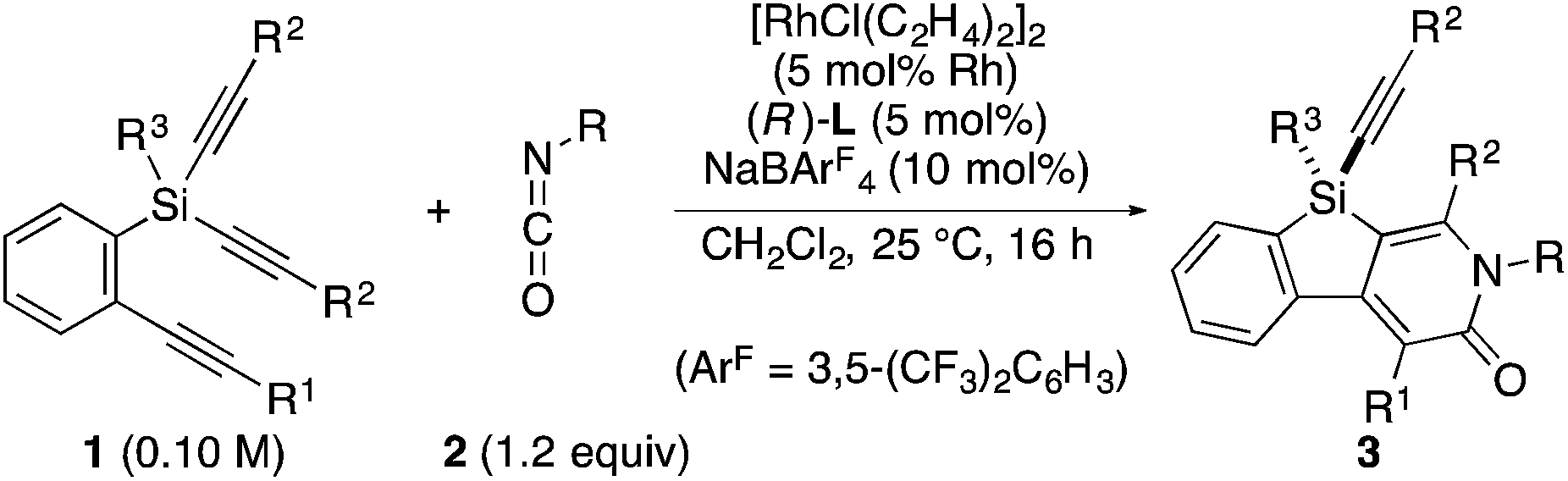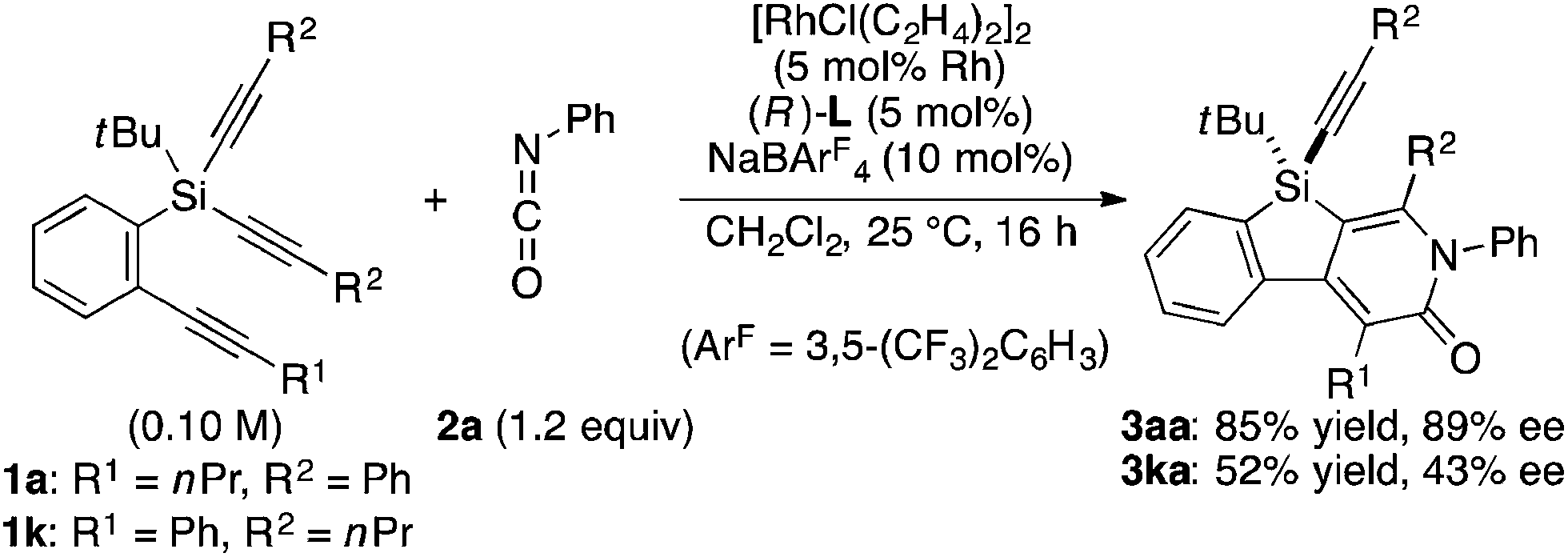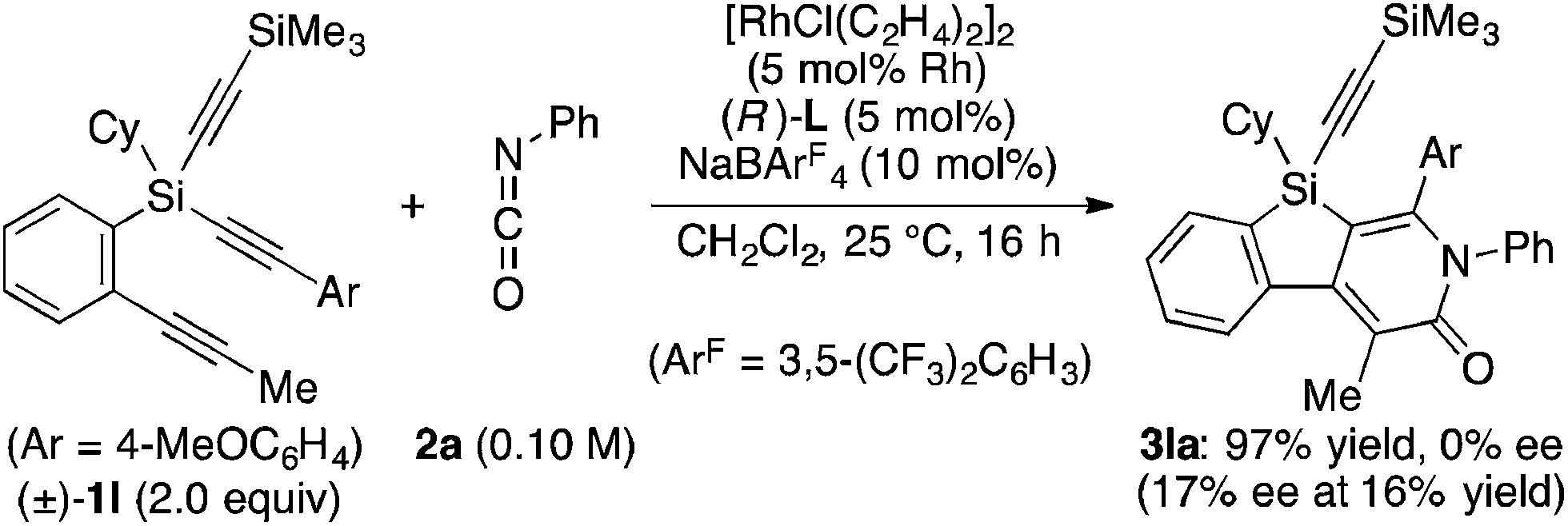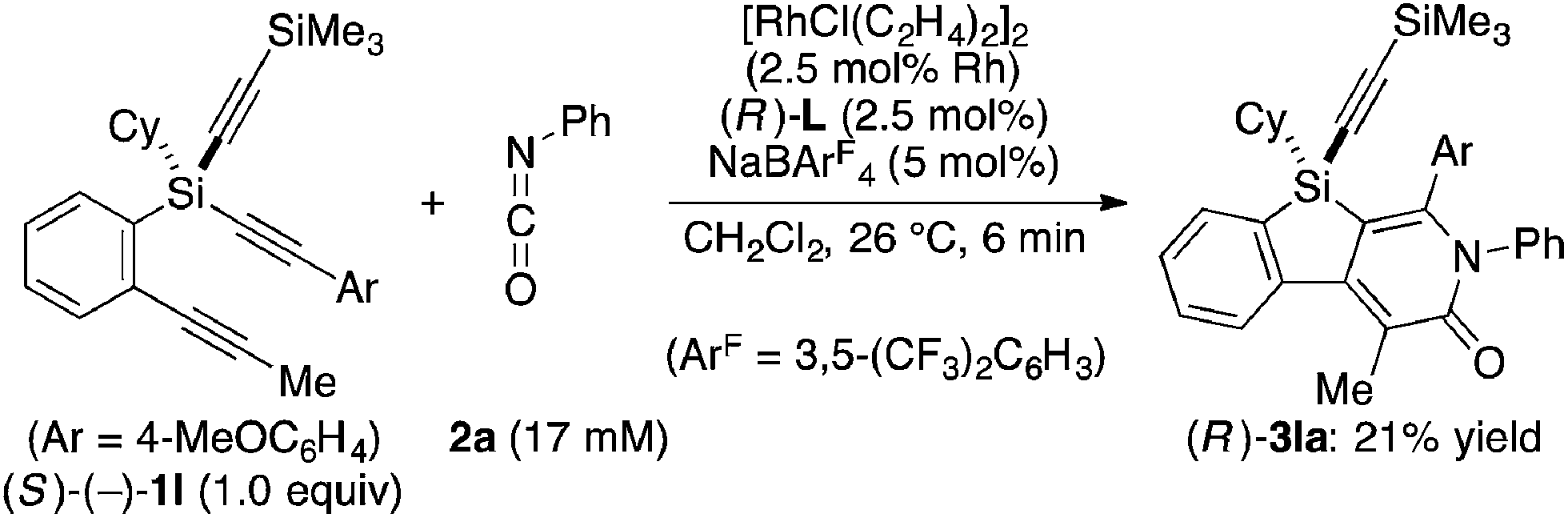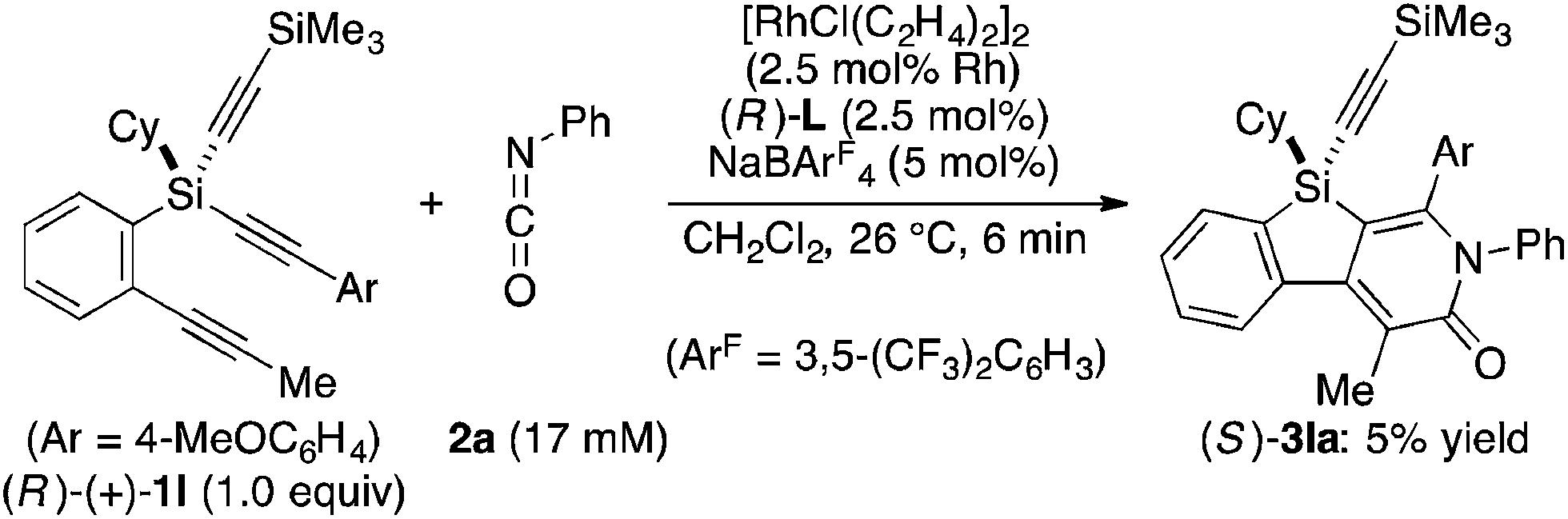DOI:
10.1039/C5SC03767K
(Edge Article)
Chem. Sci., 2016,
7, 1205-1211
Rhodium-catalyzed asymmetric synthesis of silicon-stereogenic silicon-bridged arylpyridinones†
Received
5th October 2015
, Accepted 6th November 2015
First published on 9th November 2015
Abstract
A rhodium-catalyzed regio- and enantioselective synthesis of silicon-stereogenic silicon-bridged arylpyridinones has been developed through [2 + 2 + 2] cycloaddition of silicon-containing prochiral triynes with isocyanates. High yields and enantioselectivities have been achieved by employing an axially chiral monophosphine ligand, and this process could be applied to catalytic asymmetric synthesis of silicon-stereogenic chiral polymers for the first time. The reaction mechanism of the present catalysis has also been experimentally investigated to establish a reasonable catalytic cycle, advancing the mechanistic understanding of the rhodium-catalyzed pyridinone synthesis by [2 + 2 + 2] cycloaddition reactions.
Introduction
Asymmetric catalysis represents one of the most efficient approaches for the preparation of enantioenriched chiral compounds, and extensive research has been made in developing various catalytic asymmetric reactions during the past decades.1 While most of them are directed toward the synthesis of carbon-stereogenic compounds, only limited structures are accessible for enantioenriched silicon-stereogenic compounds through asymmetric catalysis.2,3 Because organosilanes are widely utilized in various fields of research, broadening the scope of accessible silicon-stereogenic enantioenriched organosilanes would be highly desirable. Among the known catalytic enantioselective methods for the creation of silicon stereocenters, most typical approach is the desymmetrization of prochiral dihydrodiorganosilanes by way of enantioposition-selective hydrosilylation or other Si–H bond functionalization reactions.4 In contrast, the use of other types of prochiral organosilanes as substrates has been much less explored and such examples started to appear only very recently,5 many of which are intramolecular processes.
As one of the rare examples of intermolecular reactions, we recently reported a rhodium-catalyzed asymmetric synthesis of silicon-stereogenic dibenzosiloles (silicon-bridged biaryls) by enantioselective [2 + 2 + 2] cycloaddition of prochiral triynes with internal alkynes.5h,6 By taking advantage of the convergent nature of this intermolecular approach, we decided to further explore the enantioselective synthesis of a new family of silicon-stereogenic silicon-bridged π-conjugated compounds based on this strategy. Specifically, in this article, we describe the development of a catalytic regio- and enantioselective synthesis of silicon-stereogenic silicon-bridged arylpyridinones, dihydrobenzosilolopyridinones, by rhodium-catalyzed [2 + 2 + 2] cycloaddition of prochiral silicon-containing triynes with isocyanates, including the investigation of its mechanistic aspects.7,8
Results and discussion
Reaction development
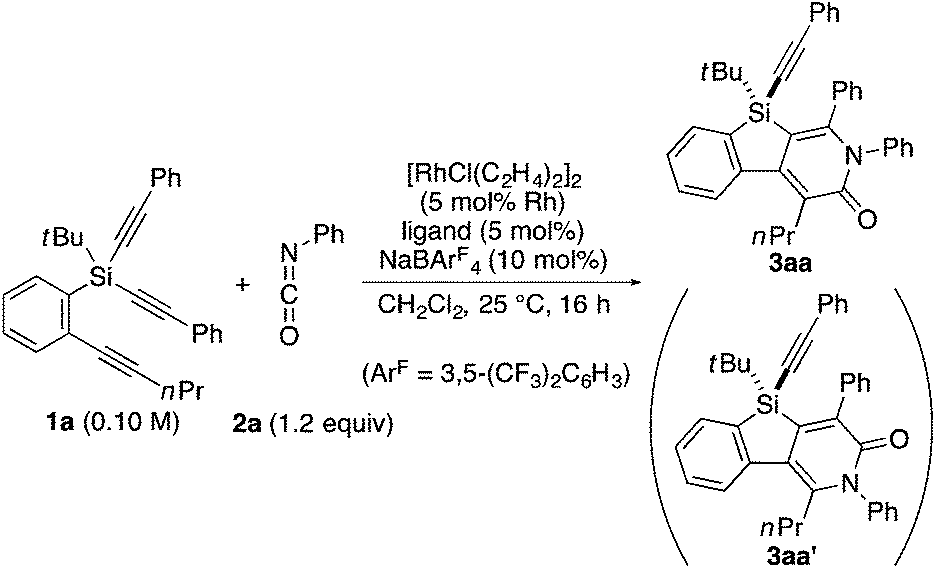
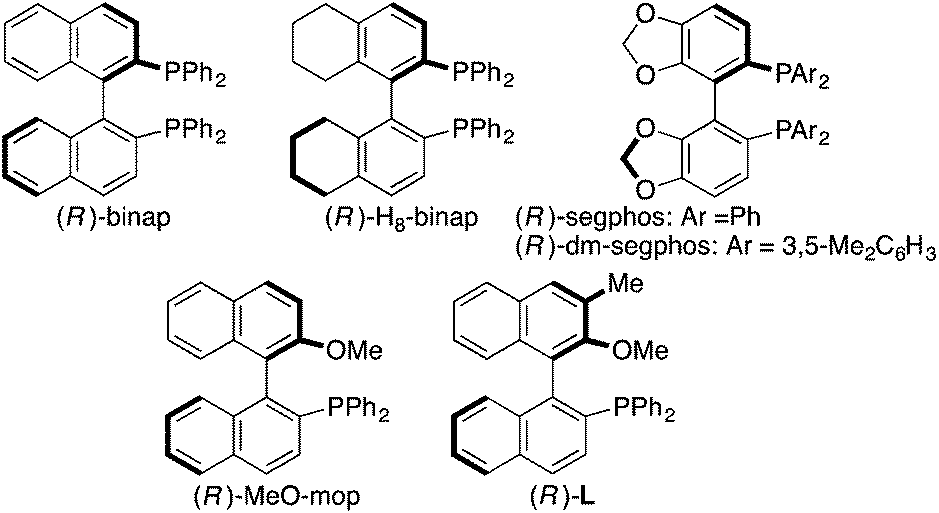
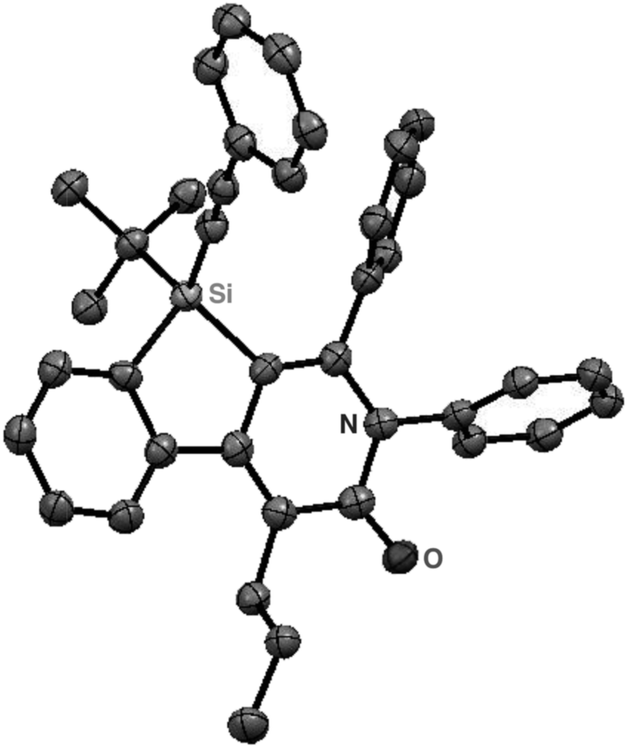
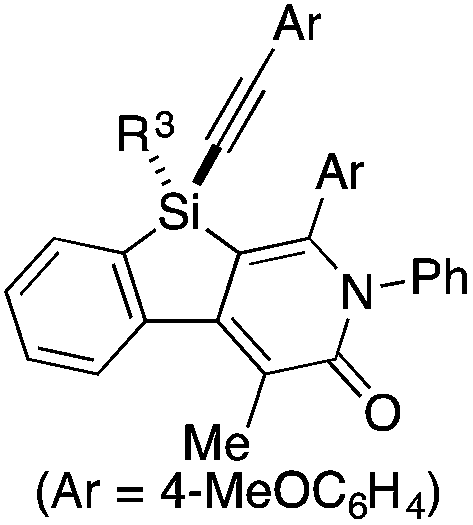
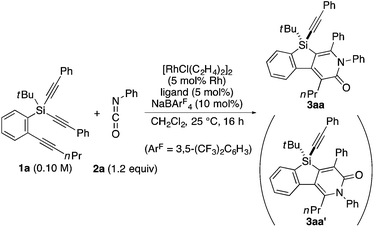
As a starting point, we employed prochiral triyne 1a as a model substrate for the cationic rhodium-catalyzed [2 + 2 + 2] cycloaddition with phenyl isocyanate (2a) in the presence of (R)-binap9 as the ligand at 25 °C (Table 1, entry 1). Although potentially two regioisomers 3aa and 3aa′ could be obtained depending on the orientation of isocyanate 2a, the reaction selectively provided only 3aa in 93% yield, albeit with low enantiomeric excess (14% ee). The change of ligand to (R)-H8-binap10 gave similarly low enantioselectivity (13% ee; entry 2), whereas the use of (R)-segphos11 improved the enantioselectivity to 76% ee (entry 3). But, the ee was not further improved by using (R)-dm-segphos (68% ee; entry 4). In comparison to these axially chiral bisphosphine ligands, axially chiral monophosphine ligand (R)-MeO-mop12 gave 3aa with higher enantioselectivity (80% ee; entry 5), and the use of (R)-L having a methyl group at the 3′-position13 further improved the enantioselectivity of 3aa to 89% ee in 85% yield (entry 6). It is worth noting that only 3aa was obtained without forming its regioisomer 3aa′ for all of these reactions, and the structure of 3aa including the absolute configuration was firmly established by X-ray crystallographic analysis with Cu-Kα radiation as shown in Fig. 1 after recrystallization of the product obtained in entry 6.14
Table 1 Rhodium-catalyzed asymmetric [2 + 2 + 2] cycloaddition of 1a with 2a: ligand effect
|
|
| Entry |
Ligand |
Yielda (%) |
eeb (%) |
|
Isolated yield.
Determined by chiral HPLC on a Chiralpak IA column with hexane/2-propanol = 95/5.
|
| 1 |
(R)-binap |
93 |
14 |
| 2 |
(R)-H8-binap |
89 |
13 |
| 3 |
(R)-segphos |
86 |
76 |
| 4 |
(R)-dm-segphos |
93 |
68 |
| 5 |
(R)-MeO-mop |
86 |
83 |
| 6 |
(R)-L |
85 |
89 |
|
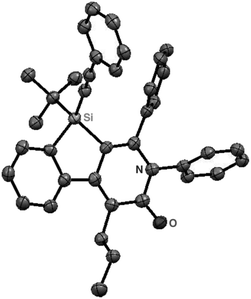 |
| | Fig. 1 X-Ray crystal structure of (R)-3aa (Flack parameter = 0.04(4)). Hydrogen atoms are omitted for clarity. | |
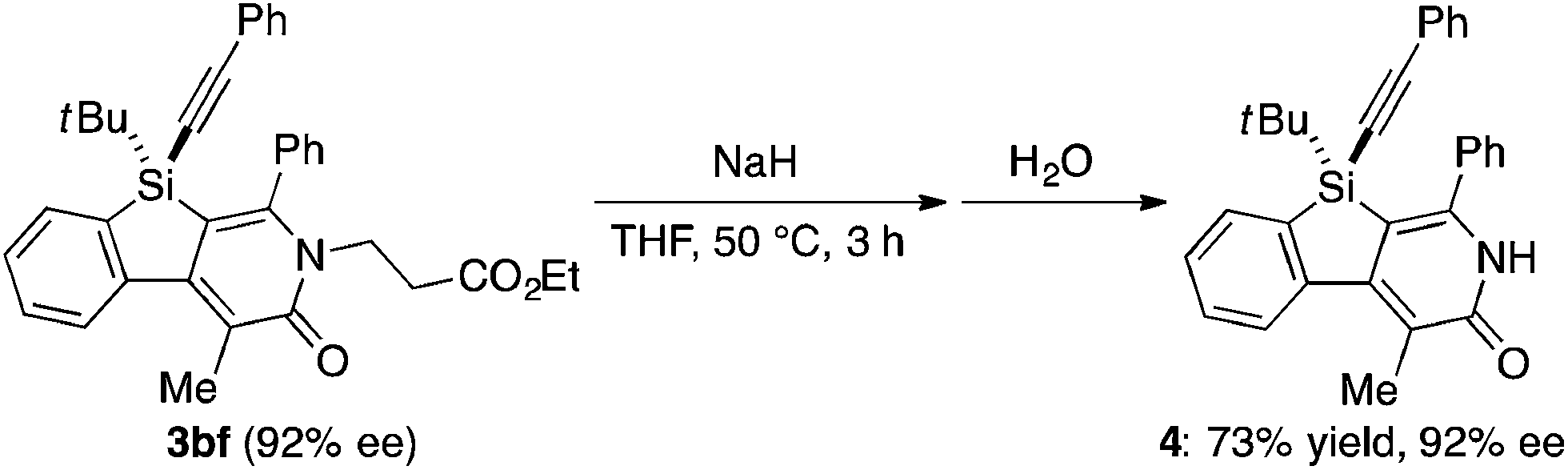
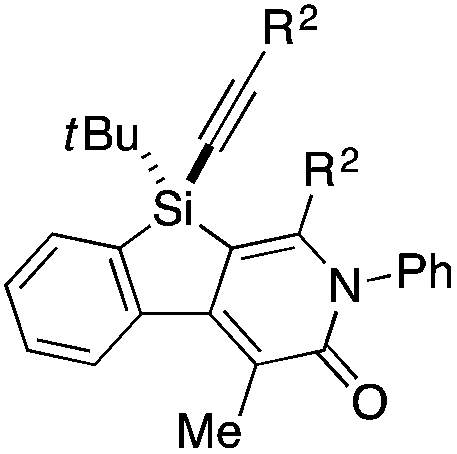
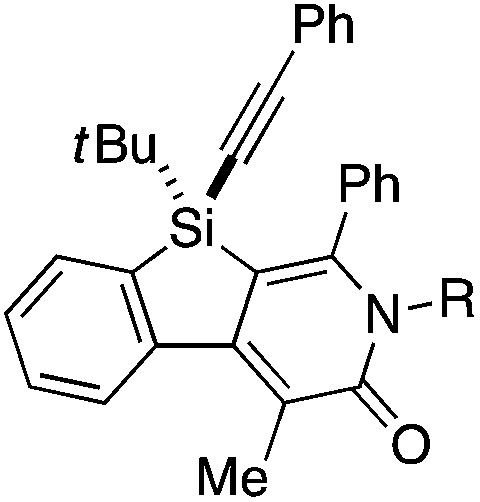
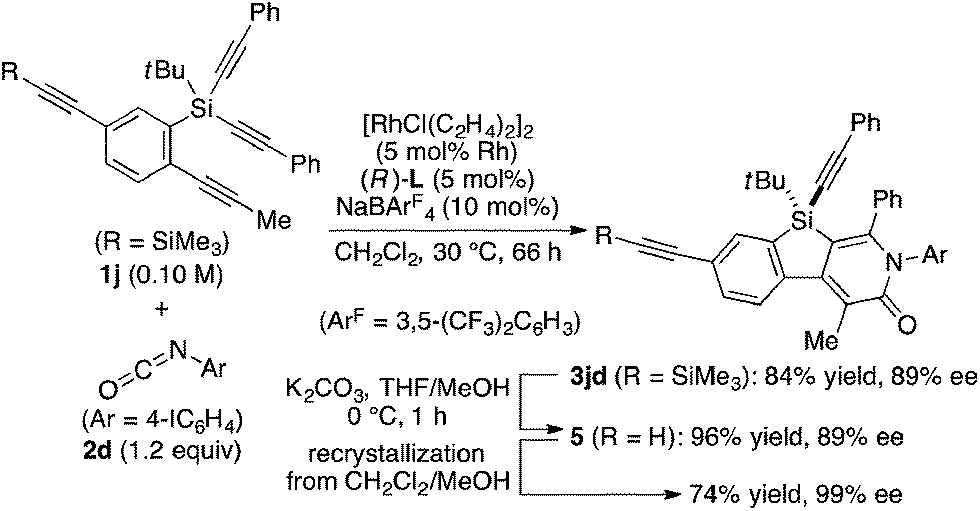
Under the conditions described in Table 1, entry 6, various silicon-stereogenic dihydrobenzosilolopyridinones (silicon-bridged arylpyridinones) 3 can be synthesized with high yields and enantioselectivities as summarized in Table 2. Thus, 1-pentynyl group of 1a (entry 1) can be replaced by other alkynyl groups such as 1-propynyl (1b), 4-methyl-1-pentynyl (1c), and unsubstituted ethynyl (1d) groups to give compounds 3ba–3da in 76–85% yield with 88–91% ee (entries 2–4). Replacement of tert-butyl group on the silicon atom by less bulky cyclohexyl group (1e) also provides the corresponding product 3ea with relatively high ee of 86% (entry 5), but the reaction of alkoxy-substituted substrate 1f gives almost racemic product 3fa under the present reaction conditions (entry 6). Triynes having two of the same substituted phenylethynyl groups on the silicon atom (1g and 1h) are also suitable substrates for the reaction with isocyanate 2a to give 3ga and 3ha with high enantiomeric excesses (91% ee; entries 7 and 8), but the use of an alkylethynyl-substituted variant (1i) results in the formation of product 3ia with lower enantioselectivity (54% ee; entry 9). With regard to the isocyanate, not only aryl isocyanates (2b–2d) but also alkyl isocyanates (2e and 2f) possessing functional groups such as halides and esters can be efficiently employed in the present catalysis to give 3bb–3bf in uniformly high yields (82–93% yield) with 91–92% ee (entries 10–14). It is worth mentioning again that all of these reactions proceed with complete regioselectivity irrespective of the substrate combination. In addition to these N-arylated or N-alkylated products 3, N-H compound 4 can also be accessed by treatment of ethoxycarbonylethyl-substituted compound 3bf (92% ee) with NaH followed by protonation via a retro-Michael addition reaction (73% yield, 92% ee; eqn (1)).15
| | 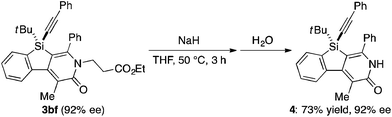 | (1) |
Table 2 Scope of rhodium-catalyzed asymmetric synthesis of silicon-bridged arylpyridinones 3
Synthesis of a silicon-stereogenic chiral polymer
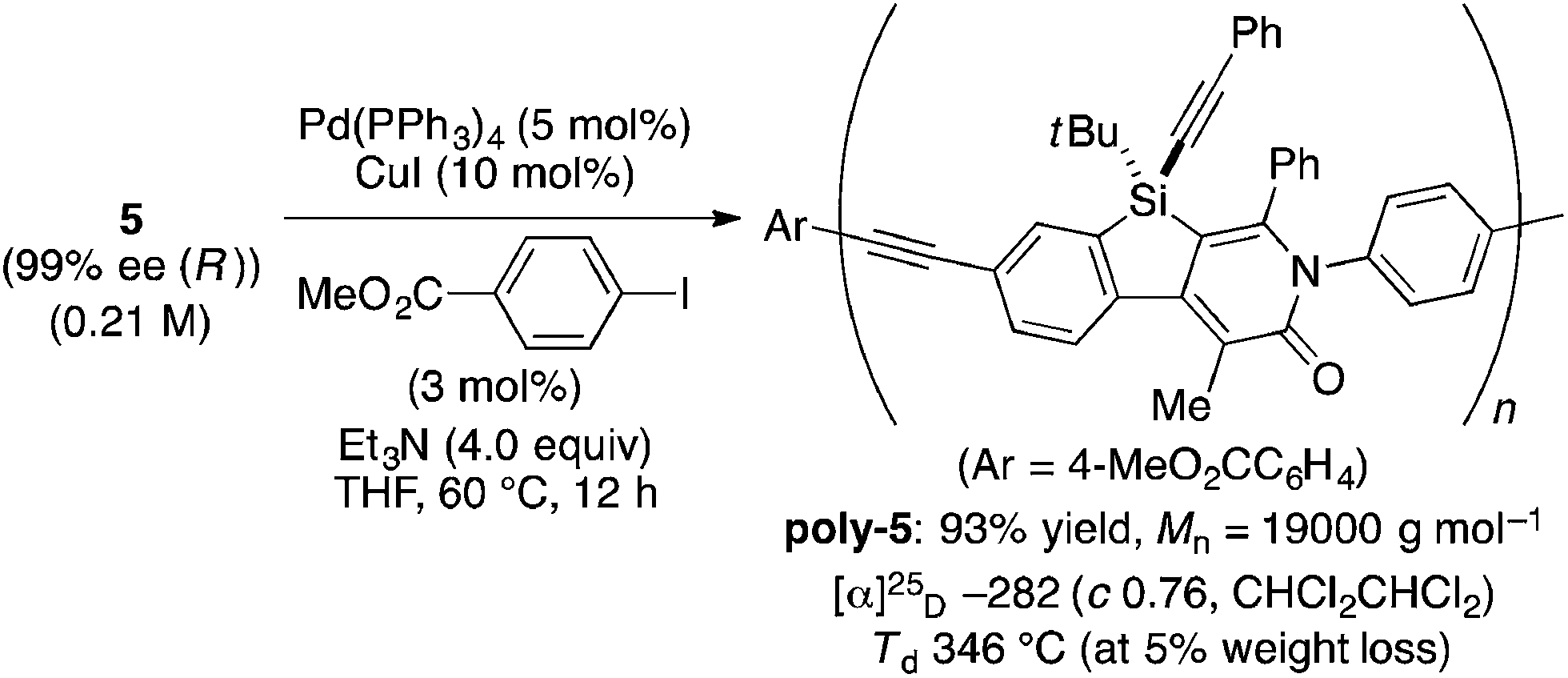
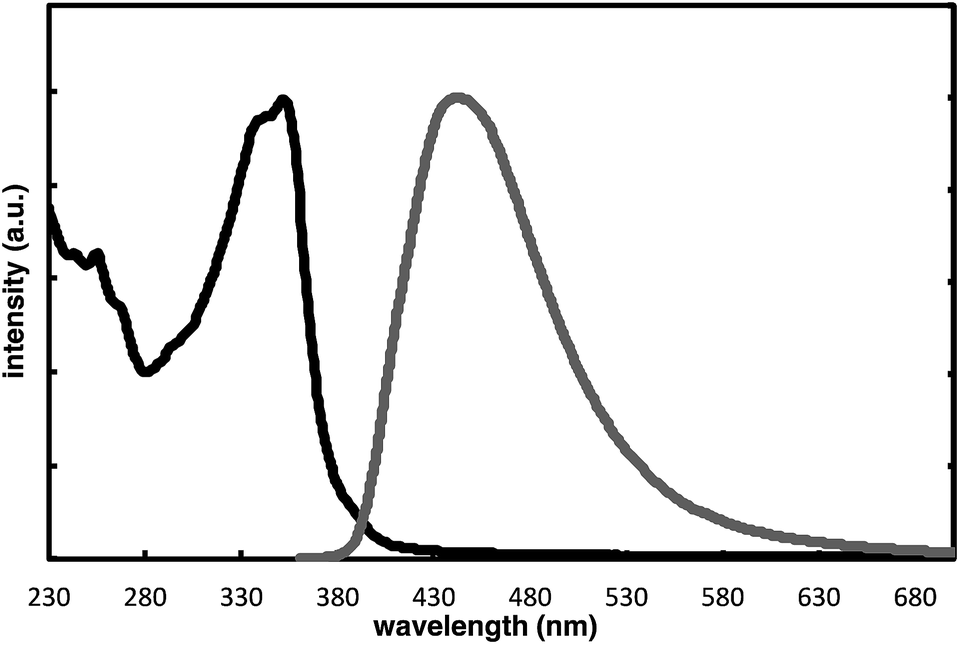
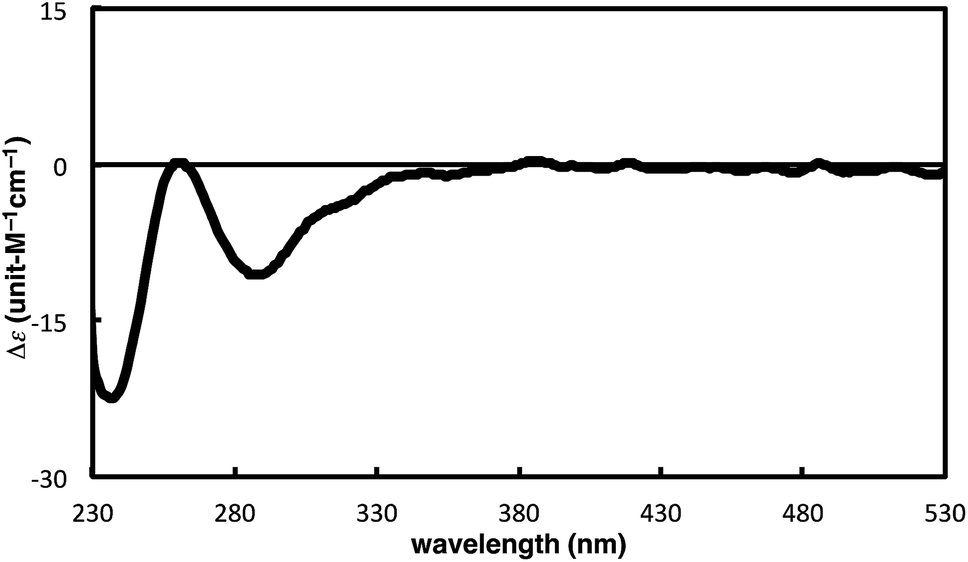
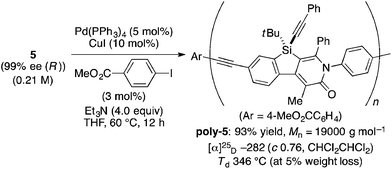
The complete regioselectivity in the present [2 + 2 + 2] cycloaddition led us to investigate the preparation of enantioenriched chiral polymers based on the silicon-stereogenic center of silicon-bridged arylpyridinones. For example, the asymmetric [2 + 2 + 2] cycloaddition reaction of trimethylsilylethynyl-substituted compound 1j with 4-iodophenyl isocyanate (2d) under Rh/(R)-L catalysis gave product 3jd in 84% yield with 89% ee (eqn (2)). Subsequent removal of the trimethylsilyl group followed by recrystallization led to bifunctional monomer 5 with 99% ee in a good overall yield. The Sonogashira coupling polymerization of 5 in the presence of 3 mol% of methyl 4-iodobenzoate as an initiator successfully afforded poly-5 in 93% yield with control of the number average molecular weight (Mn = 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, i.e., degree of polymerization = 35; eqn (3)).16 As far as we are aware, this represents the first example of the synthesis of silicon-stereogenic chiral polymers based on the catalytic asymmetric construction of the silicon stereocenter.17 The UV-vis and fluorescence spectra of poly-5 are shown in Fig. 2: the UV-vis absorption and emission band maxima are at 352 nm and 440 nm, respectively, both of which are significantly red-shifted compared to the reported silicon-stereogenic chiral conjugated polymer.17a We also examined the CD spectrum of poly-5 and found that it showed negative Cotton effects at 287 nm (Δε = −10.7 unit-M−1 cm−1) and 237 nm (Δε = −22.4 unit-M−1 cm−1) (Fig. 3).
000 g mol−1, i.e., degree of polymerization = 35; eqn (3)).16 As far as we are aware, this represents the first example of the synthesis of silicon-stereogenic chiral polymers based on the catalytic asymmetric construction of the silicon stereocenter.17 The UV-vis and fluorescence spectra of poly-5 are shown in Fig. 2: the UV-vis absorption and emission band maxima are at 352 nm and 440 nm, respectively, both of which are significantly red-shifted compared to the reported silicon-stereogenic chiral conjugated polymer.17a We also examined the CD spectrum of poly-5 and found that it showed negative Cotton effects at 287 nm (Δε = −10.7 unit-M−1 cm−1) and 237 nm (Δε = −22.4 unit-M−1 cm−1) (Fig. 3).| | 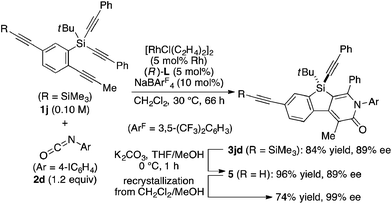 | (2) |
| |  | (3) |
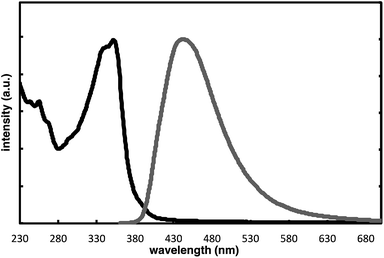 |
| | Fig. 2 Normalized UV-vis (black line; at 1.7 × 10−5 unit-M) and fluorescence spectra (gray line (ΦF = 0.04); at 1.7 × 10−5 unit-M; excited at 350 nm) of poly-5 in CH2Cl2 at 25 °C. | |
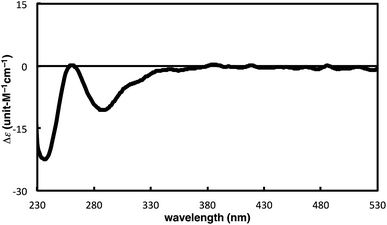 |
| | Fig. 3 CD spectrum of poly-5 (at 1.7 × 10−5 unit-M) in CH2Cl2 at 25 °C. | |
Mechanistic considerations
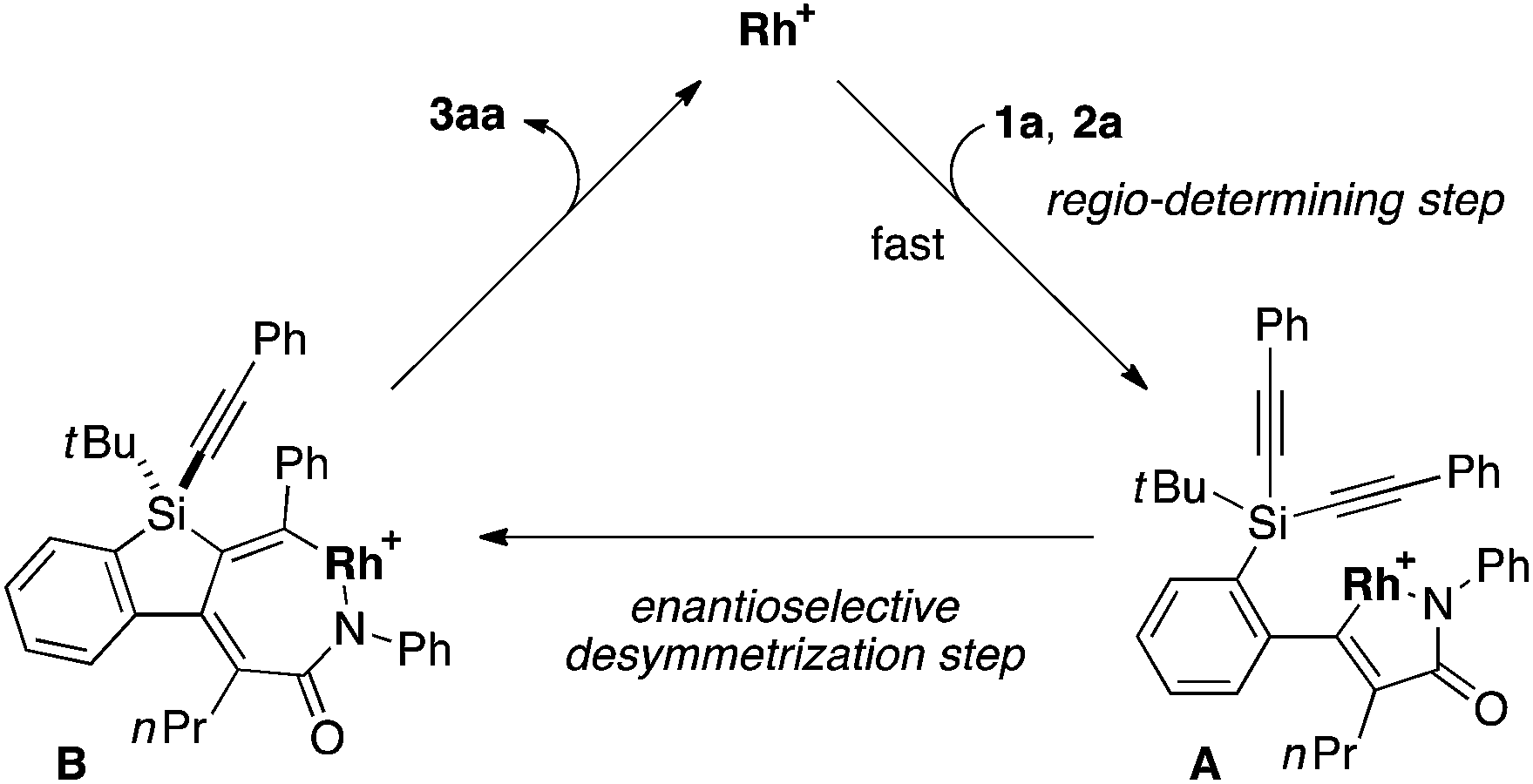
To gain some insight into the origin of regioselective formation of compounds 3 in the present catalysis, we conducted control experiments using triynes 1a and 1k, which possess opposite substitution patterns on arylalkynes (R1) and silylalkynes (R2) with each other, in the reaction of isocyanate 2a under Rh/(R)-L catalysis (eqn (4)). As was also described in Table 2, entry 1, the reaction of 1a with 2a selectively gave 3aa as the sole regioisomer in 85% yield with 89% ee. The use of 1k in place of 1a under otherwise the same conditions turned out to give product 3ka as the sole regioisomer as well, although the yield and ee became somewhat lower (52% yield, 43% ee). These results indicate that the proximal substituents of the alkynes that engage in the C–C or C–N bond-formation with an isocyanate do not influence the regioselectivity of this process. Instead, the regioselectivity is probably controlled by the reactivity difference between arylalkyne and silylalkyne of triyne 1. Based on these results, a proposed catalytic cycle for the reaction of 1a with 2a is illustrated in Scheme 1. Thus, initial oxidative cyclization of alkyne on the benzene ring of 1a and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of 2a with cationic rhodium(I) species gives five-membered rhodacycle A having a Rh–N bond to set the regiochemistry in the product formation.7d,18 Subsequent intramolecular insertion of one of the alkynes on silicon into the Rh–C bond of A takes place to give seven-membered rhodacycle B. The enantioselectivity of the silicon stereocenter is presumably determined at this insertion step. Rhodacycle B then undergoes reductive elimination to provide compound 3aa along with regeneration of cationic rhodium(I) species.
N of 2a with cationic rhodium(I) species gives five-membered rhodacycle A having a Rh–N bond to set the regiochemistry in the product formation.7d,18 Subsequent intramolecular insertion of one of the alkynes on silicon into the Rh–C bond of A takes place to give seven-membered rhodacycle B. The enantioselectivity of the silicon stereocenter is presumably determined at this insertion step. Rhodacycle B then undergoes reductive elimination to provide compound 3aa along with regeneration of cationic rhodium(I) species.| | 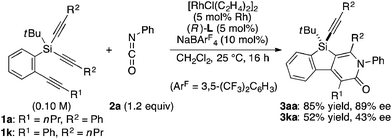 | (4) |
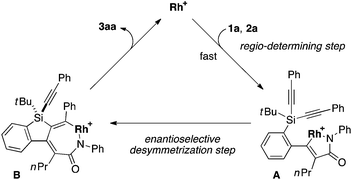 |
| | Scheme 1 Proposed catalytic cycle for the rhodium-catalyzed asymmetric [2 + 2 + 2] cycloaddition of 1a with 2a to give 3aa (Rh = Rh((R)-L)). | |
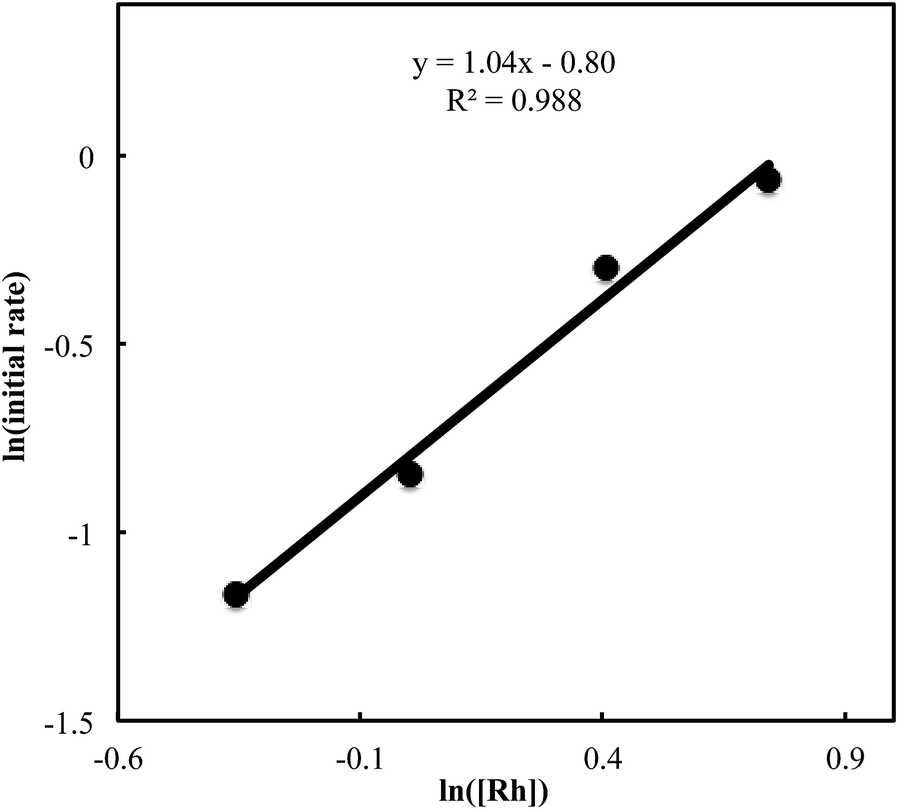
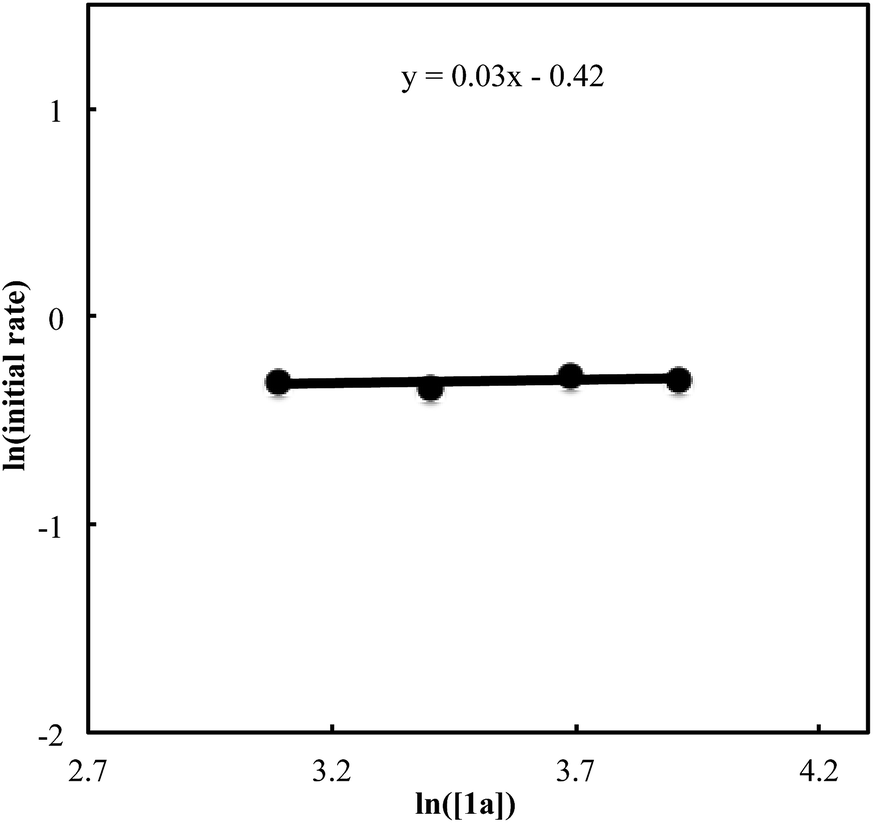
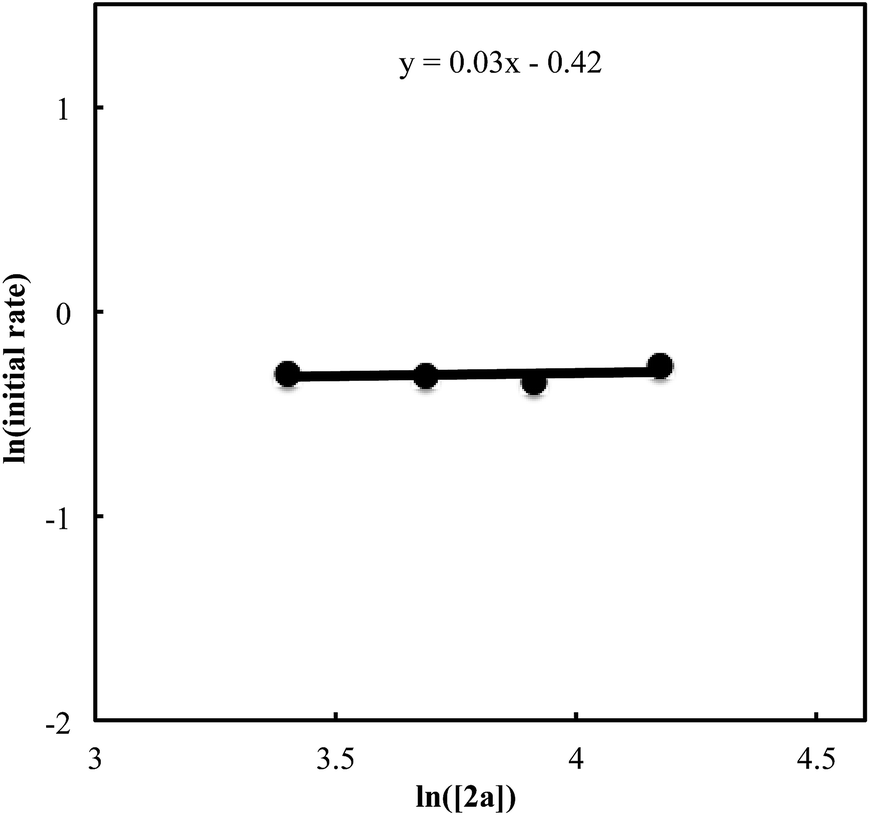
We also carried out a series of kinetic experiments to gain more detailed understanding for the reaction of triyne 1a with isocyanate 2a in the presence of [RhCl(C2H4)2]2/(R)-L/NaBArF4 as the catalyst in CH2Cl2 at 28 °C. As shown in Fig. 4, the reaction rate shows first-order dependency on the concentration of rhodium catalyst. In contrast, the initial concentrations of triyne 1a and isocyanate 2a have no influence on the initial rate of the production of 3aa (Fig. 5 and 6), indicating that the reaction is zero-order in both [1a] and [2a]. These experimental results are consistent with the proposed catalytic cycle in Scheme 1, and the oxidative cyclization step to form intermediate A takes place rapidly (zero-order in both [1a] and [2a]). The turnover-limiting step is one of the subsequent intramolecular processes, either the insertion step to form intermediate B or the reductive elimination step from B (first-order in [Rh]).
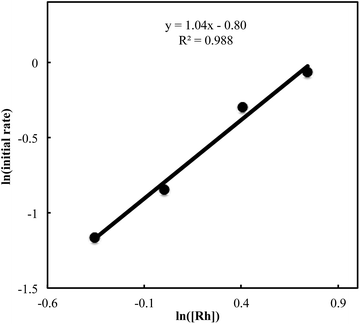 |
| | Fig. 4 Ln plot of the initial rate (mM min−1) vs. concentration of Rh catalyst (mM) ([Rh]0 = 0.7–2.1 mM, [1a]0 = 50 mM, [2a]0 = 50 mM). | |
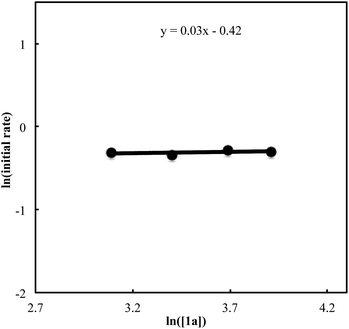 |
| | Fig. 5 Ln plot of the initial rate (mM min−1) vs. concentration of triyne 1a (mM) ([Rh]0 = 1.5 mM, [1a]0 = 22–50 mM, [2a]0 = 50 mM). | |
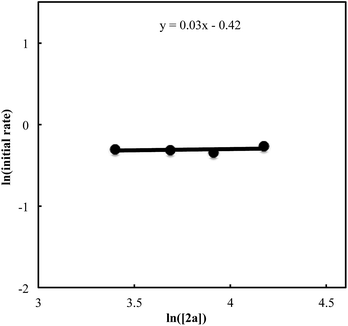 |
| | Fig. 6 Ln plot of the initial rate (mM min−1) vs. concentration of isocyanate 2a (mM) ([Rh]0 = 1.5 mM, [1a]0 = 30 mM, [2a]0 = 30–65 mM). | |
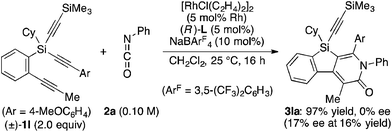
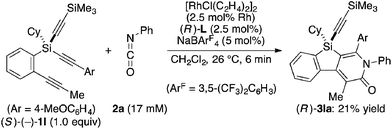
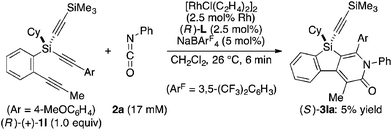
Attempted kinetic resolution of triyne (±)-1l gave further information for the mechanism of the present catalysis. As shown in eqn (5), the reaction of (±)-1l (2.0 equiv.) with phenyl isocyanate 2a in the presence of Rh/(R)-L selectively gave product 3la in 97% yield based on 2a (49% yield based on (±)-1l) by incorporating the 4-methoxyphenylethynyl group on silicon into the pyridinone framework with trimethylsilylethynyl group intact, and 3la thus obtained was found to be completely racemic. In contrast, enantiopure (S)-(−)-1l, the matched enantiomer, reacted with 2a at least 4.2 times faster than its opposite enantiomer (R)-(+)-1l under the catalysis of Rh/(R)-L (eqn (6) and (7)). These results indicate that the initial oxidative cyclization step in Scheme 1 occurs irreversibly and non-stereoselectively, and the subsequent enantio-discriminating insertion step is most likely the turnover-limiting step.
| |  | (5) |
| |  | (6) |
| |  | (7) |
Conclusions
In summary, we have developed a rhodium-catalyzed regio- and enantioselective synthesis of silicon-stereogenic silicon-bridged arylpyridinones through [2 + 2 + 2] cycloaddition of silicon-containing prochiral triynes with isocyanates. High yields and enantioselectivities have been achieved by employing axially chiral monophosphine (R)-L as the ligand, and this process could be applied to the synthesis of silicon-stereogenic chiral polymers by way of a catalytic asymmetric construction of the silicon stereocenter for the first time. We have also investigated the mechanistic aspects of the present catalysis to establish a reasonable catalytic cycle based on a series of control experiments and kinetic studies, which represents a rare example of the experimental mechanistic study for the rhodium-catalyzed synthesis of pyridinones via [2 + 2 + 2] cycloaddition reactions.
Acknowledgements
Support has been provided in part by a Grant-in-Aid for Challenging Exploratory Research, the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank Prof. Takuzo Aida at The University of Tokyo for the measurement of fluorescence and CD spectra. We thank Prof. Yoshiaki Nishibayashi and Dr Kazunari Nakajima at The University of Tokyo for the measurement of optical rotations. We thank Dr Shingo Ito for X-ray crystallographic analysis.
Notes and references
-
(a)
Comprehensive Asymmetric Catalysis I–III, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer-Verlag, New York, 1999 Search PubMed;
(b)
Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley-VCH, New York, 2nd edn, 2000 Search PubMed;
(c)
New Frontiers in Asymmetric Catalysis,ed. K. Mikami and M. Lautens, John Wiley & Sons, New Jersey, 2007 Search PubMed;
(d)
Phosphorus Ligands in Asymmetric Catalysis 1–3,ed. A. Börner, Wiley-VCH, Weinheim, 2008 Search PubMed;
(e)
Multicatalyst System in Asymmetric Catalysis, ed. J. Zhou, John Wiley & Sons, New Jersey, 2015 Search PubMed.
- For reviews on catalytic asymmetric synthesis of silicon-stereogenic organosilanes, see:
(a) L.-W. Xu, L. Li, G.-Q. Lai and J.-X. Jiang, Chem. Soc. Rev., 2011, 40, 1777 RSC;
(b) L.-W. Xu, Angew. Chem., Int. Ed., 2012, 51, 12932 CrossRef CAS PubMed;
(c) R. Shintani, Asian J. Org. Chem., 2015, 4, 510 CrossRef CAS.
- Stoichiometric synthesis of silicon-stereogenic compounds has been somewhat more investigated. For pioneering work, see:
(a)
L. H. Sommer,Stereochemistry, Mechanism and Silicon; an Introduction to the Dynamic Stereochemistry and Reaction Mechanisms of Silicon Centers, McGraw-Hill, New York, 1965 Search PubMed For selected recent examples, see:
(b) C. Strohmann, J. Hörnig and D. Auer, Chem. Commun., 2002, 766 RSC;
(c) M. Trzoss, J. Shao and S. Bienz, Tetrahedron: Asymmetry, 2004, 15, 1501 CrossRef CAS;
(d) S. Rendler, G. Auer and M. Oestreich, Angew. Chem., Int. Ed., 2005, 44, 7620 CrossRef CAS PubMed;
(e) S. Rendler, G. Auer, M. Keller and M. Oestreich, Adv. Synth. Catal., 2006, 348, 1171 CrossRef CAS;
(f) S. Rendler, M. Oestreich, C. P. Butts and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2007, 129, 502 CrossRef CAS PubMed;
(g) K. Igawa, N. Kokan and K. Tomooka, Angew. Chem., Int. Ed., 2010, 49, 728 CrossRef CAS PubMed;
(h) J. O. Bauer and C. Strohmann, Angew. Chem., Int. Ed., 2014, 53, 720 CrossRef CAS PubMed.
-
(a) R. J. P. Corriu and J. J. E. Moreau, Tetrahedron Lett., 1973, 14, 4469 CrossRef;
(b) T. Hayashi, K. Yamamoto and M. Kumada, Tetrahedron Lett., 1974, 15, 331 CrossRef;
(c) R. J. P. Corriu and J. J. E. Moreau, J. Organomet. Chem., 1975, 85, 19 CrossRef CAS;
(d) R. J. P. Corriu and J. J. E. Moreau, J. Organomet. Chem., 1976, 120, 337 CrossRef CAS;
(e) T. Ohta, M. Ito, A. Tsuneto and H. Takaya, J. Chem. Soc., Chem. Commun., 1994, 2525 RSC;
(f) Y. Yasutomi, H. Suematsu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 4510 CrossRef CAS PubMed;
(g) Y. Kurihara, M. Nishikawa, Y. Yamanoi and H. Nishihara, Chem. Commun., 2012, 48, 11564 RSC;
(h) K. Igawa, D. Yoshihiro, N. Ichikawa, N. Kokan and K. Tomooka, Angew. Chem., Int. Ed., 2012, 51, 12745 CrossRef CAS PubMed See also:
(i) K. Tamao, K. Nakamura, H. Ishii, S. Yamaguchi and M. Shiro, J. Am. Chem. Soc., 1996, 118, 12469 CrossRef CAS;
(j) D. R. Schmidt, S. J. O'Malley and J. L. Leighton, J. Am. Chem. Soc., 2003, 125, 1190 CrossRef CAS PubMed;
(k) Y. Kuninobu, K. Yamauchi, N. Tamura, T. Seiki and K. Takai, Angew. Chem., Int. Ed., 2013, 52, 1520 CrossRef CAS PubMed.
-
(a) K. Igawa, J. Takada, T. Shimono and K. Tomooka, J. Am.
Chem. Soc., 2008, 130, 16132 CAS;
(b) R. Shintani, K. Moriya and T. Hayashi, J. Am. Chem. Soc., 2011, 133, 16440 CrossRef CAS PubMed;
(c) R. Shintani, K. Moriya and T. Hayashi, Org. Lett., 2012, 14, 2902 CrossRef CAS PubMed;
(d) R. Shintani, H. Otomo, K. Ota and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 7305 CrossRef CAS PubMed;
(e) R. Shintani, E. E. Maciver, F. Tamakuni and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 16955 CrossRef CAS PubMed;
(f) M. Onoe, K. Baba, Y. Kim, Y. Kita, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2012, 134, 19477 CrossRef CAS PubMed;
(g) X. Lu, L. Li, W. Yang, K. Jiang, K.-F. Yang, Z.-J. Zheng and L.-W. Xu, Eur. J. Org. Chem., 2013, 5814 CrossRef CAS;
(h) R. Shintani, C. Takagi, T. Ito, M. Naito and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 1616 CrossRef CAS PubMed;
(i) Y. Naganawa, T. Namba, M. Kawagishi and H. Nishiyama, Chem.–Eur. J., 2015, 21, 9319 CrossRef CAS PubMed;
(j) R. Shintani, H. Kurata and K. Nozaki, Chem. Commun., 2015, 51, 11378 RSC.
- For recent reviews on the rhodium-catalyzed [2 + 2 + 2] cycloaddition reactions, see:
(a) K. Tanaka, Chem.–Asian J., 2009, 4, 508 CrossRef CAS PubMed;
(b) N. Weding and M. Hapke, Chem. Soc. Rev., 2011, 40, 4525 RSC;
(c) Y. Shibata and K. Tanaka, Synthesis, 2012, 44, 323 CrossRef CAS;
(d) D. L. J. Broere and E. Ruijter, Synthesis, 2012, 44, 2639 CrossRef CAS;
(e) K. Tanaka, Y. Kimura and K. Murayama, Bull. Chem. Soc. Jpn., 2015, 88, 375 CrossRef CAS.
- For examples of pyridinone synthesis by rhodium-catalyzed [2 + 2 + 2] cycloaddition reactions:
(a) S. T. Flynn, S. E. Hasso-Henderson and A. W. Parkins, J. Mol. Catal., 1985, 32, 101 CrossRef CAS;
(b) K. Tanaka, A. Wada and K. Noguchi, Org. Lett., 2005, 7, 4737 CrossRef CAS PubMed;
(c) G. Nishida, N. Suzuki, K. Noguchi and K. Tanaka, Org. Lett., 2006, 8, 3489 CrossRef CAS PubMed;
(d) T. Kondo, M. Nomura, Y. Ura, K. Wada and T. Mitsudo, Tetrahedron Lett., 2006, 47, 7107 CrossRef CAS;
(e) K. Tanaka, Y. Takahashi, T. Suda and M. Hirano, Synlett, 2008, 1724 CrossRef CAS;
(f) K. M. Oberg, E. E. Lee and T. Rovis, Tetrahedron, 2009, 65, 5056 CrossRef CAS PubMed;
(g) Y. Komine and K. Tanaka, Org. Lett., 2010, 12, 1312 CrossRef CAS PubMed;
(h) M. Augé, M. Barbazanges, A. T. Tran, A. Simonneau, P. Elley, H. Amouri, C. Aubert, L. Fensterbank, V. Gandon, M. Malacria, J. Moussa and C. Ollivier, Chem. Commun., 2013, 49, 7833 RSC;
(i) M. Augé, A. Feraldi-Xypolia, M. Barbazanges, C. Aubert, L. Fensterbank, V. Gandon, E. Kolodziej and C. Ollivier, Org. Lett., 2015, 17, 3754 CrossRef PubMed For a review:
(j) K. Tanaka, Heterocycles, 2012, 85, 1017 CrossRef CAS.
- Other transition-metal catalysts have also been used for pyridinone synthesis by [2 + 2 + 2] cycloaddition. Cobalt:
(a) P. Hona and H. Yamazaki, Tetrahedron Lett., 1977, 1333 Search PubMed;
(b) R. A. Earl and K. P. C. Vollhardt, J. Am. Chem. Soc., 1983, 105, 6991 CrossRef CAS;
(c) R. A. Earl and K. P. C. Vollhardt, J. Org. Chem., 1984, 49, 4786 CrossRef CAS;
(d) P. Diversi, G. Ingrosso, A. Lucherini and S. Malquori, J. Mol. Catal., 1987, 40, 267 CrossRef CAS;
(e) L. V. R. Boñaga, H.-C. Zhang, D. A. Gauthier, I. Reddy and B. E. Maryanoff, Org. Lett., 2003, 5, 4537 CrossRef PubMed;
(f) L. V. R. Boñaga, H.-C. Zhang, A. F. Moretto, H. Ye, D. A. Gauthier, J. Li, G. C. Leo and B. E. Maryanoff, J. Am. Chem. Soc., 2005, 127, 3473 CrossRef PubMed;
(g) D. D. Young and A. Deiters, Angew. Chem., Int. Ed., 2007, 46, 5187 CrossRef CAS PubMed;
(h) D. D. Young, J. A. Teske and A. Deiters, Synthesis, 2009, 3785 CAS;
(i) L. L. Lv, X. F. Wang, Y. C. Zhu, X. W. Liu, X. Q. Huang and Y. C. Wang, Organometallics, 2013, 32, 3837 CrossRef CAS Nickel:
(j) H. Hoberg and B. W. Oster, Synthesis, 1982, 324 CrossRef CAS;
(k) H. Hoberg and B. W. Oster, J. Organomet. Chem., 1983, 252, 359 CrossRef CAS;
(l) H. A. Duong, M. J. Cross and J. Louie, J. Am. Chem. Soc., 2004, 126, 11438 CrossRef CAS PubMed;
(m) H. A. Duong and J. Louie, J. Organomet. Chem., 2005, 690, 5098 CrossRef CAS Ruthenium:
(n) Y. Yamamoto, H. Takagishi and K. Itoh, Org. Lett., 2001, 3, 2117 CrossRef CAS PubMed;
(o) S. Alvarez, S. Medina, G. Domínguez and J. Pérez-Castells, J. Org. Chem., 2013, 78, 9995 CrossRef CAS PubMed Iridium:
(p) G. Onodera, M. Suto and R. Takeuchi, J. Org. Chem., 2012, 77, 908 CrossRef CAS PubMed.
- H. Takaya, K. Mashima, K. Koyano, M. Yagi, H. Kumobayashi, T. Taketomi, S. Akutagawa and R. Noyori, J. Org. Chem., 1986, 51, 629 CrossRef CAS.
- X. Zhang, K. Mashima, K. Koyano, N. Sayo, H. Kumobayashi, S. Akutagawa and H. Takaya, Tetrahedron Lett., 1991, 32, 7283 CrossRef CAS.
- T. Saito, T. Yokozawa, T. Ishizaki, T. Moroi, N. Sayo, T. Miura and H. Kumobayashi, Adv. Synth. Catal., 2001, 343, 264 CrossRef CAS.
-
(a) T. Hayashi, Acc. Chem. Res., 2000, 33, 354 CrossRef CAS PubMed;
(b) Y. Uozumi, A. Tanahashi, S.-Y. Lee and T. Hayashi, J. Org. Chem., 1993, 58, 1945 CrossRef CAS.
- B. Saha and T. V. RajanBabu, J. Org. Chem., 2007, 72, 2357 CrossRef CAS PubMed.
- CCDC-1428796 contains the supplementary crystallographic data for this paper.
-
(a) R. Shintani, T. Ito, M. Nagamoto, H. Otomo and T. Hayashi, Chem. Commun., 2012, 48, 9936 RSC See also:
(b) T. M. Ha, B. Yao, Q. Wang and J. Zhu, Org. Lett., 2015, 17, 1750 CrossRef CAS PubMed.
- Polymerization of 5 in the absence of initiator also proceeded smoothly to give highly insoluble polymers, presumably due to the formation of polymers with much higher molecular weight. In order to enable solution-phase analyses of the polymers, we conducted polymerization using 3 mol% of methyl 4-iodobenzoate to obtain polymers with a controlled number average molecular weight.
- Optically active silicon-stereogenic chiral polymers containing silicon atoms in the main chain have been reported through the chiral HPLC separation of racemic monomers:
(a) Y. Kawakami, M. Omote, I. Imae and E. Shirakawa, Macromolecules, 2003, 36, 7461 CrossRef CAS;
(b) D. Zhou and Y. Kawakami, Macromolecules, 2005, 38, 6902 CrossRef CAS Optically active silicon-stereogenic chiral polymers containing silicon atoms in the side chain have been reported through the fractional recrystallization of diastereomeric monomers:
(c) B. Z. Tang, X. Wan and H. S. Kwok, Eur. Polym. J., 1998, 34, 341 CrossRef CAS.
- Related azametallacycle intermediates have also been proposed for other transition–metal catalyst systems. See ref. 8d, k, and m.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures. CCDC 1428796. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc03767k |
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence