula of 1240/λonset, λonset is the absorption onset of the film spectrum. | |||||||
| 4a | 475 (1.55), 504 (1.73), 683 (1.39) | 537, 733 | 779 | 0.49 | −5.19 | −3.59 | 1.60 |
| 4b | 484 (1.61), 505 (1.61), 720 (1.53) | 526, 726 | 790 | 0.45 | −5.15 | −3.60 | 1.55 |
| 4c | 472 (1.64), 502 (1.95), 696 (1.81) | 520, 735 | 780 | 0.42 | −5.12 | −3.52 | 1.60 |
There is no significant changes in Soret bands due to minor electronic perturbations between the porphyrin ring and either meso-aliphatic or aromatic substituents.29 In comparison with 4a, the absorption maxima of the Q bands in 4b and 4c exhibit a significant red-shift of 38 and 14 nm, respectively, which indicates a stronger intramolecular charge transfer (ICT) between the donor and acceptor units in 4b and 4c. On the other side, their absorption maxima for both the Soret and Q bands in the thin films were significantly red-shifted, which indicates that a strong π–π intermolecular interaction exists in the solid state due to the optically active J-aggregation.30
Cyclic voltammetry (CV) was performed to investigate the electrochemical properties of 4a–c and the electrochemical data are shown in Table 1. The HOMO energy levels of 4a–c were calculated to be −5.19, −5.15 and −5.12 eV, respectively. Furthermore, the optical band gaps (Eg) for 4a–c were estimated from the absorption spectra in the films with values of 1.60, 1.55 and 1.60 eV, respectively. Therefore the potential levels of the LUMO for 4a–c were obtained from LUMO = HOMO − Eg, with values of −3.59, −3.60 and −3.52 eV, respectively. The HOMO levels of 4a–c are located within the band gap of PC61BM, while the LUMO levels are sufficiently higher (around −3.5 eV) than that of PC61BM (−4.0 eV). The results indicate that 4a–c are compatible with the commonly used acceptor material PC61BM or PC71BM in BHJ OSCs.
2.3 Photovoltaic properties
Subsequently, BHJ OSCs using 4a–c as donor materials were fabricated and tested under AM1.5 illumination, 100 mW cm−2 in a device structure of ITO/PEDOT:PSS/SM:PC61BM (or PC71BM)/PFN/Al. Following the conventional practice for fabricating solution processed SM BHJ OSCs, the active layers were spin-coated from their chlorobenzene solutions with 1 vol% pyridine as an additive and a film thickness of approximately 80 nm was obtained. The optimized J–V curves are reported in Fig. 2a, and the photovoltaic performances are summarized in Table 2. With the optimized weight ratio of 4a to PC61BM at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the best but low PCE of 1.42% was obtained, together with a short circuit current (JSC) of 6.44 mA cm−2, an open-circuit voltage (VOC) of 0.76 V, and a fill factor (FF) of 28.9%. The optimized weight ratio of 4b to PC61BM was also found to be 1:1, yielding a moderate PCE of 4.55% with VOC of 0.80 V, JSC of 10.09 mA cm−2 and FF of 56.3%. In contrast, the device based on 4c/PC61BM (1:1, w/w) received an impressive PCE of 4.98%, with a VOC of 0.89 V, a FF of 52.1%, and a significantly higher JSC of 10.72 mA cm−2 due to its good solubility, high light harvesting properties and strong π–π stacking intermolecular interaction.
:1, the best but low PCE of 1.42% was obtained, together with a short circuit current (JSC) of 6.44 mA cm−2, an open-circuit voltage (VOC) of 0.76 V, and a fill factor (FF) of 28.9%. The optimized weight ratio of 4b to PC61BM was also found to be 1:1, yielding a moderate PCE of 4.55% with VOC of 0.80 V, JSC of 10.09 mA cm−2 and FF of 56.3%. In contrast, the device based on 4c/PC61BM (1:1, w/w) received an impressive PCE of 4.98%, with a VOC of 0.89 V, a FF of 52.1%, and a significantly higher JSC of 10.72 mA cm−2 due to its good solubility, high light harvesting properties and strong π–π stacking intermolecular interaction.
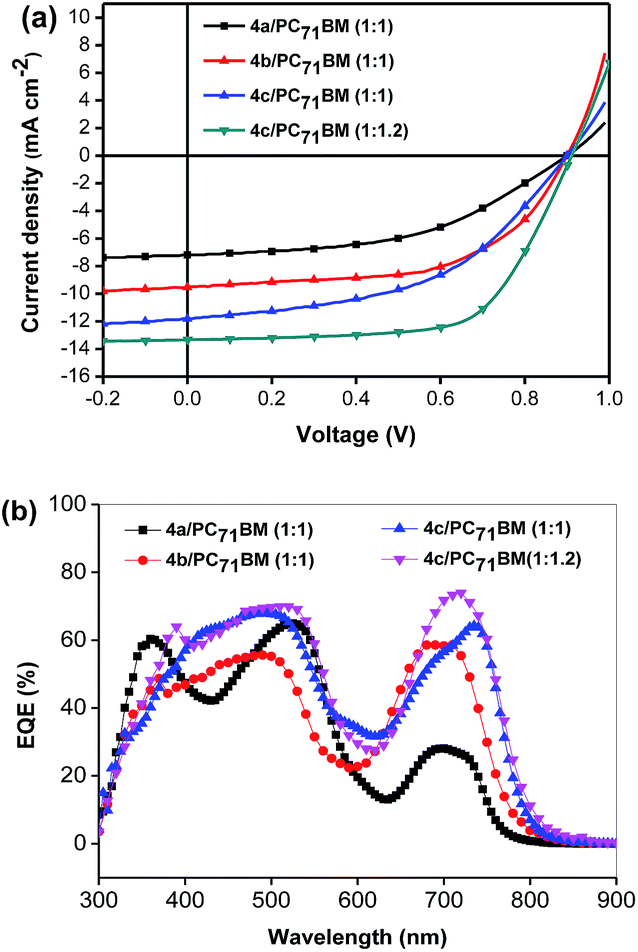 | ||
| Fig. 2 The best J–V curves (a) and EQEs (b) of devices based on 4a–c/PC71BM. | ||
| Device | Additive | Annealing time at 90 °C (min) | J SC (mA cm−2) | V OC (V) | FF (%) | PCE (%) |
|---|---|---|---|---|---|---|
|
a
4a–c/PC61BM or 4a–c/PC71BM with a 1:1 weight ratio in solar cells with conventional structures based on fresh cells.
b
4c/PC71BM with a 1:1.2 weight ratio in solar cells with conventional structures based on fresh cells.
c
4c/PC71BM with a 1:1.2 weight ratio in solar cells with inverted structures based on fresh cells.
d
4c/PC71BM with a 1:1.2 weight ratio in solar cells with inverted structures with encapsulation based on aging for 3 days.
e
4c/PC71BM with a 1:1.2 weight ratio in solar cells with inverted structures with encapsulation based on aging for 10 days.
f
4c/PC71BM with a 1:1.2 weight ratio in solar cells with inverted structures with encapsulation based on based on aging for 30 days.
|
||||||
| 4a/PC61BMa | 1% pyridine | 10 | 6.44 | 0.76 | 28.98 | 1.42 |
| 4a/PC71BMa | 1% pyridine | 10 | 7.20 | 0.90 | 48.12 | 3.21 |
| 4b/PC61BMa | 1% pyridine | 10 | 10.09 | 0.80 | 56.33 | 4.55 |
| 4b/PC71BMa | 1% pyridine | 10 | 10.14 | 0.90 | 55.60 | 5.07 |
| 4c/PC61BMa | 1% pyridine | 10 | 10.72 | 0.89 | 52.12 | 4.98 |
| 4c/PC71BMa | 1% pyridine | 10 | 12.14 | 0.89 | 48.07 | 5.20 |
| 4c/PC71BMa | 1% pyridine | 0 | 4.84 | 0.92 | 29.18 | 1.30 |
| 4c/PC71BMa | 1% pyridine | 5 | 12.28 | 0.91 | 54.67 | 6.11 |
| 4c/PC71BMa | 1% pyridine | 20 | 12.22 | 0.90 | 50.35 | 5.54 |
| 4c/PC71BMb | 1% pyridine | 5 | 13.32 | 0.91 | 63.60 | 7.70 |
| 4c/PC71BMc | 1% pyridine | 0 | 6.26 | 0.892 | 38.1 | 2.12 |
| 4c/PC71BMc | 1% pyridine | 5 | 12.89 | 0.901 | 65.1 | 7.55 |
| 4c/PC71BMd | 1% pyridine | — | 12.87 | 0.910 | 64.5 | 7.55 |
| 4c/PC71BMe | 1% pyridine | — | 12.87 | 0.882 | 59.2 | 6.62 |
| 4c/PC71BMf | 1% pyridine | — | 12.53 | 0.876 | 57.0 | 6.26 |
To further optimize their photovoltaic performance in BHJ OSCs, PC71BM was used as the electron acceptor for its higher absorption coefficient in the visible range. As expected, the active layers based on 4a–c/PC71BM (1:1, w/w) yielded improved device performances with PCEs of 3.21%, 5.07% and 5.20%, and JSC of 7.2, 10.14, and 12.14 mA cm−2 for 4a, 4b and 4c (Table 2), respectively. Specifically, the devices with PC71BM as the acceptor exhibit an exceptionally higher VOC of 0.90 V, 0.90 V and 0.89 V for 4a, 4b and 4c, respectively. It's not surprising that the morphologies and thus performance of state-of-the-art donor small molecules are sensitive to the choice of fullerene. Peng and co-workers reported that devices based on DPP-porphyrin small molecules performed better with PC61BM as the acceptor rather than PC71BM.24–26 The possible reason is that the solubility of PC71BM in some organic solvents is not as good as that of PC61BM, which leads to an unfavorable morphology and deteriorates the performance of OSCs.31,32 During the device optimization, it was found that the performance of the cells based on 4c could be further improved by reducing the annealing time. When the annealing time is 5 min, the device yielded an impressive PCE of 6.11%, with a JSC of 12.28 mA cm−2, a VOC of 0.91 V, and a FF of 54.6%. Since 4c shows a much better solubility than 4a or 4b in organic solvent, the weight ratio of 4c to PC61BM was further optimized to be 1:1.2, yielding the highest PCE of 7.70% with a VOC of 0.91 V, a JSC of 13.32 mA cm−2 and a FF of 63.6%. In general, the energy loss (Eloss), which connects Eg and VOC together and is defined as Eloss = Eg − eVOC, is one of the most important parameters to evaluate solar cells. In this study, an exceptionally high VOC of 0.91 V was obtained for the optimized cell based on 4c, in spite of a high energy loss of 0.69 eV. By comparison, Peng and his co-workers reported a DPP-porphyrin small molecule with a very low energy band gap of 1.37 eV and an open circuit voltage of 0.78 V obtained in BHJ OSCs, corresponding to a very low energy loss of 0.59 eV.26
The external quantum efficiency (EQE) spectra of the optimized devices based on 4a–c are shown in Fig. 2b. The EQE curve of 4c/PC71BM (w/w, 1:1.2) with 1% pyridine exhibits efficient photo-electron conversion efficiency from 400 to 800 nm, with the highest EQE value reaching 73% at 735 nm. The calculated JSC integrated from the EQE for 4c is 13.32 mA cm−2 shows a mismatch of about 5% compared to the JSC from the J–V measurement. In comparison, the EQE values of the devices based on 4a or 4b are below 50% in the range of 620–800 nm, resulting in lower JSC values. This is also consistent with the weaker absorptions of 4a and 4b in solution, as compared to 4c. Definitely, the incorporation of the alkyl side chains could indeed improve significantly the photo-electron conversion efficiency.
To circumvent the degradation problem in the conventional structure, the inverted structure has been intensively investigated in polymer-based BHJ solar cells.33–37 Very recently, some SMs have also been explored and have performed well in inverted device architectures.38–42 Hence, an inverted architecture of ITO/Ca/SM:PC71BM/MoO3/Ag has been applied to 4c, which yielded a PCE of 7.55%, corresponding to a JSC of 12.89 mA cm−2, a VOC of 0.91 V, and a FF of 65.1% under the optimized conditions (Fig. 3 and Table 2). It should be noted that the inverted photovoltaic devices were processed and characterized under ambient atmosphere (see ESI†). Although the inverted structure shows a comparable performance to that of the conventional one, the former exhibits impressive stability with the same PCE of 7.55% measured after 3 days of storage in air and retains 83% of the original value even after storage in air for 30 days.
 | ||
| Fig. 3 The J–V curves (top) and EQE plots (bottom) of the inverted devices with 4c/PC71BM (1:1.2, w/w) as-cast (black line) and after annealing for 5 minutes at 90 °C (red line). | ||
2.4 Morphology and mobility
To understand the effect of different peripheral substitutions on the porphyrin ring, the morphology of the active layers of 4a–c/PC71BM (1:1, w/w%) was investigated by tapping-mode atomic force microscope (AFM). Additionally, height and phase images were taken from the blend films as shown in Fig. 4. It should be noted that the blend films of 4a–c/PC71BM were prepared with 1% pyridine as an additive, using a procedure identical to that for the active layers in the devices. The values of root-means-square (RMS) roughness are 0.852, 0.780 and 0.567 nm for the blend films 4a–c/PC71BM, respectively. The lowest RMS roughness of 4c/PC71BM indicates that 4c has the best miscibility with PC71BM and may form a finer interpenetrating network to facilitate both exciton separation and charge transport. As expected, the blend film of 4c/PC71BM exhibits a better donor/acceptor interpenetrating network with much smaller phase separation domains (Fig. 4f), which is beneficial for efficient exciton dissociation and charge transporting, and should lead to an increased JSC and FF in BHJ OSCs.43,44 In contrast, surface relief and a larger domain size were observed for the 4a/PC71BM and 4b/PC71BM films, which indicates the poor film-forming characteristics due to the perpendicular aromatic peripheral substitutions on the porphyrin ring. Without a doubt, the unique characteristics of 10,20-bis(5,15-alkyl) substituted porphyrin are expected to form blend films possessing more efficient intermolecular π–π stacking and more suitable surface morphology for BHJ OSCs.
 | ||
| Fig. 4 Tapping mode AFM height (a–c) and phase images (d–f) of 4a/PC71BM, 4b/PC71BM, and 4c/PC71BM blend films of the best devices. | ||
As shown in Fig. 5, the phase and height images were taken from the blend films of 4c/PC71BM (1:1.2, w/w%) in inverted devices as-casted and after annealing at 90 °C for 5 minutes. The values of the root-means-square (RMS) roughness are 4.48 and 0.88 nm for the blend films 4c/PC71BM, as-cast and after annealing, respectively. Without a doubt, annealing at an appropriate temperature is necessary for the formation of high quality films with nanoscale amorphous domains that produce efficient charge separation and improved solar cell performances.
 | ||
| Fig. 5 Tapping mode AFM phase images (a and b) and height (c and d) of 4c/PC71BM (1:1.2, w/w%) in the inverted devices. | ||
Since the values of JSC and FF are generally reflected by the charge transport properties of the photoactive film, the hole mobilities of the porphyrin/fullerene blend films were measured for a device configuration of ITO/PEDOT:PSS/active layer/MoO3/Al, and were estimated using the space charge limited current (SCLC) model. The J–V characteristics of the polymers and the porphyrin/PC71BM blend films are shown in Fig. S1.† The hole mobilities of the 4a–c/PC71BM blend films were determined to be 1.57 × 10−5 cm2 V−1 s−1 (4a), 8.48 × 10−5 cm2 V−1 s−1 (4b), and 2.18 × 10−4 cm2 V−1 s−1 (4c). Obviously, all the 4a–c/PC71BM blend films show reasonably high hole mobilities appropriate for photovoltaic devices. Notably, the device based on 4c/PC71BM exhibits the highest mobility of about one order of magnitude higher than that based on 4a/PC71BM. This is consistent with the highest JSC values of 13.32 mA cm−2 and the best photovoltaic performance based on the active layer of 4c/PC71BM (1:1.2, w/w%). Without a doubt, the enhanced charge transport property is ascribed to the stronger intermolecular π–π stacking interaction and better surface morphology of the blend film of 5,15-dialkylated porphyrin-cored small molecule.45 At the same time, ultraviolet photoelectron spectroscopy (Fig. S2†) shows that the work functions of the annealed layer of ITO/PEDOT:PSS/4a–c:PC71BM decrease gradually from 3.75 eV for 4a, 3.66 eV for 4b, to 3.44 eV for 4c, which also accounts for the device efficiency improvement with the increasing JSC from 7.20 mA cm−2 for 4a and 10.14 mA cm−2 for 4b, to 12.14 mA cm−2 for 4c (Table 2).46
3. Conclusion
In summary, three new porphyrin-based small molecules 4a–c were prepared, in which 3-ethylrhodanine as a terminal electron deficient unit was introduced into aromatic and aliphatic 10,20-meso-substituted porphyrin rings as the central building block via the 5,15-meso-phenylethynyl linker. Although the aliphatic 10,20-bis(2-hexylnonyl) substitution in 4c has little influence on the optical absorption in solution, it significantly changes the optical and crystallization properties of the films. Based on 4c/PC71BM (1:1.2, w/w%) which has stronger intermolecular π–π stacking and more suitable surface morphology, the device possesses a much higher JSC, corresponding to the highest PCE of 7.70%. Moreover, the inverted structure based on 4c/PC71BM (1:1.2, w/w%) shows a comparable performance to that of the conventional one, and its stability was also investigated revealing the same PCE of 7.55% measured after 3 days of storage and retaining 83% of the original value even after storage in air for 30 days. The primary results demonstrate that porphyrin derivatives can play a more important role as donors in BHJ OSCs by the judicious optimization of their molecular structures and careful device engineering.
4. Experimental section
4.1 Materials and methods
3,5-Di(dodecyloxyl)benzaldehyde, 4-octyloxybenzaldehyde, 2-hexyldecanal and 1,4-dibromo-2,5-bis(hexyloxy)benzaldehyde were prepared according to the literature procedures, and characterized by comparing their 1H NMR and 13C NMR spectra with those found in the literature.47 [6,6]-Phenyl-C71-butyric acid methyl ester (PC71BM) was purchased from Nano-C. All other reagents were used as received without further purification, unless stated otherwise.4.2 General procedure for the preparation of 4a–c
To a solution of 3a–c (0.137 mmol) in CHCl3 (20 mL), two drops of piperidine and 3-ethylrhodanine (228.3 mg, 1.370 mmol) were added, and the resulting solution was refluxed for 12 h under argon. The reaction was quenched with water (30 mL) and extracted with CHCl3 (3 × 20 mL). The organic layer was dried over Na2SO4 and evaporated to dryness. The solid residue was purified first on a silica gel column and second by preparative thin layer chromatography using a CHCl3 as the eluent. Further recrystallization from hexane and CHCl3 mixed solvent five times afforded the target compounds 4a–c as a black solid.Acknowledgements
We thank the National Natural Science Foundation of China (NSFC) (Grant No. 91222201, 91333206, 51473139, 51473053 and 91333206), Hong Kong Research Grants Council (HKBU 22304115, HKBU 203011) and Hong Kong Baptist University (FRG2/14-15/034, FRG1/14-15/058 and FRG2/13-14/083) for the financial support. W.-K. Wong and W.-Y. Wong acknowledge the support from the Areas of Excellence Scheme, University Grants Committee, Hong Kong SAR ([AoE/P-03/08]). X. Zhu also thanks The Science, Technology and Innovation Committee of Shenzhen Municipality (JCYJ20150630164505504) for financial support. X. B. Peng thanks International Science & Technology Cooperation Program of China (2013DFG52740) for the financial support. X. Z. Wang thanks the project of Innovation Platform Open Foundation of University of Hunan Province (14K092) for the financial support.References
- Y. Li, Acc. Chem. Res., 2012, 45, 723 CrossRef CAS PubMed.
- Y. Lin, Y. Li and X. Zhan, Chem. Soc. Rev., 2012, 41, 4245 RSC.
- J. Zhou, Y. Zuo, X. Wan, G. Long, Q. Zhang, W. Ni, Y. Liu, Z. Li, G. He and C. Li, J. Am. Chem. Soc., 2013, 135, 8484 CrossRef CAS PubMed.
- Y. Liu, C.-C. Chen, Z. Hong, J. Gao, Y. M. Yang, H. Zhou, L. Dou, G. Li and Y. Yang, Sci. Rep., 2013, 3, 3356 Search PubMed.
- W. Liu, A. Tang, J. Chen, Y. Wu, C. Zhan and J. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 22496 CAS.
- L. Xiao, H. Wang, K. Gao, L. Li, C. Liu, X. Peng, W.-Y. Wong, W.-K. Wong and X. Zhu, Chem.–Asian J., 2015, 10, 1513 CrossRef CAS PubMed.
- Y. Lin, L. Ma, Y. Li, Y. Liu, D. Zhu and X. Zhan, Adv. Energy Mater., 2013, 3, 1166 CrossRef CAS.
- J. Liu, Y. Sun, P. Moonsin, M. Kuik, C. M. Proctor, J. Lin, B. B. Hsu, V. Promarak, A. J. Heeger and T. Q. Nguyen, Adv. Mater., 2013, 25, 5898 CrossRef CAS PubMed.
- J. Yuan, L. Xiao, B. Liu, Y. Li, Y. He, C. Pan and Y. Zou, J. Mater. Chem. A, 2013, 1, 10639 CAS.
- E. W. Snedden, A. P. Monkman and F. B. Dias, Adv. Mater., 2013, 25, 1930 CrossRef CAS PubMed.
- A. Zitzler-Kunkel, M. R. Lenze, N. M. Kronenberg, A.-M. Krause, M. Stolte, K. Meerholz and F. Würthner, Chem. Mater., 2014, 26, 4856 CrossRef CAS.
- F. Silvestri, M. D. Irwin, L. Beverina, A. Facchetti, G. A. Pagani and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 17640 CrossRef CAS PubMed.
- Y. Chen, Y. Zhu, D. Yang, Q. Luo, L. Yang, Y. Huang, S. Zhao and Z. Lu, Chem. Commun., 2015, 51, 6133 RSC.
- F. Silvestri, M. D. Irwin, L. Beverina, A. Facchetti, G. A. Pagani and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 17640 CrossRef CAS PubMed.
- Y. Sun, G. C. Welch, W. L. Leong, C. J. Takacs, G. C. Bazan and A. J. Heeger, Nat. Mater., 2012, 11, 44 CrossRef CAS PubMed.
- T. S. van der Poll, J. A. Love, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2012, 24, 3646 CrossRef CAS PubMed.
- V. Gupta, A. K. K. Kyaw, D. H. Wang, S. Chand, G. C. Bazan and A. J. Heeger, Sci. Rep., 2013, 3, 1965 Search PubMed.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886 CrossRef CAS PubMed.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2014, 9, 35 CrossRef.
- J. Kesters, P. Verstappen, M. Kelchtermans, L. Lutsen, D. Vanderzande and W. Maes, Adv. Energy Mater., 2015, 5, 1500218 Search PubMed.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. s. Torres, Chem. Rev., 2014, 114, 12330 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W. G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- L. Li, Y. Huang, J. Peng, Y. Cao and X. Peng, J. Mater. Chem. A, 2013, 1, 2144 CAS.
- H. Qin, L. Li, F. Guo, S. Su, J. Peng, Y. Cao and X. Peng, Energy Environ. Sci., 2014, 7, 1397 CAS.
- K. Gao, L. Li, T. Lai, L. Xiao, Y. Huang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russell, R. A. J. Janssen and X. Peng, J. Am. Chem. Soc., 2015, 137, 7282 CrossRef CAS PubMed.
- T. S. Balaban, A. Eichhöfer and J. M. Lehn, Eur. J. Org. Chem., 2000, 21, 4047 CrossRef.
- S. Chen, L. Xiao, X. Zhu, X. Peng, W.-K. Wong and W.-Y. Wong, Chem. Commun., 2015, 51, 14439 RSC.
- P. Thamyongkit, M. Speckbacher, J. R. Diers, H. L. Kee, C. Kirmaier, D. Holten, D. F. Bocian and J. S. Lindsey, J. Org. Chem., 2004, 69, 3700 CrossRef CAS PubMed.
- Y. Ren, A. M. Hiszpanski, L. Whittaker-Brooks and Y.-L. Loo, ACS Appl. Mater. Interfaces, 2014, 6, 14533 CAS.
- Z. Ma, E. Wang, K. Vandewal, M. R. Andersson and F. Zhang, Appl. Phys. Lett., 2011, 99, 143302 CrossRef.
- M. Williams, N. R. Tummala, S. G. Aziz, C. Risko and J.-L. Brédas, J. Phys. Chem. Lett., 2014, 5, 3427 CrossRef CAS PubMed.
- Y. Sun, J. H. Seo, C. J. Takacs, J. Seifter and A. J. Heeger, Adv. Mater., 2011, 23, 1679 CrossRef CAS PubMed.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591 Search PubMed.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S.-W. Tsang, T.-H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115 CrossRef CAS.
- S.-H. Liao, H.-J. Jhuo, Y.-S. Cheng and S.-A. Chen, Adv. Mater., 2013, 25, 4766 CrossRef CAS PubMed.
- A. R. B. M. Yusoff, D. Kim, H. P. Kim, F. K. Shneider, W. J. da Silva and J. Jang, Energy Environ. Sci., 2015, 8, 303 CAS.
- X. Tong, B. E. Lassiter and S. R. Forrest, Org. Electron., 2010, 11, 705 CrossRef CAS.
- F. J. Zhang, D. W. Zhao, Z. L. Zhuo, H. Wang, Z. Xu and Y. S. Wang, Sol. Energy Mater. Sol. Cells, 2010, 94, 2416 CrossRef CAS.
- A. K. K. Kyaw, D. H. Wang, V. Gupta, J. Zhang, S. Chand, G. C. Bazan and A. J. Heeger, Adv. Mater., 2013, 25, 2397 CrossRef CAS PubMed.
- D. Y. Liu, F. Qu, X. N. Zhao and J. M. You, J. Phys. Chem. C, 2015, 119, 17979 CAS.
- G. Long, B. Wu, X. Yang, B. Kan, Y.-c. Zhou, L.-c. Chen, X. Wan, H.-l. Zhang, T. C. Sum and Y. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 21245 CAS.
- W. L. Leong, G. C. Welch, J. Seifter, J. H. Seo, G. C. Bazan and A. J. Heeger, Adv. Energy Mater., 2013, 3, 356 CrossRef CAS.
- M. S. Su, C. Y. Kuo, M. C. Yuan, U. Jeng, C. J. Su and K. H. Wei, Adv. Mater., 2011, 23, 3315 CrossRef CAS PubMed.
- L. Zhou, Z.-X. Xu, Y. Zhou, Y. Feng, X.-G. Zhou, H.-F. Xiang and V. Roy, Chem. Commun., 2012, 48, 5139 RSC.
- Z. A. Page, Y. Liu, V. V. Duzhko, T. P. Russell and T. Emrick, Science, 2014, 34, 441 CrossRef PubMed.
- R. Miyakoshi, K. Shimono, A. Yokoyama and T. Yokozawa, J. Am. Chem. Soc., 2006, 128, 16012 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04783h |
| ‡ H. W., L. X., L. Y. and S. C. contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |