
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross-linking and other structural proteomics techniques: how chemistry is enabling mass spectrometry applications in structural biology
Alexander
Leitner

Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, Auguste-Piccard-Hof 1, 8093 Zurich, Switzerland. E-mail: leitner@imsb.biol.ethz.ch
First published on 26th April 2016
Abstract
The biological function of proteins is heavily influenced by their structures and their organization into assemblies such as protein complexes and regulatory networks. Mass spectrometry (MS) has been a key enabling technology for high-throughput and comprehensive protein identification and quantification on a proteome-wide scale. Besides these essential contributions, MS can also be used to study higher-order structures of biomacromolecules in a variety of ways. In one approach, intact proteins or protein complexes may be directly probed in the mass spectrometer. Alternatively, various forms of solution-phase chemistry are used to introduce modifications in intact proteins and localizing these modifications by MS analysis at the peptide level is used to derive structural information. Here, I will put a spotlight on the central role of chemistry in such mass spectrometry-based methods that bridge proteomics and structural biology, with a particular emphasis on chemical cross-linking of protein complexes.
1. Introduction
Proteins and protein complexes have been studied by X-ray crystallography and NMR spectroscopy for decades, and more recently, electron microscopy (EM) has increased in relevance because of technical advances that allow to obtain structures at higher resolution.1,2 However, very large protein assemblies are often not accessible by these techniques for a variety of reasons. For example, the protein amounts or concentrations required for crystallization studies or NMR experiments may not be easily obtainable. A high degree of conformational flexibility and/or compositional heterogeneity may decrease the practically achievable resolution in EM. Therefore, an analytical technique that is able to deal with limited sample amounts and tolerates some degree of heterogeneity is of great interest to provide complementary information for structural biologists. This is where mass spectrometry (MS) comes into play.Over the last decades, mass spectrometry has made enormous contributions to the identification, quantitation and characterization at the level of individual proteins up to whole proteomes (Fig. 1).3–6 Today's proteomic research is closely tied to mass spectrometry as the central instrumental analytical technology for a number of reasons:
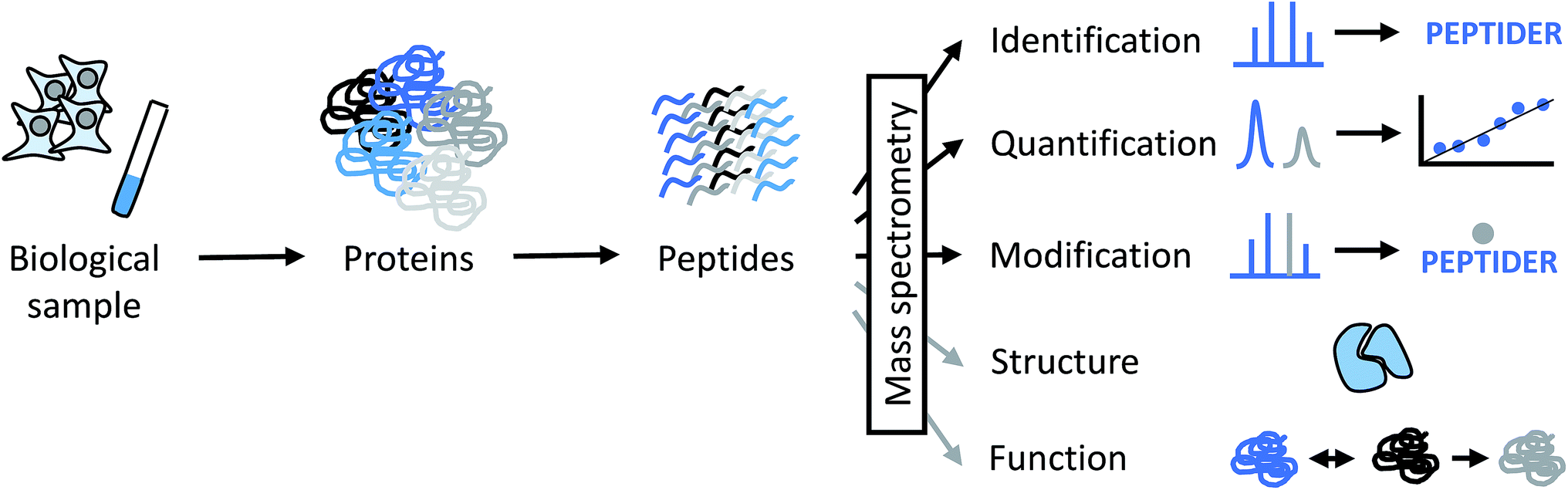 | ||
| Fig. 1 Different levels of information that can be derived from MS-based proteomics experiments directly (black arrows) or indirectly (grey arrows). PEPTIDER refers to a hypothetical amino acid sequence of a peptide, the grey dot above the sequence symbolizes a modification. Structural analysis by MS is discussed in more detail in this perspective. Functional properties of proteins can by derived indirectly from MS data by obtaining data about subcellular localization, distribution between different cell types, abundance changes in response to biological perturbations, and involvement in protein–protein interactions, among others. | ||
(1) The availability of electrospray ionization (ESI) as a robust method to generate ions from solutions, thus allowing hyphenation with solution-phase separation techniques, in particular high-performance liquid chromatography;
(2) the substantial improvements in sensitivity and acquisition speed of modern mass spectrometers and the possibility for the routine acquisition of mass spectra at high resolution and mass accuracy without sacrificing sensitivity;
(3) the predictable fragmentation behavior of peptide cations in tandem mass spectrometry, thereby allowing automated peptide sequencing using a variety of computational approaches, such as database searching or spectral library searching.
Since the early days of protein and peptide analysis by MS, chemical reactions have been important contributors to the evolution of the technique (see, for example, the historical perspective from Biemann).7 More recently, chemical modification (or “tagging”) of reactive groups in peptides and proteins has facilitated proteome analysis through the improvement of detection sensitivity, the introduction of affinity tags for enrichment purposes, and by enabling various quantitative workflows through (stable) isotope labeling.8–10 Therefore, we are currently at a stage where MS-based proteome analysis allows the nearly comprehensive protein profiling of organisms such as yeast,11,12 the identification of more than 10′000 proteins in samples of human origin,13,14 and quantitative profiling of large protein populations in diverse biological contexts. Such comprehensive and robust data sets are increasingly combined with other “omics” data at the gene, transcript or metabolite level and integrated in systems biology approaches.
While the above mentioned applications are very well accepted by biologists, mass spectrometry is also able to provide structural information about proteins. Information derived from mass spectra of proteins has been correlated with their solution phase structural properties more than 20 years ago. Chowdhury et al.15 and Loo et al.16 monitored the ESI charge state distribution (CSD) of small proteins such as cytochrome c, ubiquitin and lysozyme under non-denaturing and denaturing conditions. Different solvent systems were found to affect the distribution and this was attributed to conformational changes taking place in solution that were reflected in the number of charges carried by the protein ions. Shortly thereafter, Katta and Chait showed that the non-covalent complex between myoglobin and its heme group could be transferred into the gas phase and detected by MS.17 The deconvolution of CSDs to distinguish conformational states was later proposed by Dobo and Kaltashov.18
In the following decades, a number of MS-based methods have been developed that provide structural information about proteins and protein complexes.19 On their own, they typically do not provide sufficient information to derive a model of a protein, let alone a protein complex at reasonably high resolution. However, the combination of several structural mass spectrometry (or “structural proteomics”) methods or the combination of such methods with techniques such as EM, NMR spectroscopy or small-angle X-ray scattering and/or computational methods can be extremely powerful. This has led to the emergence of “hybrid” or “integrative” structural biology methods20 with contributions from various MS techniques. Some, but not all of them involve the solution-phase chemical manipulation of proteins and protein complexes in some form and will be the focus of this perspective article. The goal is to emphasize how both chemistry and MS contribute in this area to dissect key issues in (structural) biology.
2. Structural proteomics and the role of chemistry
Conceptually, MS-based structural proteomics techniques can be classified in two main categories according to the species that are actually studied in the mass spectrometer (Fig. 2).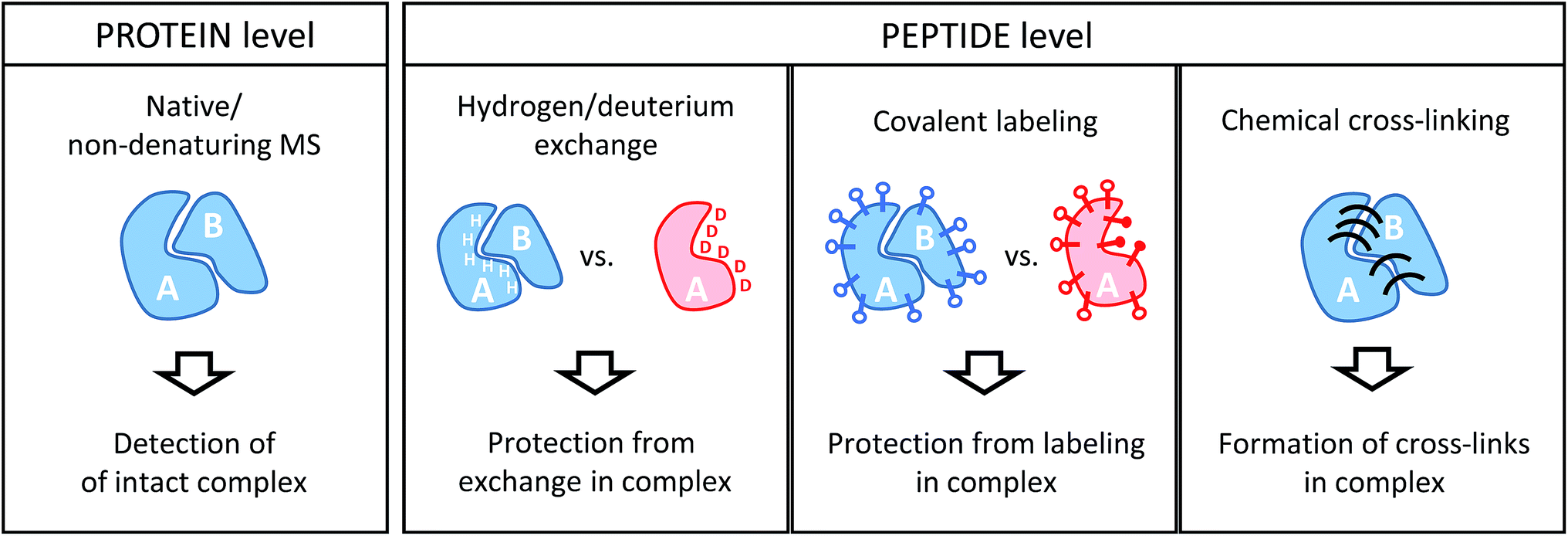 | ||
| Fig. 2 Schematic illustration of the information derived from structural proteomics/structural MS techniques using a hypothetic binary complex. | ||
2.1. Protein-level readout
Intact protein complexes are studied in “native” or “non-denaturing” MS approaches.21–23 In this case, the aim is to retain the integrity of complexes upon transfer from the solution-phase into the high vacuum of the mass spectrometer for further study. This is facilitated by careful optimization of ionization and ion transmission parameters and, frequently, dedicated instrument design.24–26 Acquiring a mass spectrum of a protein complex under non-denaturing conditions allows the elucidation of subunit stoichiometries through the determination of the mass of the intact assembly. This is a unique advantage compared to the structural proteomics methods that work at the peptide level. Ionized complexes may then be further manipulated in the mass spectrometer, for example through dissociation into sub-complexes.27 This way, subunit connectivity can be inferred from tandem mass spectra. Alternatively, complexes may already be disrupted in solution by solvent additives. In addition, native MS analysis can be combined with ion mobility spectrometry, a gas-phase separation technique that can yield shape information of ions in form of their collisional cross-section (CCS).28 Both complex stoichiometry and CCSs serve as valuable input for hybrid modeling approaches.29Although powerful in principle, the mass spectrometric analysis of intact proteins remains challenging and low throughput. For example, instrumental settings need to be optimized for every sample and not all proteins and complexes can be ionized from MS-compatible solutions. Therefore, native MS continues to be limited to relatively few expert research groups, and alternative strategies overcome some of the limitations by working at the peptide level.
2.2. Peptide-level readout
All other structural proteomics techniques have in common that although information on the protein or protein complex level is obtained, protein-derived peptides are analyzed in the mass spectrometer. This is possible because different types of chemical reactions that conserve the native structure are performed on the sample in solution. This “encoded” information is then retained throughout further sample processing that includes a proteolytic cleavage step whereby proteins are cleaved into peptides by means of an enzyme (protease). This step has the advantage that peptides are generally more amenable to mass spectrometric analysis. Such a “bottom-up” approach comes, however, with the drawbacks of added sample complexity (one protein creates many peptides) and potential ambiguity during data analysis (peptide sequences may not be unique for a particular protein). Because of the limited number of proteins in a typical sample studied by structural proteomics methods, these issues are usually less of a concern than in more conventional proteomics studies.For the remainder of this article, I will focus on methods that involve MS analyses on the peptide level, which includes three main chemical approaches: the exchange of labile hydrogen atoms with deuterium atoms in hydrogen/deuterium exchange (H/D exchange or HDX) experiments; the covalent modification of amino acid residues (typically, functional groups in the side chains) in various covalent labeling (CL) methods; and chemical cross-linking (XL), where two spatially proximate amino acid side chains are covalently coupled. The three methods and their practical application to protein complexes will be discussed in detail in the following sections.
3. Hydrogen/deuterium exchange
HDX takes advantage of the exchange of labile hydrogens in proteins with hydrogens from the surrounding solvent. If H2O is replaced by D2O the resulting exchange is associated with a mass increase (approximately 1.006 Da per D atom) that can be detected by mass spectrometry. Among other parameters (such as temperature and pH), the exchange rates are dependent on the structure of the protein(s) under investigation. While exchange of side-chain hydrogens is so fast that it cannot be monitored by conventional MS-based methods, the exchange of backbone amide hydrogens is slower and more dependent on the structure or, more specifically, the hydrogen bonding or solvation patterns of the analytes. This makes HDX-MS a versatile tool to monitor conformational changes in proteins or protein complexes.30–333.1. The HDX workflow
The analytical workflow for HDX-MS (Fig. 3) is influenced by the reversible nature of the deuterium incorporation. Prolonged exposure of a deuterated sample to 1H in normal water will lead to “back exchange”. This effect is minimized at low temperatures, low pH (with a minimum at approximately 2.5) and by keeping the exposure time to a minimum. Therefore, the processing of samples derived from HDX experiments needs to be optimized accordingly.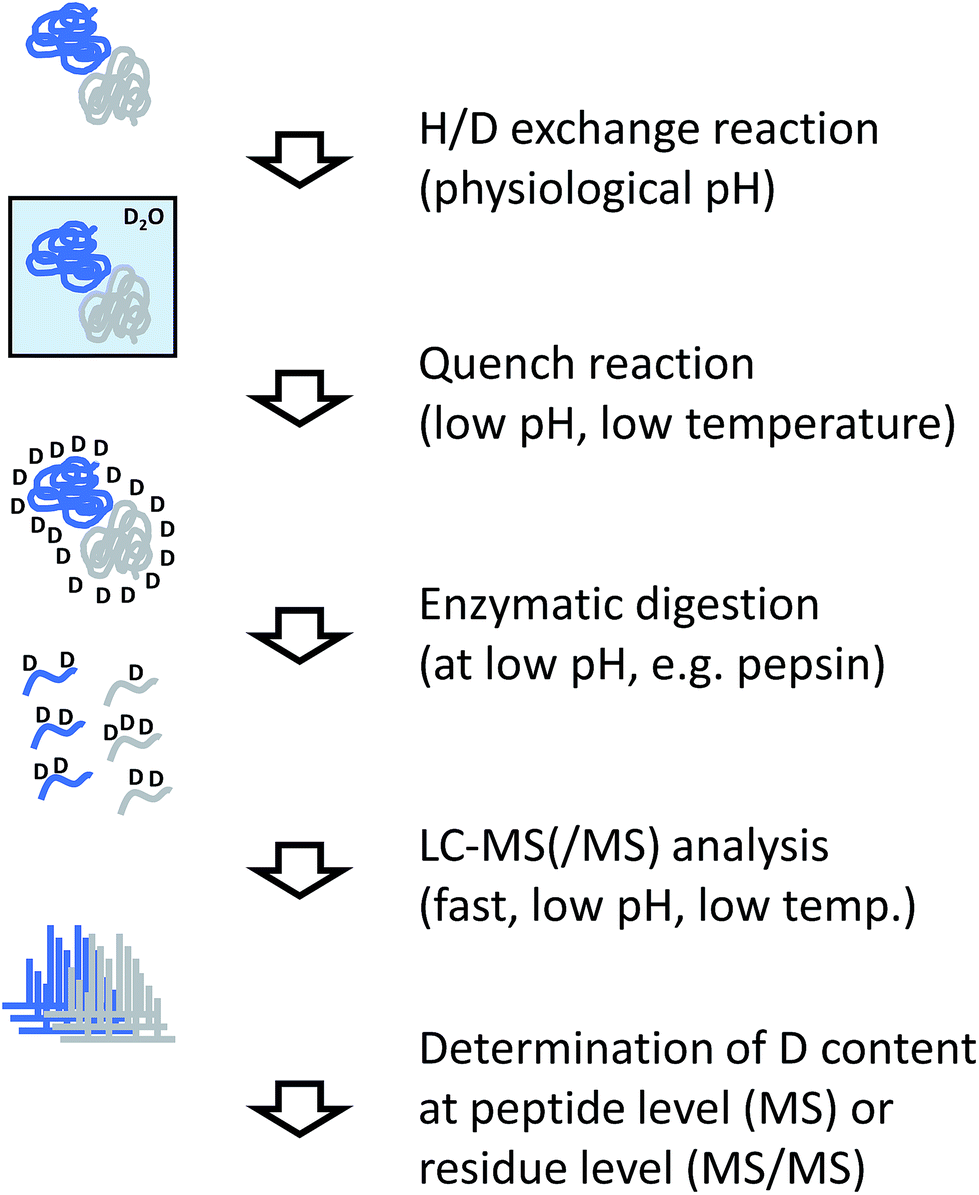 | ||
| Fig. 3 Experimental workflow for hydrogen/deuterium exchange/MS. | ||
In most applications of HDX, labeled proteins are enzymatically digested into peptides, similar to conventional proteomics workflows or the covalent labeling and cross-linking methods discussed in the following sections. In the case of HDX, proteases are needed that work at low temperature and low pH, requirements that are fulfilled by pepsin which is the most commonly used protease for this purpose. To reduce processing times and accelerate the cleavage reaction, the enzyme is frequently used in immobilized form in a dedicated column reactor that is directly integrated into the HPLC system. The pepsin reactor and other components of the HPLC system such as valves or chromatography columns for trapping and separating the peptide mixture are cooled to temperatures close to 0 °C by placing them in ice baths or dedicated refrigerators.34 Sub-zero °C chromatography set-ups have also been reported recently.35
Incorporation of deuterium in the protein/peptide sequence is a gradual process. Accordingly, the natural isotope distributions are shifted by the number of hydrogens that are exchanged. While this pattern encodes the structural information, it complicates peptide identification so that the identity of peptides is usually determined in a separated experiment using unlabeled sample. In addition pepsin and other proteases used for HDX-MS such as nepenthesin36 are relatively unspecific and cleave at many different residues. This is advantageous because deuterium incorporation can be localized more accurately through overlapping peptides. However, the complexity of peptide mixtures resulting from unspecific digests can be considerably higher than if more specific proteases would be used.
The amount of deuterium incorporated at the peptide level can be determined by dedicated software packages.37,38 To increase the spatial resolution from the peptide level to the individual amino acid level, data from overlapping peptides are combined, or tandem MS (MS/MS) is performed on the deuterated peptides. Conventional collisional activation of peptides may lead to “scrambling” of the deuteration pattern,39 which can be avoided by employing electron-mediated fragmentation methods such as electron transfer dissociation.40,41
An alternative to the relatively cumbersome bottom-up strategy in HDX-MS would be to avoid the digestion step and perform MS/MS on the intact protein level. Although attractive in theory, practical applications are still limited and restricted to smaller proteins, for which sufficient sequence coverage can be obtained.42
3.2. Representative applications
HDX-MS is suitable to study many different aspects of protein structure and dynamics, and recent applications of the technique have been summarized by Engen and co-workers.33 These applications include protein folding and aggregation as well as the comparison of different conformational states of proteins, for example states induced by interaction with ligands or with other proteins. In the latter case, surfaces involved in binding will experience a change in the exchange pattern (also schematically shown in Fig. 2), although conformational changes are not necessarily restricted to the immediate binding region.A noteworthy application to individual proteins is the analysis of biopharmaceuticals where HDX adds another level of MS-derived information beyond sequence confirmation and the characterization of artefactual and post-translational modifications.32 Protein–ligand interactions (such as drug binding) and protein–protein interactions (such as antibody–antigen complexes) are also commonly examined.32,33 As a consequence, the method is well accepted and established in the pharmaceutical industry.
As the focus of this article is on the analysis of larger protein assemblies, I will restrict myself to highlighting some noteworthy applications of HDX-MS in this area. G-Protein-coupled receptors (GPCRs) are an important protein family involved in signal transduction. Chung et al. used HDX to study the structure of bovine G protein alone and in complex with the β2 adrenergic receptor in its agonist-bound state,43 and Shukla et al. examined the recruitment of β-arrestin to GPCR complexes using a combination of HDX-MS and EM.44 These two techniques were also combined to create a pseudo-atomic model of the COPII vesicle cage, whereby HDX data were used to localize contact regions.45 HDX-MS can also be extended to very large systems such as the GroEL chaperonin.46,47 This 14-mer, 800 kDa complex is the biggest system studied in detail by HDX to date, and changes in the conformational dynamics of this assembly upon ATP binding were successfully monitored.46 Folding of a substrate protein inside the folding chamber of GroEL/GroES was also recently probed by HDX.47
As noted in the Introduction, diverse types of MS information can be included in integrative modeling strategies. Recently, the Bonvin and Schriemer groups introduced Mass Spec Studio,48 a software package that allows the combination of HDX and chemical labeling data with protein modeling. In this work, the concept of data-independent HDX-MS/MS49 was also first introduced, which may be an interesting expansion of HDX-MS methods. Application of this new approach to the interaction of mitotic centromere-associated kinesin with microtubules was shown.48,49
4. Covalent labeling
Covalent labeling (CL) involves the introduction of irreversible modifications at reactive side chains to obtain information about solvent or surface exposed residues in proteins and protein complexes. This provides conceptually similar information to HDX workflows. The advantage of irreversibility is that sample handling is less restricted and the processing steps are more similar to conventional bottom-up proteomics techniques. With the exception of hydroxyl radicals, CL probes are larger than water, therefore extensive labeling increases the risk of inducing conformational changes.50CL encompasses different labeling strategies depending on the type of chemistry involved. Labeling can be restricted to a single or a few different amino acids if specific labeling reagents are used, however in this case the obtainable spatial resolution is limited. Alternatively, the simultaneous modification of many residue types is possible by oxidative labeling, which is also frequently described as “footprinting”.
4.1. Labeling chemistries
The most commonly used reagents for covalent labeling of primary amines are anhydrides (such as acetic anhydride51) and active esters (such as N-hydroxysuccinimide acetate, NHSA52). Recently, the amine-reactive quantification reagent, tandem mass tag (TMT), has been used for the same purpose.53,54 Glycine ethyl ester (GEE) in combination with coupling reagents such as carbodiimides is used for modifying carboxyl groups.55 The more promiscuous reagent, diethyl pyrocarbonate (DEPC) has been shown to modify several amino acid residues (His, Lys, Tyr, Ser, Thr and Cys).56 Because DEPC leads to modifications on different residues, higher special resolution of modification patterns may be achieved.
Different residues are known to react with ˙OH at different rate constants, with Cys, Trp and Tyr being the most reactive residues.61 However, in practice every amino acid with the exception of Gly can be modified. Insertion of a single oxygen atom with the corresponding mass shift of +16 Da is the most common reaction product, but other mass shifts such as +14 and +32 are also observed.61
4.2. The covalent labeling workflow
Independent of the actual labeling chemistry, samples from CL labeling experiments are most commonly analyzed in a traditional bottom-up workflow (Fig. 4). Due to the irreversible nature of the modification, proteases that are commonly used in mass spectrometry-based proteomics, such as trypsin, can be used to digest modified proteins into peptides. The resulting peptide mixtures are then separated by (reversed-phase) chromatography and sequenced by tandem mass spectrometry. Modified amino acids are identified with the help of database search software through their specific mass shifts compared to unmodified residues.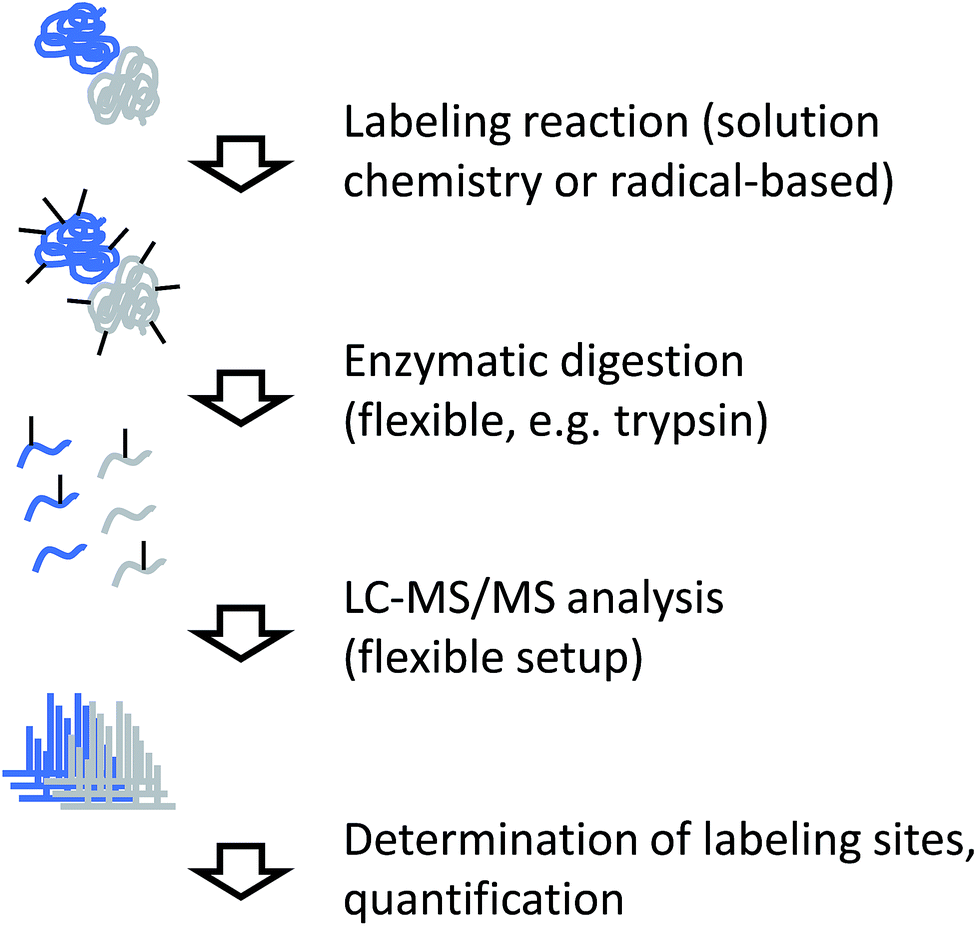 | ||
| Fig. 4 Experimental workflow for covalent labeling/MS. | ||
A few points have to be considered for the practical interpretation of CL data: for example, if the labeling chemistry targets the same residue like the protease, enzymatic cleavage is typically prevented. Therefore, the resulting peptides will be different between unmodified sample and modified sample in such cases, making quantitative interpretation of the data less straightforward. In addition, unspecific labeling approaches such as radical footprinting can generate many different reaction products. This means that even samples of low complexity can yield peptide mixtures of relatively high complexity. Furthermore, it may not always be possible to exactly pinpoint the modification site within a peptide sequence due to insufficient spectral quality or chromatographic co-elution of differently labeled forms of the same peptide. The diversity of reaction products from footprinting requires the specification of many modification sites during database search, which remains computationally challenging.
4.3. Representative applications
Because of the rapid timescale of the reaction in comparison to other chemical modification methods discussed here, radical footprinting is particularly suited to monitor rapid dynamic events such as protein folding.62 However, application of footprinting to larger protein assemblies has also been reported. Chance and co-workers, for example, have used synchrotron-generated hydroxyl radicals to probe the role of structural water molecules in GPCRs such as rhodopsin.63 The same group also studied conformational differences during gating of a potassium channel64 and upon photoactivation of rhodopsin.65 Recently, conformational dynamics in the proton-coupled zinc transporter YiiP were also examined.66 Gross and co-workers applied their FPOP technique (for “fast chemical oxidation of proteins”, i.e. hydroxyl radical generation by laser photolysis) for epitope mapping in antigen–antibody complexes,67 among others. Recently, the first steps towards the application of FPOP in large complexes68 and even whole cells69 have been reported by the group of Jones.Noteworthy applications of conventional chemical labeling include the application of GEE labeling to study Tyr kinase dimerization70 and to the structural analysis of the cyanobacterial orange carotenoid protein.71 Lys-specific labeling using the TMT reagent has been applied to antibody–antigen complexes involving therapeutic antibodies.54 An elegant example of the combination of several labeling methods was reported by the Vachet group.72 In this work, labeling of lysine residues with NHSA, of arginine residues with 2,3-butanedione and less specific labeling with DEPC have been applied to study the dimerization of β2-microglobulin.
5. Cross-linking
Cross-linking (XL) reactions involve the covalent coupling of two functional groups within a protein or between two different proteins. XL can be used to stabilize protein–protein interactions during purification procedures and therefore facilitate the detection of transient or low affinity interaction partners, and unspecific reagents such as formaldehyde or glutaraldehyde are frequently used for this purpose.73,74 However, the introduction of cross-links at specific residues also provides spatial information at different levels if the actual linked residues are identified (by MS), as outlined in Fig. 5.75–77 The structure of the cross-linker defines a spatial restraint, that means is defines an upper limit of how far apart the two reactive sites can be in space.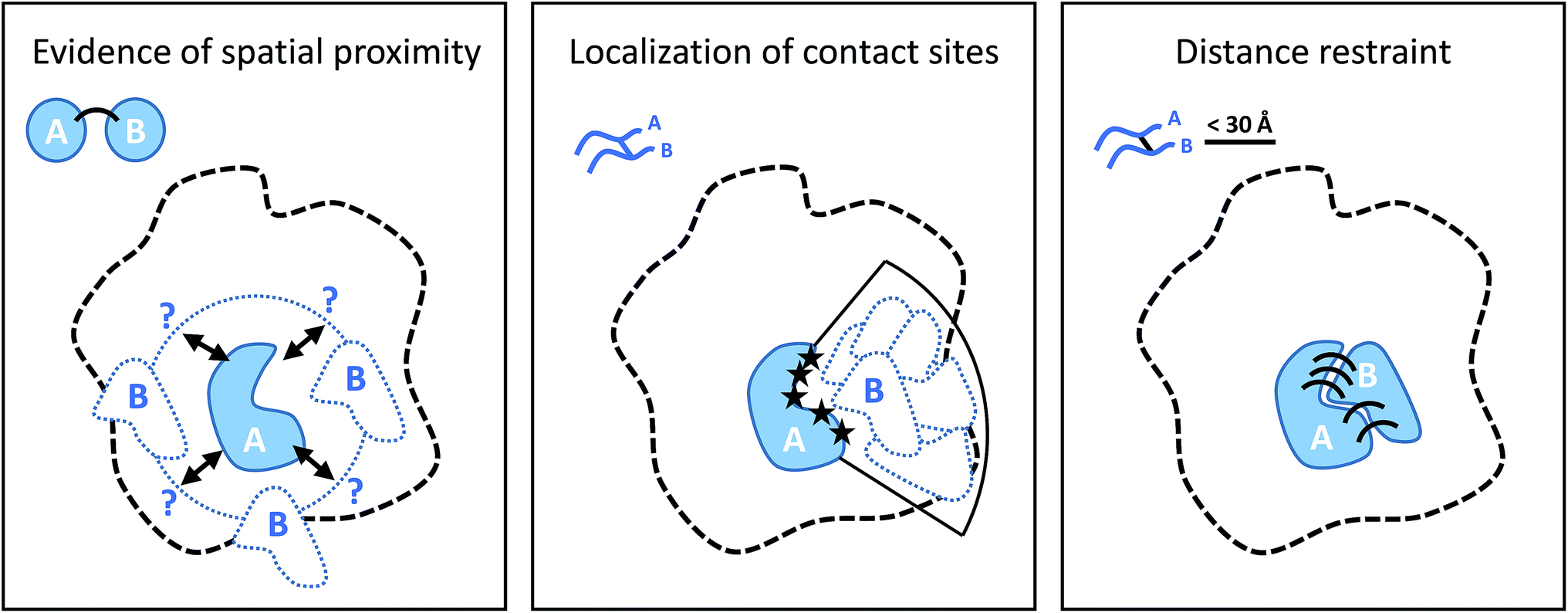 | ||
| Fig. 5 Different levels of spatial information derived from cross-linking experiments. From left to right: the presence of cross-links puts subunit B in the proximity of subunit A; localization of the contact (cross-linking) sites on the two proteins narrows down the possible location of B relative to A; defining an upper bound of the distance between cross-linked residue as specified by cross-linker structure positions and orients the subunits according to their binding interface. | ||
5.1. Cross-linking chemistry
Although many different cross-linking chemistries have been proposed,78–80 there are only a few that are actually widely used for structural proteomics applications. Generally, the reaction conditions for cross-linking are restricted in terms of pH, temperature, and solvent/buffer composition in order to preserve the structure of the proteins. Cross-linking reactions should proceed fast enough to allow the formation of the desired products in a time frame of minutes, although reaction times can be extended up to a few hours if cross-linking is carried out at lower temperatures (4 °C). The extent of cross-linking is controlled by adjusting various experimental parameters such as the protein or reagent concentrations, and by rapid quenching of the reaction.A practical consideration is that the reactions should target abundant functional groups, which practically restricts the list of candidates to primary amines (Lys) and carboxylic acids (Asp/Glu). In practice, the reactions will also target the proteins' N- and C-termini. An emerging, but highly interesting alternative is the use of photochemistry by using reagents with aziridines and other photochemically activated moieties that are less specific towards a particular side chain.81,82
Some popular XL chemistries are shown in Fig. 6. Most commonly, homobifunctional reagents are used, although heterobifunctional reagents are also available and combine, for example, an amine-reactive succinimide ester group with a photoreactive moiety.83 Zero-length cross-linking is a special case, because – as the name implies – amino and carboxyl groups are directly linked with the help of a coupling reagent (such as carbodiimides), and no spacer is introduced. In all other cases, the structure of the reagent indirectly determines the reactivity because the longer the spacer of the reagent, the more likely it is that two reactive groups come into contact to form a cross-link. At the same time, an increased spacer length will reduce the accuracy of the restraint. Therefore, the choice of the reagent is a compromise between number of cross-links and spatial accuracy.
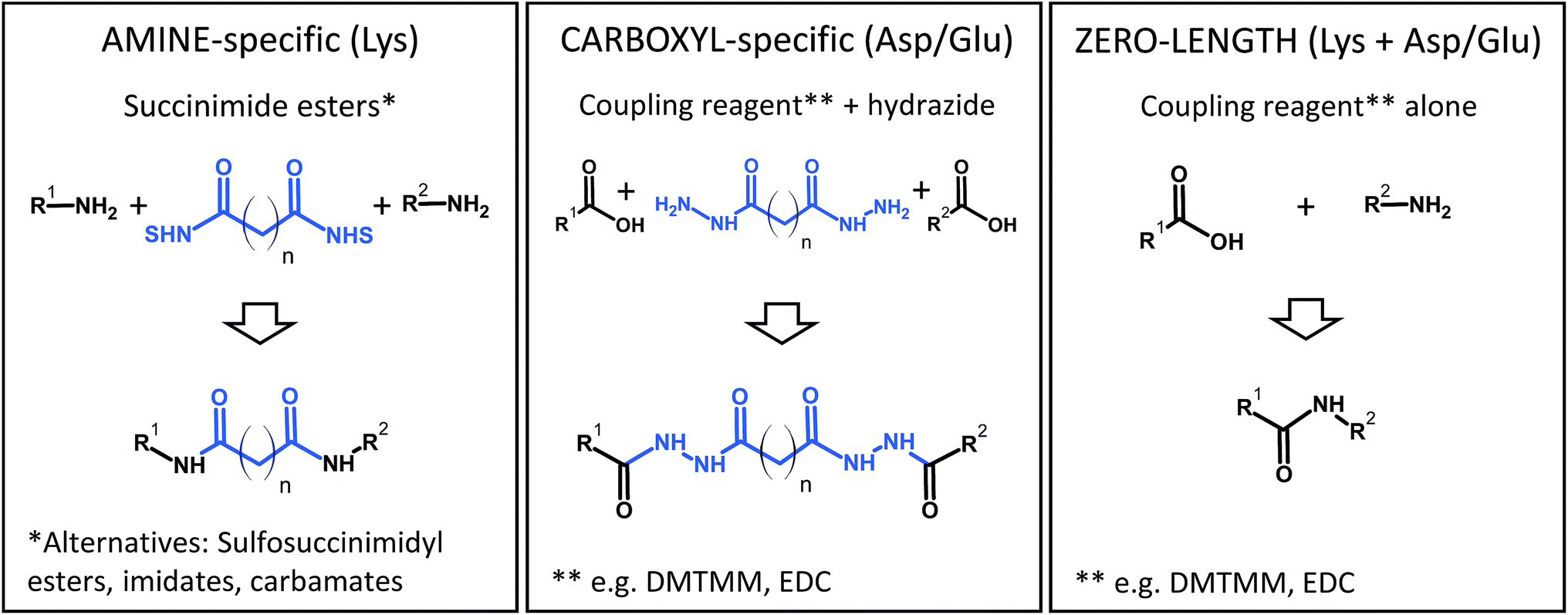 | ||
| Fig. 6 Common chemistries to couple reactive residues in chemical cross-linking. DMTMM, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride; EDC, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride; NHS, N-hydroxysuccinimide. | ||
To facilitate the identification of cross-links in XL-MS experiments, different variants of reagents have been proposed. For example, reagents that carry affinity tags such as biotin84–86 allow the enrichment of cross-linked peptides from complex peptide mixtures, thereby facilitating their detection and identification. Compounds with specific fragmentation properties in the mass spectrometer (cleavable cross-linkers)85–91 have been used to extend the application range of XL-MS to whole proteomes. Finally, reagents that are used in two different isotope labeled variants impart a unique signature for identification by mass spectrometry and improve data analysis.86,87,92,93 Such functionalized reagents are frequently not commercially available, which limits their more widespread use. In addition, affinity tags can make reagents bulkier and can have a negative impact on MS analysis, for example by affecting ionization and peptide fragmentation.
5.2. Cross-linking workflow
The generic workflow of an XL-MS experiment is summarized in Fig. 7. Similar to covalent labeling approaches, many steps of the experimental procedure are the same as for a conventional MS-based proteomics strategy. For more focused applications as discussed here, the sample can either be a highly purified or reconstituted protein complex, or a lower purity complex prepared by methods such as affinity purification (“pull-down”). The cross-linking reaction is commonly carried out for tens of minutes. Depending on the actual chemistry involved, the reaction is stopped by chemical quenching or by removal of the reagents (e.g. by gel filtration). At this point, the structural information is encoded in the form of covalent bonds and subsequent processing steps can be performed under denaturing conditions.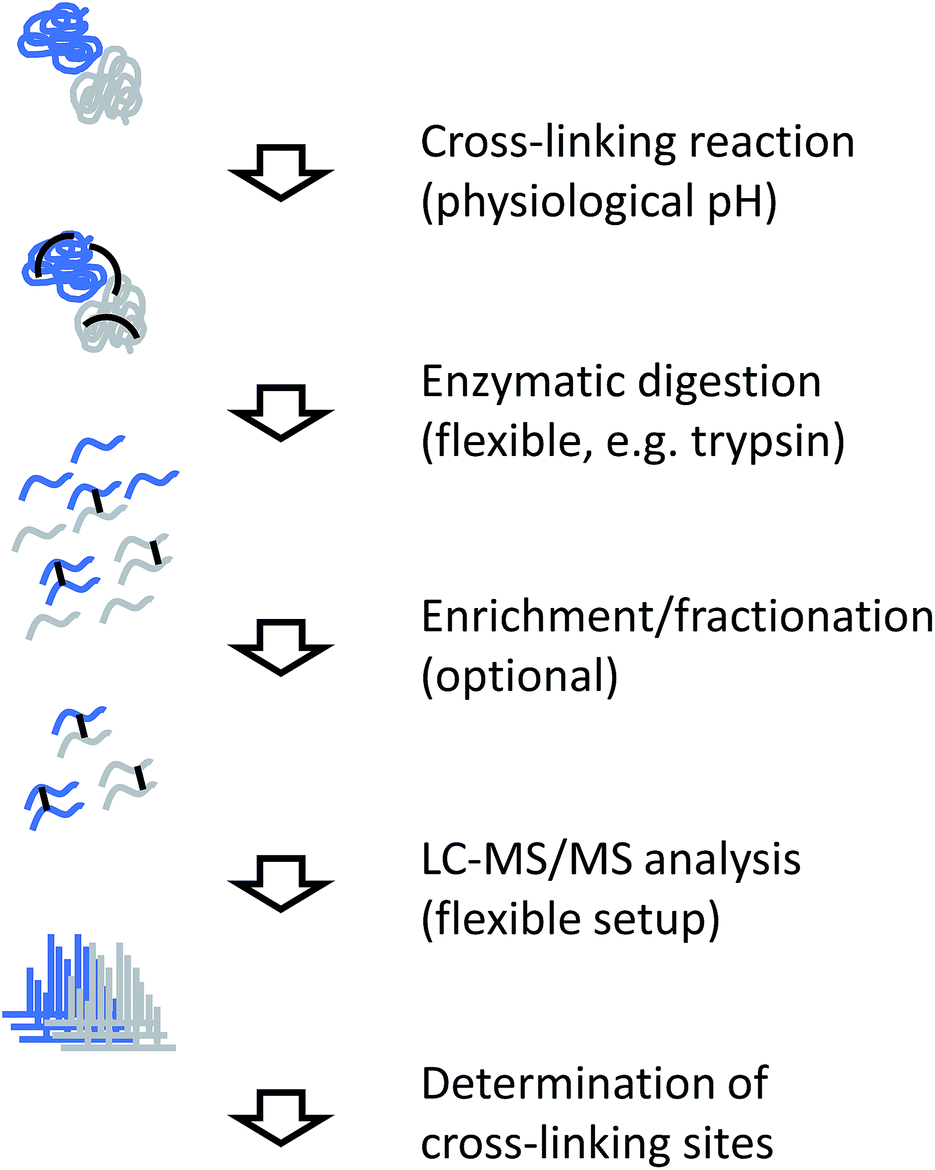 | ||
| Fig. 7 Experimental workflow for chemical cross-linking/MS. | ||
Cross-linked samples are then either directly subjected to proteolysis or individual constituents of the sample are separated by SDS-PAGE. Such a gel-based separation step also facilitates the removal of non-cross-linked subunits and/or undesired oligomeric reaction products, although recovery from in-gel digestion may be less efficient than in-solution processing and highly cross-linked complexes do not enter the gel. For proteolysis, trypsin is the most commonly used enzyme, although many alternative proteases used in proteomics research have also been employed in cross-linking studies.94,95 For example, less specific proteases that cleave at many different sites increase the chance of identifying a particular cross-link,95 although the complexity of the sample increases notably.
The resulting peptide mixture will consist of a majority of unmodified or modified, but not cross-linked peptides, where only one end of the reagent has reacted with the target functional group (termed “dead-end links” or “mono-links”). Cross-linked peptides only represent a small fraction of the total peptide pool. Prior to mass spectrometric analysis, cross-linked peptides can be enriched further, for example by size exclusion chromatography,94 ion exchange chromatography,96,97 or purification through an affinity tag.84–86 Finally, the sample is subjected to LC-MS/MS analysis.
Different strategies to analyze MS/MS data from cross-linking experiments exist and they depend mainly on the design of the cross-linking reagent. Non-cleavable linkers result in complex spectra to which both connected peptide chains contribute fragment ions. This requires the use of dedicated analysis software for interpretation,98–101 although some general proteomics database search programs have also been adapted for this purpose.102,103 Cleavable linkers include a preferential fragmentation site so that in an initial fragmentation event, individual peptide chains are released and subjected to further fragmentation in a second step. This approach requires a different computational approach for data analysis.104,105 Many variation of both workflows exist, although no direct comparison of the two approaches using identical samples has been reported in the literature. Despite the more complicated data analysis involved, non-cleavable linkers seem to be in more widespread use at the moment.
The output of the data analysis step is essentially a list of connected peptides. In most cases, the precise location of the cross-linking sites within these peptide sequences can be determined as well. Importantly, a measure of the “quality” of the identification (essentially the statistical certainty) should be available, although not many programs are currently able to produce such information, and the reported error rates may not always be accurate.
5.3. Representative applications
As shown in Fig. 5, cross-linking data provides different levels of structural or spatial information. As a consequence, the practical use of XL-derived restraints varies widely. For small complexes (or even individual proteins), only a limited number of practically useful cross-links may be obtained with a standard workflow. A combination of different experimental protocols may be used in such cases to increase the number of restraints. For example, there may not be enough lysines close to the binding interface between two proteins, which may be compensated by applying different cross-linking chemistries. For example, Raasch et al. recently applied a carboxyl group-specific chemistry106 to cross-link the 2-oxoglutarate dehydrogenase subunits OdhA and OdhI from Corynebacterium glutamicum, which did not yield any contacts using Lys-specific cross-linking.107 A second possibility is that Lys–Lys cross-links are formed but have unfavorable properties for MS analysis, for example because the peptides are too long or too short. A solution to this problem would be the use of additional specific or non-specific proteases, as mentioned above, which increases the chance the cross-link will be observable by MS.The first study that made use of cross-linking data for structural modeling purposes used a lysine reactive reagent to derive intra-molecular cross-link contacts on bovine fibroblast growth factor 2.108 However, it took nearly another decade to apply XL-MS to larger assemblies. In 2010, Rappsilber and co-workers applied the technique to study the subunit organization of RNA polymerase II in combination with a transcription initiation factor.109 Sinz provides an excellent account of the evolution of XL-MS during this decade.110
In the following years, the number of applications to larger protein complexes (composed of four or more subunits) has increased enormously.77,111 This has been facilitated by technical improvements of the cross-linking workflow (introduction of enrichment methods, faster and more sensitive mass spectrometers, new software), but also by the increased acceptance of XL-MS and its integration into hybrid structural biology methods.29,112 In particular, the combination of XL-MS and EM has been exploited in many cases, because cross-linking restraints facilitate the placement of individual subunits or even domains in EM maps. Therefore, despite the limited spatial resolution of individual cross-link restraints with typical upper limits in the range of 20–30 Å,113 the method adds an important level of information because a large number of such restraints – up to several hundred112 – can be obtained for bigger systems.
Selected recent highlights of XL-MS applications to study the architecture of large protein complexes include the 26S proteasome,114 RNA polymerases,109,115,116 chromatin remodeling complexes,117,118 photosystem complexes119 and chaperones,120,121 among many others. Ribosomes that consist of almost 100 individual proteins are the largest systems that have been studied by cross-linking approaches as individual entities.112,122 However, the concept has been expanded to protein interaction networks, as exemplified by cross-linking of affinity purified sub-complexes in the protein phosphatase 2A network.123
6. Conclusions and outlook
In the preceding chapters, I have summarized the three main methods that combine chemical labeling and mass spectrometric analysis for the structural analysis of proteins and protein complexes. Not surprisingly, each method has particular advantages, but also limitations.HDX-MS is unique in the sense that the chemical probe that is used for labeling is the smallest one possible, thereby ensuring minimal structural perturbations. Different experimental set-ups make this method extremely versatile and the study of large protein assemblies is only a small, but certainly growing part. The method is mature enough to be widely used in industry and applications such as epitope mapping will be of growing importance, given the increasing relevance of antibodies as biopharmaceuticals. Applications to large protein assemblies remain somewhat limited, but with further improvements in instrumentation and software tools, HDX-MS will also provide important insights into structural changes in these bigger systems.
CL-MS methods are easier to implement experimentally, but experimental conditions have to be chosen carefully to prevent unwanted structural perturbations, for example through excessive modification. Conventional in-solution labeling has been used for decades, even before modern structural biology techniques were routinely used. I feel that these labeling methods remain underappreciated compared to H/D exchange and cross-linking, despite their simplicity. Radical footprinting methods have been shown to be competitive with HDX, although the user base is still relatively small and special instrumentation (or access to synchrotrons) is required to perform the experiments. The extension of the concept to whole cells is a very exciting development.
XL-MS has increasingly been adopted in integrative structural biology projects and appears to be the most appropriate technique among the three for larger assemblies at the moment. However, it cannot be considered a mature or routine technique right now. One reason might be the diversity of cross-linking chemistries, reagent designs and software tools. This diversity may be intimidating to newcomers, and objective comparisons of different experimental workflows are lacking at the moment. However, the number of applications reported in the literature has been growing enormously in recent years, and the strength of the technique is its straightforward integration into emerging “hybrid” methods in structural biology.
This is also the direction where the other methods will be heading in the near future, so that together with native MS approaches, chemical labeling based structural MS methods will become even more widely adopted than at the moment.
The field is highly interdisciplinary: developing new reagents and probes will be the task for chemists and chemical biologists, while analytical scientists and bioinformaticians can contribute new experimental and computational workflows. Performing research at the interface of so many disciplines comes with its own challenges; many reports in the literature are only proof-of-principle experiments on simple model systems. Therefore, it is crucial for researchers in the field to forge collaborations with biologists to apply these methods to relevant research questions and to help to increase further adoption of structural mass spectrometry techniques.
Acknowledgements
I apologize to researchers whose work could not be cited due to space constraints. AL acknowledges support from the European Union COST Action BM 1403 (Mass Spectrometry and Related Methods for Structural Biology).References
- X.-c. Bai, G. McMullan and S. H. W. Scheres, Trends Biochem. Sci., 2015, 40, 49–57 CrossRef CAS PubMed.
- Y. Cheng, Cell, 2015, 161, 450–457 CrossRef CAS PubMed.
- A. F. M. Altelaar, J. Munoz and A. J. R. Heck, Nat. Rev. Genet., 2013, 14, 35–48 CrossRef CAS PubMed.
- Y. Zhang, B. R. Fonslow, B. Shan, M.-C. Baek and J. R. Yates III, Chem. Rev., 2013, 113, 2343–2394 CrossRef CAS PubMed.
- Z. Zhang, S. Wu, D. L. Stenoien and L. Pasa-Tolic, Annu. Rev. Anal. Chem., 2014, 7, 427–454 CrossRef CAS PubMed.
- M. Larance and A. I. Lemond, Nat. Rev. Mol. Cell Biol., 2015, 16, 269–280 CrossRef CAS PubMed.
- K. Biemann, J. Proteomics, 2014, 107, 62–70 CrossRef CAS PubMed.
- A. Leitner and W. Lindner, Proteomics, 2006, 6, 5418–5434 CrossRef CAS PubMed.
- M. Bantscheff, S. Lemeer, M. M. Savitski and B. Küster, Anal. Bioanal. Chem., 2012, 404, 939–965 CrossRef CAS PubMed.
- N. Rauniyar and J. R. Yates III, J. Proteome Res., 2014, 13, 5293–5309 CrossRef CAS PubMed.
- N. Nagaraj, N. A. Kulak, J. Cox, N. Neuhauser, K. Mayr, O. Hoerning, O. Vorm and M. Mann, Mol. Cell. Proteomics, 2012, 11, M111.013722 Search PubMed.
- A. S. Hebert, A. L. Richards, D. J. Bailey, A. Ulbrich, E. E. Coughlin, M. S. Westphall and J. J. Coon, Mol. Cell. Proteomics, 2014, 13, 339–347 CAS.
- N. Nagaraj, J. R. Wisniewski, T. Geiger, J. Cox, M. Kircher, J. Kelso, S. Pääbo and M. Mann, Mol. Syst. Biol., 2011, 7, 548 CrossRef PubMed.
- M. Beck, A. Schmidt, J. Malmstroem, M. Claassen, A. Ori, A. Szymborska, F. Herzog, O. Rinner, J. Ellenberg and R. Aebersold, Mol. Syst. Biol., 2011, 7, 549 CrossRef PubMed.
- S. K. Chowdhury, V. Katta and B. T. Chait, J. Am. Chem. Soc., 1990, 112, 9012–9013 CrossRef CAS.
- J. A. Loo, R. R. Ogorzalek Loo, H. R. Udseth, C. G. Edmonds and R. D. Smith, Rapid Commun. Mass Spectrom., 1991, 5, 101–105 CrossRef CAS PubMed.
- V. Katta and B. T. Chait, J. Am. Chem. Soc., 1991, 113, 8534–8535 CrossRef CAS.
- A. Dobo and I. A. Kaltashov, Anal. Chem., 2001, 73, 4763–4773 CrossRef CAS PubMed.
- L. Konermann, S. Vahidi and M. A. Sowole, Anal. Chem., 2014, 86, 213–232 CrossRef CAS PubMed.
- A. B. Ward, A. Sali and I. A. Wilson, Science, 2013, 339, 913–915 CrossRef CAS PubMed.
- G. R. Hilton and J. L. P. Benesch, J. R. Soc., Interface, 2012, 9, 801–816 CrossRef CAS PubMed.
- A. Konijnenberg, A. Butterer and F. Sobott, Biochim. Biophys. Acta, 2013, 1834, 1239–1256 CrossRef CAS PubMed.
- J. Snijder and A. J. R. Heck, Annu. Rev. Anal. Chem., 2014, 7, 43–64 CrossRef CAS PubMed.
- F. Sobott, H. Hernández, M. G. McCammon, M. A. Tito and C. V. Robinson, Anal. Chem., 2002, 74, 1402–1407 CrossRef CAS PubMed.
- R. J. Rose, E. Damoc, E. Denisov, A. Makarov and A. J. R. Heck, Nat. Methods, 2012, 9, 1084–1086 CrossRef CAS PubMed.
- S. Mehmood, T. M. Allison and C. V. Robinson, Annu. Rev. Phys. Chem., 2015, 66, 453–474 CrossRef CAS PubMed.
- J. A. Loo, B. Berhane, C. S. Kaddis, K. M. Wooding, Y. Xie, S. L. Kaufman and I. V. Chernushevich, J. Am. Soc. Mass Spectrom., 2005, 16, 998–1008 CrossRef CAS PubMed.
- B. T. Ruotolo, J. L. P. Benesch, A. M. Sandercock, S.-J. Hyung and C. V. Robinson, Nat. Protoc., 2008, 3, 1139–1152 CrossRef CAS PubMed.
- A. Politis, F. Stengel, Z. Hall, H. Hernández, A. Leitner, T. Walzthoeni, C. V. Robinson and R. Aebersold, Nat. Methods, 2014, 11, 403–406 CrossRef CAS PubMed.
- L. Konermann, J. Pan and Y.-H. Liu, Chem. Soc. Rev., 2011, 40, 1224–1234 RSC.
- A. J. Percy, M. Rey, K. M. Burns and D. C. Schriemer, Anal. Chim. Acta, 2012, 721, 7–21 CrossRef CAS PubMed.
- H. Wei, J. Mo, L. Tao, R. J. Russell, A. A. Tymiak, G. Chen, R. E. Iacob and J. R. Engen, Drug Discovery Today, 2014, 19, 95–102 CrossRef CAS PubMed.
- G. F. Pirrone, R. E. Iacob and J. R. Engen, Anal. Chem., 2015, 87, 99–118 CrossRef CAS PubMed.
- T. E. Wales, K. E. Fadgen, G. C. Gerhardt and J. R. Engen, Anal. Chem., 2008, 80, 6815–6820 CrossRef CAS PubMed.
- J. Pan, S. Zhang, C. E. Parker and C. H. Borchers, J. Am. Chem. Soc., 2014, 136, 13065–13071 CrossRef CAS PubMed.
- M. Rey, M. Yang, K. M. Burns, Y. Yu, S. P. Lees-Miller and D. C. Schriemer, Mol. Cell. Proteomics, 2013, 12, 464–472 CAS.
- B. D. Pascal, S. Willis, J. L. Lauer, R. R. Landgraf, G. M. West, D. Marciano, S. Novick, D. Goswami, M. J. Chalmers and P. R. Griffin, J. Am. Soc. Mass Spectrom., 2012, 23, 1512–1521 CrossRef CAS PubMed.
- T. E. Wales, M. J. Eggerton and J. R. Engen, Methods Mol. Biol., 2013, 1007, 263–288 CAS.
- T. J. D. Jørgensen, H. Gårdsvoll, M. Ploug and P. Roepstorff, J. Am. Chem. Soc., 2005, 127, 2785–2793 CrossRef PubMed.
- K. D. Rand, C. M. Adams, R. A. Zubarev and T. J. D. Jørgensen, J. Am. Chem. Soc., 2008, 130, 1341–1349 CrossRef CAS PubMed.
- K. D. Rand, M. Zehl, O. N. Jensen and T. J. D. Jørgensen, Anal. Chem., 2009, 81, 5577–5584 CrossRef CAS PubMed.
- K. D. Rand, M. Zehl and T. J. D. Jørgensen, Acc. Chem. Res., 2014, 47, 3018–3027 CrossRef CAS PubMed.
- K. Y. Chung, S. G. F. Rasmussen, T. Liu, S. Li, B. T. DeVree, P. S. Chae, D. Calinski, B. K. Kobilka, V. L. Woods Jr and R. K. Sunahara, Nature, 2011, 477, 611–615 CrossRef CAS PubMed.
- A. K. Shukla, G. H. Westfield, K. Xiao, R. I. Reis, L.-Y. Huang, P. Tripathi-Shukla, J. Qian, S. Li, A. Blanc, A. N. Oleskie, A. M. Dosey, M. Su, C.-R. Liang, L.-L. Gu, J.-M. Shan, X. Chen, R. Hanna, M. Choi, X. J. Yao, B. U. Klink, A. W. Kahsai, S. S. Sidhu, S. Koide, P. A. Penczek, A. A. Kossiakoff, V. L. Woods Jr, B. K. Kobilka, G. Skiniotis and R. J. Lefkowitz, Nature, 2014, 512, 218–222 CrossRef CAS PubMed.
- A. J. Noble, Q. Zhang, J. O'Donnell, H. Hariri, N. Bhattacharya, A. G. Marshall and S. M. Stagg, Nat. Struct. Mol. Biol., 2013, 20, 167–173 CAS.
- Q. Zhang, J. Chen, K. Kuwajima, H.-M. Zhang, F. Xian, N. L. Young and A. G. Marshall, Sci. Rep., 2013, 3, 1247 Search PubMed.
- F. Georgescauld, K. Popova, A. J. Gupta, A. Bracher, J. R. Engen, M. Hayer-Hartl and F. U. Hartl, Cell, 2014, 157, 922–934 CrossRef CAS PubMed.
- M. Rey, V. Sarpe, K. M. Burns, J. Buse, C. A. H. Baker, M. Van Dijk, L. Wordeman, A. M. J. J. Bonvin and D. C. Schriemer, Structure, 2014, 22, 1538–1548 CrossRef CAS PubMed.
- K. M. Burns, V. Sarpe, M. Wagenbach, L. Wordeman and D. C. Schriemer, Protein Sci., 2015, 24, 1313–1324 CrossRef CAS PubMed.
- V. L. Mendoza and R. W. Vachet, Mass Spectrom. Rev., 2009, 28, 785–815 CrossRef CAS PubMed.
- E. O. Hochleitner, C. Borchers, C. Parker, R. J. Bienstock and K. B. Tomer, Protein Sci., 2000, 9, 487–496 CrossRef CAS PubMed.
- A. Ori, P. Free, J. Courty, M. C. Wilkinson and D. G. Fernig, Mol. Cell. Proteomics, 2009, 8, 2256–2265 CAS.
- E. Bloem, E. H. T. M. Ebberink, M. van den Biggelaar, C. van der Zwaan, K. Mertens and A. B. Meijer, Biochem. J., 2015, 468, 65–72 CrossRef CAS PubMed.
- J. D. Venable, C. Steckler, W. Ou, J. Grünewald, S. Agarwalla and A. Brock, Anal. Chem., 2015, 87, 7540–7544 CrossRef CAS PubMed.
- H. Zhang, J. Wen, R. Y.-C. Huang, R. E. Blankenship and M. L. Gross, Int. J. Mass Spectrom., 2012, 312, 78–86 CrossRef CAS PubMed.
- V. L. Mendoza and R. W. Vachet, Anal. Chem., 2008, 80, 2895–2904 CrossRef CAS PubMed.
- P. Liuni, S. Zhu and D. J. Wilson, Antioxid. Redox Signaling, 2014, 21, 497–510 CrossRef CAS PubMed.
- S. D. Maleknia and K. M. Downard, Chem. Soc. Rev., 2014, 43, 3244–3258 RSC.
- S. D. Maleknia, M. Brenowitz and M. R. Chance, Anal. Chem., 1999, 71, 3965–3973 CrossRef CAS PubMed.
- D. M. Hambly and M. L. Gross, J. Am. Soc. Mass Spectrom., 2005, 16, 2057–2063 CrossRef CAS PubMed.
- G. Xu and M. R. Chance, Anal. Chem., 2005, 77, 4549–4555 CrossRef CAS PubMed.
- L. Konermann, Y. Pan and B. B. Stocks, Curr. Opin. Struct. Biol., 2011, 21, 634–640 CrossRef CAS PubMed.
- T. E. Angel, S. Gupta, B. Jastrzebska, K. Palczewski and M. R. Chance, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14367–14372 CrossRef CAS PubMed.
- S. Gupta, V. N. Bavro, R. D'Mello, S. J. Tucker, C. Vénien-Bryan and M. R. Chance, Structure, 2010, 18, 839–846 CrossRef CAS PubMed.
- T. Orban, B. Jastrzebska, S. Gupta, B. Wang, M. Miyagi, M. R. Chance and K. Palczewski, Structure, 2012, 20, 826–840 CrossRef CAS PubMed.
- S. Gupta, J. Chai, J. Cheng, R. D'Mello, M. R. Chance and D. Fu, Nature, 2014, 512, 101–104 CAS.
- L. M. Jones, J. B. Sperry, J. A. Carroll and M. L. Gross, Anal. Chem., 2011, 83, 7657–7661 CrossRef CAS PubMed.
- A. Rinas and L. M. Jones, J. Am. Soc. Mass Spectrom., 2015, 26, 540–546 CrossRef CAS PubMed.
- J. A. Espino, V. S. Mali and L. M. Jones, Anal. Chem., 2015, 87, 7971–7978 CrossRef CAS PubMed.
- H. Zhang, W. Shen, D. Rempel, J. Monsey, I. Vidavsky, M. L. Gross and R. Bose, Mol. Cell. Proteomics, 2011, 10, M110.005678 Search PubMed.
- H. Liu, H. Zhang, J. D. King, N. R. Wolf, M. Prado, M. L. Gross and R. E. Blankenship, Biochim. Biophys. Acta, 2014, 1837, 1955–1963 CrossRef CAS PubMed.
- V. L. Mendoza, K. Antwi, M. A. Barón-Rodríguez, C. Blanco and R. W. Vachet, Biochemistry, 2010, 49, 1522–1532 CrossRef CAS PubMed.
- H. G. Budayeva and I. M. Cristea, in Advancements in Experimental Medicine and Biology, ed. A. G. Woods and C. C. Darie, Springer, 2014, vol. 806, pp. 263–282 Search PubMed.
- R. I. Subbotin and B. T. Chait, Mol. Cell. Proteomics, 2014, 13, 2824–2835 CAS.
- A. N. Calabrese and T. L. Pukala, Aust. J. Chem., 2013, 66, 749–759 CrossRef CAS.
- A. N. Holding, Methods, 2015, 89, 54–63 CrossRef CAS PubMed.
- F. Liu and A. J. R. Heck, Curr. Opin. Struct. Biol., 2015, 35, 100–108 CrossRef CAS PubMed.
- E. V. Petrotchenko and C. H. Borchers, Mass Spectrom. Rev., 2010, 29, 862–876 CrossRef CAS PubMed.
- D. Paramelle, G. Miralles, G. Subra and J. Martinez, Proteomics, 2013, 13, 438–456 CrossRef CAS PubMed.
- Bioconjugate Techniques, ed. G. T. Hermanson, Academic Press/Elsevier, 3rd edn, 2013 Search PubMed.
- D. Robinette, N. Neamati, K. B. Tomer and C. H. Borchers, Expert Rev. Proteomics, 2006, 3, 399–408 CrossRef CAS PubMed.
- N. D. Pham, R. B. Parker and J. J. Kohler, Curr. Opin. Chem. Biol., 2013, 17, 90–101 CrossRef CAS PubMed.
- F. Krauth, C. H. Ihling, H. H. Rüttinger and A. Sinz, Rapid Commun. Mass Spectrom., 2009, 23, 2811–2818 CrossRef CAS PubMed.
- M. Trester-Zedlitz, K. Kamada, S. K. Burley, D. Fenyö, B. T. Chait and T. W. Muir, J. Am. Chem. Soc., 2003, 125, 2416–2425 CrossRef CAS PubMed.
- X. T. Tang, G. R. Munske, W. F. Siems and J. E. Bruce, Anal. Chem., 2005, 77, 311–318 CrossRef CAS PubMed.
- E. V. Petrotchenko, J. J. Serpa and C. H. Borchers, Mol. Cell. Proteomics, 2011, 10, M110.001420 Search PubMed.
- E. V. Petrotchenko, V. K. Olkhovik and C. H. Borchers, Mol. Cell. Proteomics, 2005, 4, 1167–1179 CAS.
- A. Kao, C.-l. Chiu, D. Vellucci, Y. Yang, V. R. Patel, S. Guan, A. Randall, P. Baldi, S. D. Rychnovsky and L. Huang, Mol. Cell. Proteomics, 2011, 10, M110.002212 Search PubMed.
- C. Benda, J. Ebert, R. A. Scheltema, H. B. Schiller, M. Baumgartner, F. Bonneau, M. Mann and E. Conti, Mol. Cell, 2014, 56, 43–54 CrossRef CAS PubMed.
- M. Q. Müller, F. Dreiocker, C. H. Ihling, M. Schäfer and A. Sinz, Anal. Chem., 2010, 82, 6958–6968 CrossRef PubMed.
- E. J. Soderblom and M. B. Goshe, Anal. Chem., 2006, 78, 8059–8068 CrossRef CAS PubMed.
- D. R. Müller, P. Schindler, H. Towbin, U. Wirth, H. Voshol, S. Hoving and M. O. Steinmetz, Anal. Chem., 2001, 73, 1927–1934 CrossRef.
- J. Seebacher, P. Mallick, N. Zhang, J. S. Eddes, R. Aebersold and M. H. Gelb, J. Proteome Res., 2006, 5, 2270–2282 CrossRef CAS PubMed.
- A. Leitner, R. Reischl, T. Walzthoeni, F. Herzog, S. Bohn, F. Förster and R. Aebersold, Mol. Cell. Proteomics, 2012, 11, M111.014126 Search PubMed.
- E. V. Petrotchenko, J. J. Serpa, D. B. Hardie, M. Berjanskii, B. P. Suriyamongkol, D. S. Wishart and C. H. Borchers, Mol. Cell. Proteomics, 2012, 11, M111.013524 Search PubMed.
- O. Rinner, J. Seebacher, T. Walzthoeni, L. N. Müller, M. Beck, A. Schmidt, M. Mueller and R. Aebersold, Nat. Methods, 2008, 5, 315–318 CrossRef CAS PubMed.
- R. Fritzsche, C. H. Ihling, M. Götze and A. Sinz, Rapid Commun. Mass Spectrom., 2012, 26, 653–658 CrossRef CAS PubMed.
- T. Walzthoeni, M. Claassen, A. Leitner, F. Herzog, S. Bohn, F. Förster, M. Beck and R. Aebersold, Nat. Methods, 2012, 9, 901–903 CrossRef CAS PubMed.
- B. Yang, Y.-J. Wu, M. Zhu, S.-B. Fan, J. Lin, K. Zhang, S. Li, H. Chi, Y.-X. Li, H.-F. Chen, S.-K. Luo, Y.-H. Ding, L.-H. Wang, Z. Hao, L.-Y. Xiu, S. Chen, K. Ye, S.-M. He and M.-Q. Dong, Nat. Methods, 2012, 9, 904–906 CrossRef CAS PubMed.
- M. Götze, J. Pettelkau, S. Schaks, K. Bosse, C. H. Ihling, F. Krauth, R. Fritzsche, U. Kuhn and A. Sinz, J. Am. Soc. Mass Spectrom., 2012, 23, 76–87 CrossRef PubMed.
- M. R. Hoopmann, A. Zelter, R. S. Johnson, M. Riffle, M. J. MacCoss, T. N. Davis and R. L. Moritz, J. Proteome Res., 2015, 14, 2190–2198 CrossRef CAS PubMed.
- H. Xu, P.-H. Hsu, L. Zhang, M.-D. Tsai and M. A. Freitas, J. Proteome Res., 2010, 9, 3384–3393 CrossRef CAS PubMed.
- M. J. Trnka, P. R. Baker, P. J. J. Robinson, A. L. Burlingame and R. J. Chalkley, Mol. Cell. Proteomics, 2014, 13, 420–434 CAS.
- M. Götze, J. Pettelkau, R. Fritzsche, C. H. Ihling, M. Schäfer and A. Sinz, J. Am. Soc. Mass Spectrom., 2015, 26, 83–97 CrossRef PubMed.
- C. R. Weisbrod, J. D. Chavez, J. K. Eng, L. Yang, C. Zheng and J. E. Bruce, J. Proteome Res., 2013, 12, 1569–1579 CrossRef CAS PubMed.
- A. Leitner, L. A. Joachimiak, P. Unverdorben, T. Walzthoeni, J. Frydman, F. Förster and R. Aebersold, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9455–9460 CrossRef CAS PubMed.
- K. Raasch, M. Bocola, J. Labahn, A. Leitner, L. Eggeling and M. Bott, J. Biotechnol., 2014, 191, 99–105 CrossRef CAS PubMed.
- M. M. Young, N. Tang, J. C. Hempel, C. M. Oshiro, E. W. Taylor, I. D. Kuntz, B. W. Gibson and G. Dollinger, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5802–5806 CrossRef CAS PubMed.
- Z. A. Chen, A. Jawhari, L. Fischer, C. Buchen, S. Tahir, T. Kamenski, M. Rasmussen, L. Lariviere, J.-C. Bukowski-Wills, M. Nilges, P. Cramer and J. Rappsilber, EMBO J., 2010, 29, 717–726 CrossRef CAS PubMed.
- A. Sinz, Expert Rev. Proteomics, 2014, 11, 733–743 CrossRef CAS PubMed.
- A. Leitner, M. Faini, F. Stengel and R. Aebersold, Trends Biochem. Sci., 2016, 41, 20–32 CrossRef CAS PubMed.
- J. P. Erzberger, F. Stengel, R. Pellarin, S. Zhang, T. Schaefer, C. H. S. Aylett, P. Cimermancic, D. Boehringer, A. Sali, R. Aebersold and N. Ban, Cell, 2014, 158, 1123–1135 CrossRef CAS PubMed.
- E. D. Merkley, S. Rysavy, A. Kahraman, R. P. Hafen, V. Daggett and J. N. Adkins, Protein Sci., 2014, 23, 747–759 CrossRef CAS PubMed.
- K. Lasker, F. Förster, S. Bohn, T. Walzthoeni, E. Villa, P. Unverdorben, F. Beck, R. Aebersold, A. Sali and W. Baumeister, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1380–1387 CrossRef CAS PubMed.
- K. Murakami, M. Elmlund, N. Kalisman, D. A. Bushnell, C. M. Adams, M. Azubel, D. Elmlund, Y. Levi-Kalisman, X. Liu, B. J. Gibbons, M. Levitt and R. D. Kornberg, Science, 2013, 342 DOI:10.1126/science.1238724.
- C. Plaschka, L. Larivière, L. Wenzeck, M. Seizl, M. Hemann, D. Tegunov, E. V. Petrotchenko, C. H. Borchers, W. Baumeister, F. Herzog, E. Villa and P. Cramer, Nature, 2015, 518, 376–380 CrossRef CAS PubMed.
- A. Tosi, C. Haas, F. Herzog, A. Gilmozzi, O. Berninghausen, C. Ungewickell, C. B. Gerhold, K. Lakomek, R. Aebersold, R. Beckmann and K.-P. Hopfner, Cell, 2013, 154, 1207–1219 CrossRef CAS PubMed.
- V. Q. Nguyen, A. Ranjan, F. Stengel, D. Wei, R. Aebersold, C. Wu and A. E. Leschziner, Cell, 2013, 154, 1220–1231 CrossRef CAS PubMed.
- H. Liu, H. Zhang, D. M. Niedzwiedzki, M. Prado, G. He, M. L. Gross and R. E. Blankenship, Science, 2013, 342, 1104–1107 CrossRef CAS PubMed.
- A. Leitner, L. A. Joachimiak, A. Bracher, L. Mönkemeyer, T. Walzthoeni, B. Chen, S. Pechmann, S. Holmes, Y. Cong, B. Ma, S. Ludtke, W. Chiu, F. U. Hartl, R. Aebersold and J. Frydman, Structure, 2012, 20, 814–825 CrossRef CAS PubMed.
- L. A. Joachimiak, T. Walzthoeni, C. W. Liu, R. Aebersold and J. Frydman, Cell, 2014, 159, 1042–1055 CrossRef CAS PubMed.
- B. J. Greber, P. Bieri, M. Leibundgut, A. Leitner, R. Aebersold, D. Boehringer and N. Ban, Science, 2015, 348, 303–308 CrossRef CAS PubMed.
- F. Herzog, A. Kahraman, D. Boehringer, R. Mak, A. Bracher, T. Walzthoeni, A. Leitner, M. Beck, F.-U. Hartl, N. Ban, L. Malmström and R. Aebersold, Science, 2012, 337, 1348–1352 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |