
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGold nanoparticle superlattices: structure and cavities studied by GISAXS and PALS†
Natalia
Olichwer
a,
Tönjes
Koschine‡
b,
Andreas
Meyer
a,
Werner
Egger
c,
Klaus
Rätzke
*b and
Tobias
Vossmeyer
*a
aInstitute of Physical Chemistry, University of Hamburg, Grindelallee 117, 20146 Hamburg, Germany. E-mail: tobias.vossmeyer@chemie.uni-hamburg.de
bInstitute for Materials Science, Chair for Multicomponent Materials, Faculty of Engineering, Christian-Albrechts-University of Kiel, Kaiserstraße 2, D-24143 Kiel, Germany. E-mail: kr@tf.uni-kiel.de
cInstitut für Angewandte Physik und Messtechnik, Universität der Bundeswehr München, Werner-Heisenberg-Weg 39, 85579 Neubiberg, Germany
First published on 3rd November 2016
Abstract
The performance of nanoparticle assemblies with respect to various applications (e.g. sensors, catalysts, filtration membranes) depends on their microporosity. Here, the microcavities within the ligand matrix of superlattice films comprised of 1-dodecanethiol-stabilized gold nanoparticles (GNPs, core diameter ∼4 and ∼5.5 nm) were studied by positron annihilation lifetime spectroscopy (PALS). The superlattice composition, the size and spatial arrangement of the GNP cores were characterized by thermogravimetric analysis, transmission electron microscopy and grazing-incidence small-angle X-ray-scattering. From these data a structural model was derived to predict the sizes of the voids formed within the interstitial (tetrahedral and octahedral) sites of the superlattices. The comparison of the PALS-measured cavity sizes (0.50 to 0.74 nm) with the predicted void sizes of the interstitial sites (∼0.7 to ∼1.7 nm) and the free volume in solid dodecane (0.36 nm), previously measured by PALS, indicate that both types of cavities may contribute to the experimentally determined cavity sizes. However, the GNP core sizes had only a minor influence on the measured cavity size. Larger cavities with sizes corresponding to the voids (∼1.7 nm) expected within the octahedral sites of the superlattices comprised of ∼5.5 nm-sized GNP cores could not be detected. Assuming the intensities arising from these voids are measurable, this finding suggests that the octahedral sites are occupied by excess ligands trapped during film preparation. Apart from the voids predicted for the interstitial sites, the larger cavity sizes measured for the GNP superlattices compared to crystalline dodecane may result from some degree of disorder in the ligand arrangement.
Introduction
Assemblies of ligand-stabilized noble metal nanoparticles are of great interest because of their unique optical,1 electrical2 and mechanical3,4 properties, which all can be tuned by varying the materials' composition and structural parameters. This provides the opportunity to tailor materials with desired properties for various specific applications like resistive5–7 or optical8,9 strain and vapor sensing, catalysis,10 nanoelectromechanical systems,11,12 and surface enhanced Raman scattering.1 During the past two decades the structure–property relationships for a variety of such assemblies have been studied extensively. Besides the chemical nature and the length of the organic ligands as well as the shape and size of the particle cores, the spatial arrangement of the components within the assemblies dictates their physical properties. Thus, interest in colloidal crystals self-assembled as periodical structures13 has grown significantly. These so called superlattices exist in the form of films or faceted polyhedra13,14 revealing fcc, hcp or bcc packing.15–18 The challenge of controlling their assembly process has been addressed in multiple studies.14,16,17,19–22 For the resulting structure of the superlattice and its physical properties the ligand arrangement plays a key role. The ligand–ligand interactions determine the degree of superlattice order, the spacing between particle cores, and the lattice type.16,17,19,21–24 Accordingly, numerous theoretical and experimental studies have focused on the self-assembled ligand monolayers encapsulating the nanoparticles and their arrangement within 2D and 3D assemblies.24–34 It was shown that the ligands can be arranged in a rather disordered liquid-like state or they can form rather tightly packed bundles with crystalline-like order. Further, the ligand distribution around the cores can be uniform or regions of different ligand density, including cavities can be formed.28,29,32 Hence, in nanoparticle assemblies different ligand arrangements come along with different cavities within the ligand shells. In addition, the interstitial (tetrahedral and octahedral) sites of 3D superlattices may provide sufficient space for the formation of what we will term unoccupied voids and, thus, can contribute to the porosity of the structure.In general, detailed knowledge about the porosity of a nanoparticle superlattice is mandatory for enabling a comprehensive understanding of its structure and physical properties, which is highly relevant for several applications. For example, the elastic properties of superlattices of alkanethiol-stabilized gold nanoparticles (GNPs) were predicted to depend on the ability of the ligands to be pushed into the available free space.23 Further, cavities are able to trap or selectively let pass molecules and are thus relevant for the application of nanoparticle superlattices as nanofiltration membranes35 or as chemical sensors. Regarding chemi-resistive sensing cavities are generally presumed to play a major role in the sensing mechanism.7,36 Indeed, several studies indicated their influence on the sensing properties.37–40 For example, assemblies of thiol-stabilized cubic platinum nanoparticles, revealing a more compact structure than their spherical counterparts, showed considerably higher response amplitudes.38 Further, the perforation of assemblies of thiol-stabilized GNPs has been shown to markedly alter the chemiresistive response.39 For chemiresistive sensing in liquid phase using an assembly of thiol-stabilized GNPs it has been shown that reducing the number of cavities with diameters between 0.3 to 3 nm significantly prolongs the response time and at the same time increases the response amplitudes.40 The cavity sizes in that study were estimated using electrochemical capacitance measurements. A more widely used technique to study cavity sizes of nanoporous materials, such as zeolites, are gas adsorption (BET-) measurements. However, in general this method requires significant amounts of the sample and is, thus, not practical for characterizing cavity sizes in the present thin nanoparticle-based superlattice films.
A powerful method for studying free volume in polymers and sub-nanoscale voids in thin film materials is positron annihilation lifetime spectroscopy (PALS), a well-established technique in materials science. For metals and semiconductors vacancy concentrations and defect formation enthalpies can be detected.41,42 For amorphous polymers free volume, i.e. the unoccupied space between the chains can be characterized.43,44 Examples reported by some of the present authors include storage stability predictions in epoxide mixtures,45 characterization of nanocomposites for membrane applications,46,47 and aging of thin polymer films.48,49 Recently, Chai et al. used PALS to study thin films of oleic acid-stabilized PbSe nanocrystals with respect to surface composition.50
PALS probes the electron density distribution in materials, in particular for amorphous polymers the free volume between the atoms and chains, and vacancies in crystals. In insulating materials the ortho-positronium (o-Ps) is formed after the injection of positrons and its lifetime can be correlated to the average free volume size via a well-established model.43,44 The free volume diameters in polymers are usually in the range of 0.5 nm or less. The aim of the present study was to show in a set of proof-of-principle experiments the suitability of PALS for the detection of cavities in thin film superlattices self-assembled from 1-dodecanethiol (DDT)-stabilized GNPs. Although PALS is generally suitable for measuring cavities on various length scales, the thin film analysis required a moderated, low energy positron beam and the corresponding pulsing of the present system51,52 restricted us to measure lifetimes on the order of 10 ns and, thus, cavities smaller than a few nm. A combination of thermogravimetric analysis (TGA), transmission electron microscopy (TEM) and grazing-incidence small-angle X-ray-scattering (GISAXS) revealed the material's composition, the size and arrangement of the GNP cores and allowed us to compare the PALS-measured cavity sizes with void sizes predicted by the structural model of the nanoparticle superlattices.
Experimental
Materials
Chloro(triphenylphosphine)gold(I) (98%) was purchased from ABCR. All other chemicals were purchased from Sigma-Aldrich. They were all of analytical grade and used as received.Synthesis of gold nanoparticles (GNPs)
1-Dodecanethiol (DDT)-stabilized GNPs were prepared following the Stucky-method53 modified by Pileni and coworkers.14 A solution of tert-butylamine borane complex in toluene and a solution of 0.25 mmol chloro(triphenylphosphine)gold(I) and 1-dodecanethiol in 25 mL toluene were mixed at 100 °C and stirred for 5 min. For samples GNP4 nm1, GNP4 nm2 and GNP4 nm3 (with core diameters of ∼4 nm) 5 mmol tert-butylamine borane complex in 2 mL toluene and 500 μL 1-dodecanethiol, and for samples GNP5.5 nm1, GNP5.5 nm2 and GNP5.5 nm3 (with core diameters of ∼5.5 nm) 2.5 mmol tert-butylamine borane complex in 15 mL toluene and 125 μL 1-dodecanethiol were used. After the red GNP solution had cooled down to room temperature, 20 mL ethanol were added and the samples were centrifuged at 5095 × g and −10 °C. This precipitation step was repeated once or twice using 5 to 10 mL ethanol. The precipitate was then dried under nitrogen and dispersed in ∼2 mL toluene (particle concentration ∼0.01 to 0.1 mM). In order to test the GNP samples for residual nitrogen compounds, which are known to quench positronium43,44 generated during the PALS experiments, the chemical composition was characterized by elementary analysis and X-ray photoelectron spectroscopy (XPS). For details see the ESI (Fig. S1†).Deposition of GNP superlattice films
For all measurements (PALS, SEM, GISAXS, XPS) the samples were prepared the same way. The GNP solution was dropped onto a silicon wafer and dried at ambient conditions. PALS, GISAXS, SEM and XPS measurements were carried out within 1–3 days after depositing the nanoparticle films.Transmission electron microscopy (TEM)
For TEM imaging 10 μL of a GNP solution (∼1 μM) diluted in a solution of 1-dodecanethiol in toluene (∼1 mM) were transferred to a carbon coated copper grid. After solvent evaporation the GNP sizes were measured using a Philips CM 300 microscope, equipped with an LaB6 cathode and operated at 200 kV. For size determination 300–1000 particles were evaluated using the software ImageJ.Scanning electron microscopy (SEM)
For imaging GNP films deposited onto silicon substrates a LEO-1550 (Carl Zeiss) field-emission scanning electron microscope (FE-SEM) was used. In order to measure the cross sections of the films, the GNP-coated wafers were cleaved before the measurements. Representative SEM images can be found in the ESI.†Grazing-incidence small-angle X-ray-scattering (GISAXS)
For GISAXS measurements our custom built SAXS apparatus equipped with an Incoatec™ X-ray source IμS and Quazar Montel optics was employed. The GNP coated silicon substrates were mounted onto a high-resolution motorized goniometer. The X-ray beam had a wavelength of 0.154 nm and a focal spot size at the sample position of 0.6 mm2. The incident angle was adjusted to 0.3°. For acquiring the scattering patterns a Rayonix™ SX165 CCD-detector was used. The distance between the sample and the detector was 1.6 m. The accumulation time per GISAXS measurement was 600 s. All GISAXS measurements were performed at ambient conditions.Thermogravimetric analysis (TGA)
Aluminum oxide cups (∼70 μL) were filled with the GNP solutions. The dry mass after solvent evaporation at ambient conditions over night was ∼3 mg. TGA measurements of the GNP assemblies were carried out using a Mettler Toledo TGA 1 in the temperature range between 25 °C to 800 °C and under nitrogen. The heating rate was 10 °C min−1 and the nitrogen flow was 20 mL min−1.X-ray photoelectron spectroscopy (XPS)
XPS measurements were done with an Omicron Full Lab XPS system with a double Al/Mg anode (VG Microtech XR3E2) and a hemispherical analyzer (VSW Instruments EA 125). Survey spectra were taken with 100 eV pass energy, region spectra with 30 eV pass energy. The Al anode was operated at a power of 240 W (15 kV, 16 mA). XPS spectra are provided in the ESI (Fig. S1†).Positron annihilation lifetime spectroscopy (PALS)
Although PALS is suited for various materials, for thin films a positron source with moderated, low energy positrons has to be used in order to analyze the thin film only, and not the underlying substrate. Here, the measurements were done using the pulsed low energy positron system (PLEPS) of the neutron-induced positron sources at FRM2 in Garching, Germany.52 All samples were measured between 2 and 4 keV positron implantation energy to ensure that all positrons annihilate in the sample layer and not in the substrate. The spectra contained at least 3.5 × 106 counts. All measurements were done at room temperature (∼30 °C) and in high vacuum (10−6 mbar). Counting rates of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cts per s were achieved at a time resolution of ∼260 ps and a peak to background ratio of at least 50000 to 1. The pulsing frequency of the beam was set to 25 MHz to resolve lifetimes up to 10 ns corresponding to cavity sizes up to some nm reliably. For data evaluation the program POSWIN, which is a MATLAB integration of the POSFIT routine was used.54 Two approaches for fitting were performed. First, for each implantation energy free fits for lifetimes, intensities and background were used and, second, an average for the background was fixed and the corresponding results for the ortho-positronium (o-Ps) lifetimes were obtained with higher reliability.
000 cts per s were achieved at a time resolution of ∼260 ps and a peak to background ratio of at least 50000 to 1. The pulsing frequency of the beam was set to 25 MHz to resolve lifetimes up to 10 ns corresponding to cavity sizes up to some nm reliably. For data evaluation the program POSWIN, which is a MATLAB integration of the POSFIT routine was used.54 Two approaches for fitting were performed. First, for each implantation energy free fits for lifetimes, intensities and background were used and, second, an average for the background was fixed and the corresponding results for the ortho-positronium (o-Ps) lifetimes were obtained with higher reliability.
Results and discussion
General structural characterization of gold nanoparticle superlattices
The objective of this study was to accomplish a detailed structural characterization of superlattice films assembled from DDT-stabilized GNPs. The main focus was placed on determining the average size of free volumes and voids by PALS. Free volumes can be expected to form within the organic ligand shells of the GNPs, whereas voids are formed within interstitial (tetrahedral and octahedral) sites. Because a meaningful interpretation of these measurements requires profound knowledge about various structural features of the superlattice, we first determined the particle core sizes by TEM and the morphology of as-deposited GNP superlattice films by SEM. Furthermore, the superlattice structure and the interparticle distances were investigated by GISAXS.Representative TEM images of the DDT-stabilized GNPs used in this study are shown in Fig. 1a and e. To discern a possible influence of the GNP core sizes and, thus, the lattice cell parameters on the PALS-measured cavity sizes, we used GNP batches with two different core sizes, i.e. with mean diameters of ∼4 nm (GNP4 nm1, GNP4 nm2 and GNP4 nm3) and ∼5.5 nm (GNP5.5 nm1, GNP5.5 nm2 and GNP5.5 nm3). Detailed analyses of TEM images revealed size standard deviations below 10%. The average core diameters and standard size deviations are listed in Table 1 for all samples used in this study.
 | ||
| Fig. 1 (a) and (e) TEM images, (b), (c), (f) and (g) SEM images at two different magnifications and (d) and (h) the corresponding Fourier transforms. Figure parts (a)–(d) correspond to a film prepared from sample GNP4 nm2 and (e)–(h) to a film prepared from sample GNP5.5 nm2. The scale bars are 20 nm in (a), (e), (c) and (g) and 10 μm in (b) and (f). | ||
| GNP4 nm1 | GNP4 nm2 | GNP4 nm3 | GNP5.5 nm1 | GNP5.5 nm2 | GNP5.5 nm3 | |
|---|---|---|---|---|---|---|
| a The two values correspond to two different measurements. | ||||||
| D (nm) | 3.8 ± 0.3 | 3.9 ± 0.2 | 4.1 ± 0.2 | 5.3 ± 0.2 | 5.6 ± 0.2 | 5.4 ± 0.2 |
| δ (nm) | 1.9 | 1.9 | 1.8 | 2.0 | 2.2 | 2.2 |
| n Au (%) | 86.5 | 86.9 | 88.3 | 90.0 | 89.6 | 89.2 |
| n Au,TGA (%) | 86.1 | 86.2 | 89.8/89.8a | 83.6/86.7a | 82.7 | |
The GNP superlattice films were prepared by drop casting a GNP dispersion onto silicon substrates. Representative SEM images of the GNP films are shown in Fig. 1b, c, f and g. Further SEM images of a GNP film are provided in the ESI (Fig. S2†). In case of the ∼4 nm-sized particles a film morphology with only few cracks was obtained after evaporation of the solvent (Fig. 1b). Particles with diameters >5 nm tended to homogeneous nucleation in solution,14i.e. they spontaneously formed polyhedral supercrystals and precipitated. At the air–toluene interface, however, a film was observed. As described earlier by Pileni and coworkers55 this interfacial film is formed via a layer-by-layer growth mechanism (heterogeneous growth). Here, the particles stack perpendicular to the interface with the (111)-planes of the supercrystal oriented parallel to the interface. For film preparation, a solution aliquot close to the air–toluene interface was taken and deposited onto the silicon substrate. Depending on the amount of precipitated supercrystals and interfacial film in this aliquot, a plate-like film morphology with pronounced cracks resulting from shrinkage upon solvent evaporation was observed (Fig. 1f), or the film showed a more structured surface morphology with edges and clefts defined by the facets of fused superlattice polyhedra (ESI, Fig. S2a†). The thicknesses of the six films prepared for PALS measurements were between ∼200 and ∼900 nm (ESI, Fig. S3†). Considering a heterogeneous growth starting at the surface of the silicon substrate14 or at the toluene/air interface,55 the film surfaces observed in the SEM images (Fig. 1c, g and S2b in the ESI†) were assigned to the (111)-facets of fcc superlattices. The relatively sharp spots in the Fourier transformations of the SEM images at high magnifications (Fig. 1d, h and S2c in the ESI†) substantiate the ordered structure of these nanoparticle assemblies.
In order to characterize the GNP superlattice structures and the interparticle distances the film samples were subjected to GISAXS measurements. The GISAXS patterns (Fig. 2a) were analyzed in two different manners. Firstly, the GISAXS patterns shown in Fig. 2b were simulated using IsGISAXS.56 Here, a monolayer of hexagonally packed spheres on a substrate was assumed. As shown exemplarily for samples GNP4 nm1 and GNP5.5 nm2 by the comparison of Fig. 2a and b, the simulations and the measurements resulted in almost identical GISAXS patterns. Secondly, in addition to the reflections along the qy-direction originating from scattering by the lateral arrangement of the GNP cores, in the measured patterns (Fig. 2a) a ring-shaped signature with its center at qz = 0 and qy = 0 is observed. This ring corresponds to the conventional SAXS signature resulting from scattering by the 3D arrangement of GNPs. The appearance of the ring-shaped patterns indicates that the superlattice films were composed of crystalline domains with different orientations, however, the brighter spots within the rings indicate some preferential orientation (texture effect). Similar as described previously,36 this ring-shaped signature was analyzed by performing a line cut from center to edge of the detector along regions without the GISAXS reflections and integrating along the ring signature over a defined section (as depicted schematically in Fig. 2a left). This way, we could separate the SAXS signal and analyze the resulting graph like a conventional SAXS curve (shown in Fig. 2c). The SAXS analysis was performed using the software scatter57,58 and allowed us to determine the lattice type. As was evident from the comparison of the measured SAXS curves with the calculated scattering curves of fcc and bcc lattices (Fig. 2c), all investigated samples showed a highly ordered fcc arrangement. The standard deviation of the lattice constant was between 2 to 5% for all samples. The SAXS/GISAXS analyses confirmed that the gold cores were very uniform in shape and size. It is to note, that the center-to-center nearest neighbor distances extracted from both above mentioned methods, i.e. simulating the GISAXS pattern and analyzing the SAXS curve, were identical. However, while GISAXS is a precise method to determine lateral distances, it was not used to extract the particle sizes because the intensity ratio of the (111)- and (200)-reflection, from which in principle the particle diameters can be derived, is affected by texture effects, as outlined earlier.36 Hence, for determining the edge-to-edge distance δ between adjacent gold cores (see Table 1) the TEM-measured GNP core diameters were subtracted from the center-to-center nearest neighbor distances obtained by GISAXS measurements.
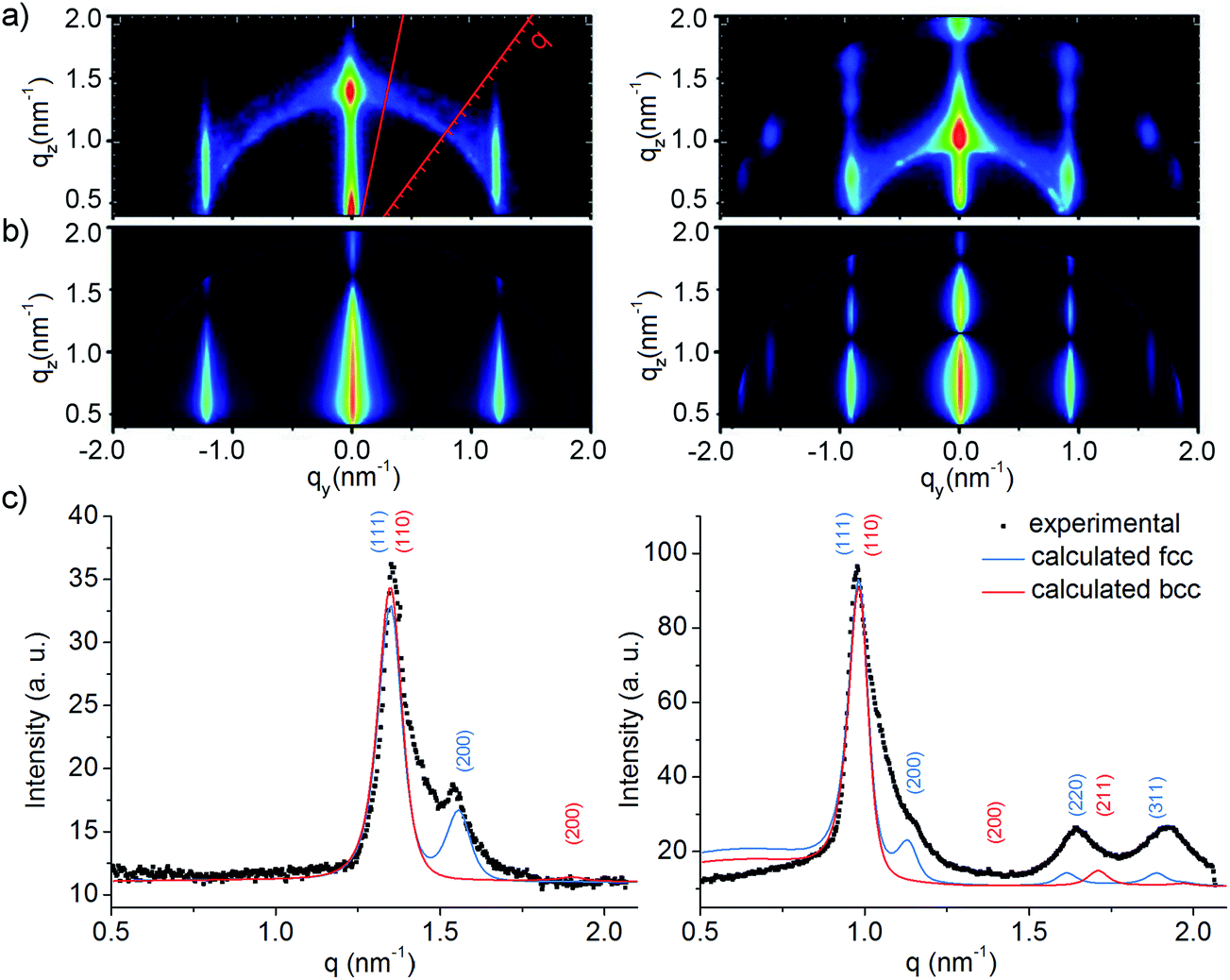 | ||
| Fig. 2 (a) Measured GISAXS patterns of superlattice films prepared from samples GNP4 nm1 (left) and GNP5.5 nm2 (right), (b) corresponding GISAXS simulations, (c) SAXS intensity profiles extracted from ring sections of the GISAXS patterns (as exemplarily indicated by red lines in Fig. 2a left) and calculated scattering curves of fcc (blue lines) and bcc superlattices (red lines). | ||
Structural model of the gold nanoparticle superlattice
In superlattices composed of ligand-stabilized GNPs two kinds of cavities are expected. Firstly, the free volume within the ligand shells with the diameter DfV, i.e. the interchain spacing between the ligands and defect sites within the shells. Secondly, voids within the interstitial sites of the close packed fcc arrangement of GNPs, i.e. within the tetrahedral and octahedral sites.Regarding the free volume within the ligand shell it is to note that self-assembled ligand monolayers (SAMs) on planar, irregular and curved metal surfaces have been studied extensively.30,31 In the most common system, i.e. alkanethiols chemi-sorbed on planar Au(111)-surfaces the surface coverage (S:Au atomic ratio) is 33%. Depending on their length the upright ligand chains are fully extended in a nearly all-trans conformation forming a quasi-crystalline structure (>C18), or they are in a semicrystalline state, or they are in a disordered liquid-like state (<C8). The distance between the alkanethiolates is ∼0.5 nm (ref. 30) and the tilt angle of the linear backbones with respect to the surface normal is 30°. However, 3D-SAMs on GNPs differ from their 2D counterpart on extended planar gold surfaces.25,31 The highly curved surface of GNP cores is characterized to a large extent by atoms located at corners or edges, resulting in a significantly higher surface coverage with ligands of ∼60%. Due to the small radius of curvature the chain density decreases from the core surface to the methyl end group of the ligands. The larger mobility at the end of the chains results in a higher degree of distorted chain conformations (end-gauche defects) causing disorder.25,26 The transition from a 3D-SAM to a 2D-SAM has been shown to occur at a core diameter of 4.4 nm for DDT-stabilized GNPs.25 For these particles an ordered arrangement of the ligands has been observed by IR spectroscopy at room temperature.
In assemblies of ligand-stabilized GNPs different ligand arrangements are possible. For the ligand arrangement in a 3D superlattice of DDT-stabilized gold nanoclusters molecular dynamics simulations predicted that at room temperature the ligands are in a crystalline-like ordered state in which they form interlocking bundles.24 In another study molecular dynamics simulations revealed that in 2D membranes of alkanethiol-stabilized GNPs (on water or in vacuum) the ligands display order on bottom and on top of the membrane, whereas the interdigitating ligands among the particles were disordered.27 Experimentally, the ligand arrangement in 2D assemblies of ligand-stabilized nanoparticles has been studied e.g. by TEM techniques. 2D superlattices of thiol-stabilized faceted silver nanoparticles (in shape of a truncated octahedron) revealed regions of increased organic density among the facets of the nanoparticles and channels of low or no organic material confined by the corners. The interparticle distances were shorter than twice the ligand length and indicated the formation of bundles of interdigitating ligands between adjacent nanoparticles.28,29 Recently, atomic-resolution TEM imaging of individual ligand molecules has been achieved by using graphene as TEM support.32–34 Diverse ligand conformations have been observed among different ligand/particle systems but also within the same system. For example, the ligands wrapped around the particle core or formed bundles between adjacent particles while leaving unoccupied voids in their vicinity. Thus, with respect to PALS measurements the different ligand arrangements are expected to contribute to the measured cavity size.
In addition to the free volume within the ligand shells, it is assumed that larger voids may be formed within the tetrahedral and octahedral sites. We estimated the size of these sites by assuming the simple geometrical model shown in Scheme 1. As derived from TEM and GISAXS data, the GNPs assembled into an fcc superlattice with an edge-to-edge distance δ between the gold cores of ∼2 nm. Comparing this interparticle distance with the length of the 1-dodecanethiol molecule in its fully extended all-trans conformation (1.8 nm)59 and considering previously reported findings,29,31 we assume that the ligands between adjacent particles are interdigitated. In the model shown in Scheme 1 an fcc lattice is formed by spheres with the radius reff, comprising the radius of the gold cores r plus half the interparticle distance: reff = r + δ/2.
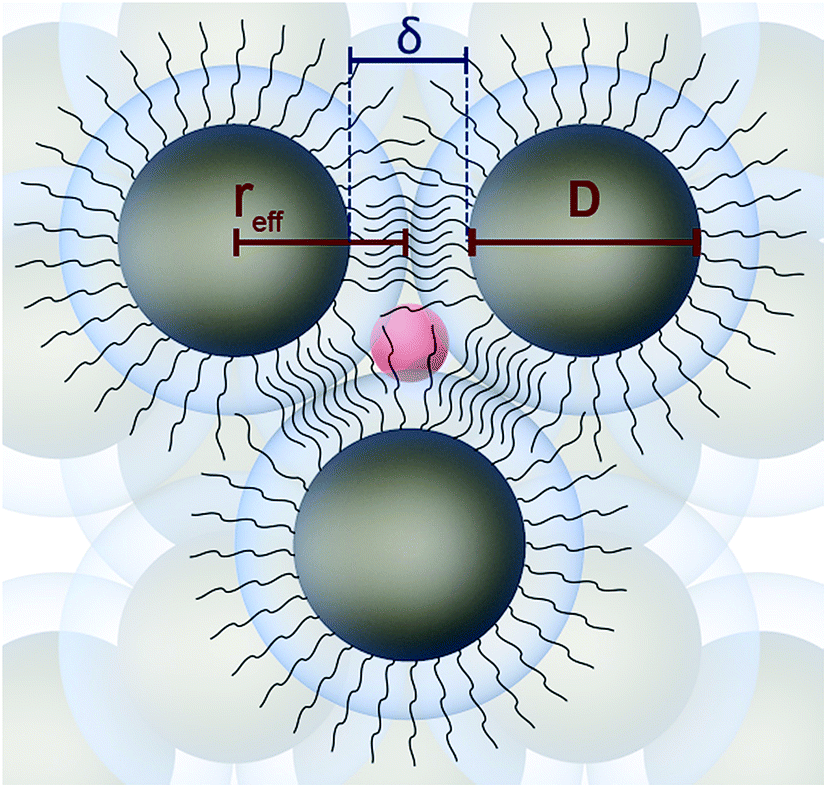 | ||
| Scheme 1 Geometrical model of the superlattice of ligand-stabilized gold nanoparticles. An fcc superlattice is formed by spheres with the radius reff = r + δ/2 (with r = ½D). The red sphere with the radius (reff × 0.225) was used to estimate the remaining void size within the tetrahedral sites after subtracting the ligand chains extending into this sphere. The same calculations were done for the octahedral sites. | ||
In order to estimate the size of voids in the interstitial sites, the radii of spheres fitting the tetrahedral (reff × 0.225, red sphere in Scheme 1) and octahedral sites (reff × 0.414) must be corrected by the ligands extending into these sites. Assuming that the ligands are in the fully extended conformation the value 1.8 nm − δ/2 must be subtracted from the sphere radius. Accordingly, the diameters of the voids in the octahedral sites DOH are ∼0.7 nm for samples with the ∼4 nm-sized GNP cores and range from 1.4 to 1.8 nm for samples with the ∼5.5 nm-sized GNP cores. For the smaller tetrahedral sites the diameters of the void spheres DTH are 0.03 nm to ∼0.4 nm for the samples with the ∼5.5 nm-sized particles. Thus, the void sizes estimated for the tetrahedral sites in these samples are similar to the defect dimensions in crystalline dodecane (0.36 nm,60 deduced from the o-Ps lifetime using eqn (1), see below), as previously measured by PALS. However, considering the tilt angle of the chains with respect to the surface normal, and depending on the conformation of the ligands directed toward the interstitial sites, the actual values of DTH and DOH could be somewhat larger. In the case of the 4 nm-sized GNP cores the fully extended ligands would overlap by ∼0.4 nm within the tetrahedral sites. Hence, in these samples the tetrahedral sites are assumed to be fully occupied by the ligands.
In order to validate the above described model based on GISAXS and TEM data, the mass fractions of gold nAu respectively organic material were calculated based on this model (similarly as has been shown in our previous work36) and compared to the mass fractions nAu,TGA determined by thermogravimetric analysis (TGA). For calculating the mass fractions it was assumed that apart from the space filled by the spherical gold cores all space within the superlattice films was filled with liquid 1-dodecanethiol (see the ESI for details†). As shown by Table 1 the values nAu based on our structural model and the TGA-measured values nAu,TGA are in excellent agreement. For samples GNP4 nm1, GNP4 nm2 and GNP5.5 nm1 the deviations between nAu and nAu,TGA were <0.5%, while for samples GNP5.5 nm2 and GNP5.5 nm3 the deviations were ≤8%. We note that taking into account the voids within the octahedral sites (with diameter DOH) when calculating nAu results in only marginal deviations (<0.2%).
Positron annihilation lifetime spectroscopy (PALS)
PALS is a generally accepted method for the characterization of defects (vacancies in crystalline materials, free volume in amorphous materials, voids, pores) in all classes of materials, in particular within polymer films. The technique is based on correlating the lifetime of positrons with the average size of cavities within the polymeric specimen. The positron lifetime is determined by measuring the time elapsed between the injection of positrons (obtained from a lab source, e.g.22Na, or a reactor) into the sample and the release of gamma rays in consequence of their annihilation with electrons. The theore-tical background of positron annihilation is well described in many textbooks.43,44Once a positron is injected into an insulating sample, it has three different possibilities for reaction and decay. According to the first decay path, the positron can be annihilated by direct impact with an electron. The lifetime of this decay within a polymer is typically ∼400 ps. Alternatively, the positron can form a bound state with an electron, which is termed “positronium”. According to the second decay path, this bound state is formed as para-positronium (p-Ps) with the positron and the electron having antiparallel spins. The p-Ps (singlet state) decays immediately and has a very short lifetime of only ∼125 ps. Finally, according to the third decay path, the bound state is formed as ortho-positronium (o-Ps) with the positron and the electron having parallel spins (triplet state). In vacuum the lifetime of the o-Ps is 142 ns and, thus, the decay is much slower than the annihilation of the free positron or the p-Ps. However, within the cavities of a specimen the decay of the o-Ps proceeds via a so called pick-off annihilation with an electron from the walls of the cavity. This pick-off annihilation depends on the local electron density distribution around the o-Ps and, thus, its lifetime can be used to determine the average free volume size. This is usually done by applying a standard quantum mechanical model originally developed by Tao and later extended by Eldrup and Jean.43,44 In this model the o-Ps is assumed to be confined within a spherical potential well with infinite walls, which are “decorated” with an electron layer. At this layer the o-Ps can pick off an electron and decay. The calculation of the overlap integral of the positronium's probability density function and the electron layer results in a direct relation between the o-Ps lifetime τ3 and the average cavity radius Rh:
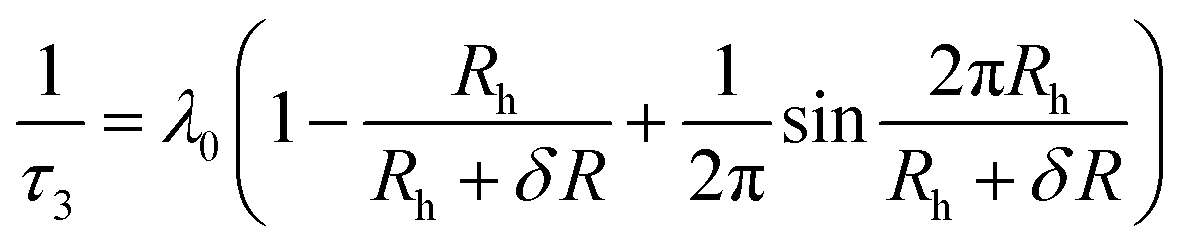 | (1) |
This equation includes the reciprocal o-Ps decay rate τ3, the spin averaged decay rate λ0 in the electron layer at the edge of the potential well, the thickness δR = 0.166 nm of the electron layer, which has been determined by using materials with a well-known pore size,61 and the average cavity radius Rh. Here, we use this equation analogously for the interstitial voids and free volumes in the sample. Additional information can be obtained from the relative signal intensities, which reflect the probabilities of the three decay possibilities (free positron, p-Ps, o-Ps) discussed in the previous paragraph. The positronium intensities (o-Ps and p-Ps) depend on the formation probability of positronium in the respective polymeric material, which are not known a priory and are often also related to the void concentration.44 Additionally, the measured o-Ps intensity is proportional to the amount of polymer “seen” by the positrons, which is in particular relevant to the very thin layers investigated here, where the intensity will be low.
The PALS spectra of the GNP superlattice films display a shape which is very similar to that known from polymer films. Fig. 3 shows a representative PALS spectrum of a superlattice film consisting of GNPs with a core size of 5.6 nm (sample GNP5.5 nm2). Due to the low amount of sample material (film thickness: ∼200 to ∼900 nm) and the corresponding low count rates, a well known artefact51 is observed between 10 to 15 ns and 20 to 25 ns. These signals originate from positrons, which are reflected from the sample and annihilate after a well known flight time somewhere in the chamber but not within the sample. This assignment is corroborated by the fact that these signals shift to longer times with decreasing acceleration energy, i.e. with decreasing velocities of injected positrons (Fig. 3, inset). In addition, simulations have proven this assumption.51 Thus, in the following we take into consideration the relevant part of the spectra, i.e. up to decay times of 10 ns. To evaluate the measured PALS spectra we first used our structural model to estimate the expected void diameters, providing void sizes in the tetrahedral sites DTH of ∼0.4 nm and in the octahedral sites DOH of ∼1.8 nm for sample GNP5.5 nm2 (Fig. 3). For the free volume within the ligand matrix DfV a value of ∼0.4 nm (ref. 60) was assumed. These cavity diameters were converted to o-Ps lifetimes according to eqn (1) and corresponding curves are shown in Fig. 3. For DfV and DTH (both ∼0.4 nm) the curve fits reasonably well to the raw data and corroborate our assumption, that the positronium is localized in the corresponding cavities. In contrast, lifetimes corresponding to DOH could not be identified. This finding will be discussed below.
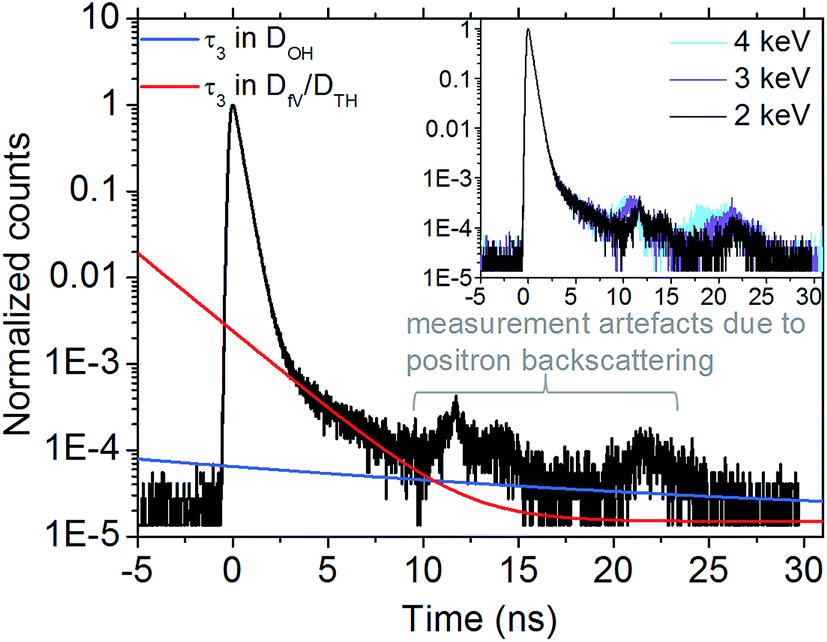 | ||
| Fig. 3 Representative PALS spectrum of a superlattice film comprised of DDT-stabilized GNPs with a core size of 5.6 nm (sample GNP5.5 nm2) recorded using an acceleration energy of 2 keV. The red and dark blue lines are simulations according to eqn (1) using free volumes in the ligand shell DfV and tetrahedral void diameters DTH of ∼0.4 nm as well as octahedral void diameters DOH of 1.8 nm, respectively. The inset shows spectra of the same sample measured with different implantation energies of the positrons, as indicated. | ||
The agreement of the calculated lifetimes with the measured PALS spectra reveals that the relevant part of the spectra for the decay of positronium is between 2 to 10 ns. As expected, within this range the spectra are not affected when varying the implantation energy of the positrons (see inset Fig. 3).
Fig. 4 shows representative PALS spectra of GNP superlattices comprised of GNPs with different core sizes. The red curve refers to sample GNP5.5 nm2 with a GNP core size of 5.6 nm, whereas the black curve shows a spectrum referring to sample GNP4 nm2 with a GNP core size of 3.9 nm. A slight difference in the slope of the curves is clearly seen, revealing somewhat longer lifetimes and, thus, somewhat larger cavity sizes for the superlattice consisting of the larger GNPs.
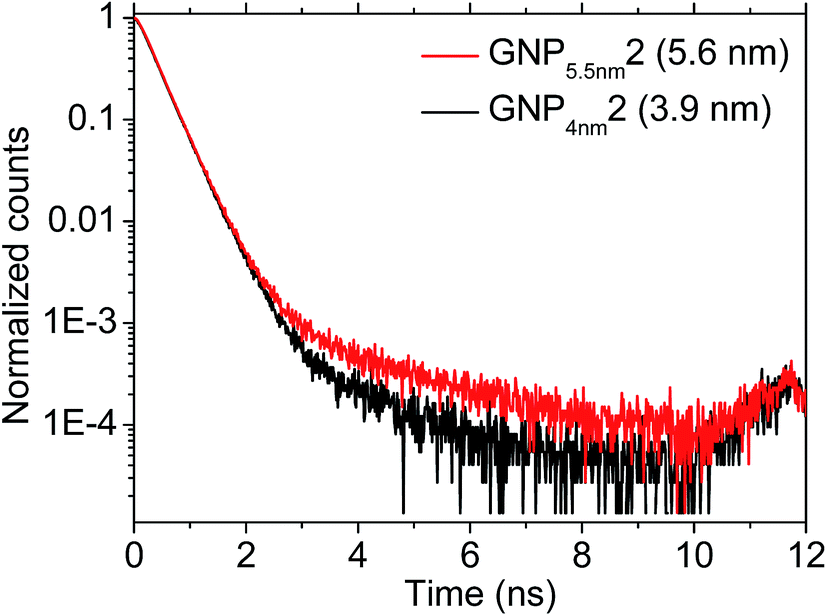 | ||
| Fig. 4 PALS spectra of two GNP superlattice films consisting of GNPs with core sizes of 5.6 nm (sample GNP5.5 nm2, red line) and 3.9 nm (sample GNP4 nm2, black line). The spectra were measured using an acceleration energy of 2 keV. | ||
We note that the o-Ps lifetimes τ3 obtained from fitting the spectra are, in general, similar to previously reported o-Ps lifetimes for oleic acid-stabilized PbSe nanoparticles.50 The lifetimes of all GNP superlattice films measured are listed in Table 2 together with the intensities I and the cavity sizes Dpals determined by converting the respective o-Ps lifetimes using eqn (1). The intensities are low, but in the expected range for a thin film composite material comprising gold and will not be discussed further.
| GNP4 nm1 | GNP4 nm2 | GNP4 nm3 | GNP5.5 nm1 | GNP5.5 nm2 | GNP5.5 nm3 | |
|---|---|---|---|---|---|---|
| o-Ps τ3 (ns) | 2.03 | 1.72 | 2.50 | 2.28 | 2.62 | 3.20 |
| o-Ps I (%) | 0.30 | 0.51 | 0.53 | 0.83 | 0.87 | 1.42 |
| D pals (nm) | 0.56 | 0.50 | 0.66 | 0.62 | 0.68 | 0.74 |
Cavity sizes
Fig. 5 shows the PALS-measured cavity diameters Dpals (Table 2) plotted vs. the structural parameter reff (see Scheme 1) for all GNP superlattices investigated. For comparison, the diameters DOH of the voids estimated for the octahedral sites using our structural model (see above) as well as the free volume sizes of crystalline (DDD,s) and liquid (DDD,l) dodecane, which have been measured previously by PALS,60 are indicated. Further, according to our structural model, the tetrahedral sites of films consisting of ∼4 nm-sized GNPs are expected to be fully occupied by the alkyl chains of the ligands, whereas in films consisting of ∼5.5 nm GNPs the remaining unoccupied voids in the tetrahedral sites are expected to have diameters similar to the free volume size determined for solid dodecane. Therefore, no values for DTH, i.e. the void diameters within tetrahedral sites, are included in Fig. 5. The cavity sizes measured by PALS for GNP films of both particle sizes are in the range defined by the free volume sizes found in solid and liquid dodecane (DDD,s = 0.36 nm, lifetime 1.1 ns and DDD,l = 0.73 nm, lifetime 3.1 ns, respectively).60 Here, it is to note that the comparison with liquid alkanes is not straight forward. On the one hand, o-Ps can form bubbles in liquid alkanes; then the o-Ps lifetime is not related to the cavity size anymore.43,44 On the other hand, as discussed above, the conformation of grafted alkanes varies from those in the liquid phase, making a direct comparison difficult. However, the measured cavity sizes Dpals for both types of films are obviously larger compared to the free volume found in solid dodecane, indicating that the free space around the gold cores is not filled by tightly packed crystalline ligand bundles throughout the entire structure. This finding can be interpreted straightforwardly by taking into account both the geometry of the superlattice and the possible spatial ligand arrangement within the organic matrix. On the one hand, regions of high ligand density are expected between adjacent particles, where the ligands interdigitate to a significant extent. On the other hand, the ligands directed toward the tetrahedral and octahedral sites, where the alkyl chains do not interdigitate (at least not significantly), may be more loosely packed. This effect is possibly more pronounced in the case of films containing the smaller ∼4 nm-sized GNP cores, because their higher surface curvature reduces the density of the ligands' alkyl chains. However, it may also be expected that the larger voids within the tetrahedral and octahedral sites in films of the larger GNPs provide space for less dense packing of the ligand matrix. Supporting this hypothesis the average cavity size of the films containing the ∼5.5 nm-sized GNP cores is in fact slightly larger than that of films comprising the ∼4 nm-sized GNPs. | ||
| Fig. 5 Experimentally (PALS) determined cavity sizes Dpals (black squares) and their respective mean values (red squares) for superlattice films comprised of ∼4 nm (left) and ∼5.5 nm-sized (right) GNPs. The diameters DOH of voids within the octahedral sites were estimated using our structural model and are indicted as blue squares. Note the break in both axes. The free volume sizes measured previously by PALS60 in crystalline DDD,s (purple) and liquid DDD,l (cyan blue) dodecane are represented by horizontal lines. For details see text. | ||
Nevertheless, in the case of the superlattice films prepared from the ∼5.5 nm-sized GNPs, all measured lifetimes indicate cavity sizes significantly smaller than the voids (DOH) expected within the octahedral sites. This observation can have two reasons. Firstly, the intensity of the o-Ps lifetime in the octahedral sites could be too low. As the probability to form positronium is unknown, the intensity cannot be estimated. Secondly, assuming that there are no technical reasons for the lack of the corresponding lifetimes, this finding suggests that the octahedral sites are at least partly occupied by excess ligand or solvent molecules, which were trapped within the 1.4 to 1.8 nm-sized cavities during film assembly.
Conclusions
In this study we used PALS to probe the microcavities in well-defined superlattice films comprised of DDT-stabilized GNPs. These superlattice films were assembled using batches of differently sized GNPs. For the films consisting of ∼4 nm-sized GNPs a structural model, based on GISAXS and TEM measurements, predicts that the space between the GNP cores is almost completely filled by the ligands' alkyl chains. While the ligands completely penetrate the tetrahedral sites, the open space left within the octahedral sites should have a diameter of only ∼0.7 nm. The average PALS-measured cavity size was (0.57 ± 0.08) nm, which is larger than the value measured by PALS for solid dodecane (0.36 nm (ref. 60)). Most likely, loosening of the ligand matrix results from disorder of the ligand arrangement and, possibly, from unoccupied voids available within the octahedral sites.For the films consisting of ∼5.5 nm-sized GNPs the structural model predicts voids of up to ∼0.4 nm within the tetrahedral sites. Such small voids are comparable to the free volume measured by PALS in crystalline dodecane (0.36 nm (ref. 60)). However, within the octahedral sites the model predicts much larger voids, with diameters of ∼1.7 nm. In contrast, our PALS measurements revealed average cavity sizes of (0.68 ± 0.06) nm, which is only somewhat larger than the cavity sizes measured for the films of ∼4 nm-sized GNPs. Therefore, we conclude that the larger voids available within the tetrahedral and, especially, within the octahedral sites leads to only somewhat less dense packing of the alkyl chains within the ligand shells, compared to the films consisting of ∼4 nm-sized GNPs. Furthermore, assuming measurable intensities of the o-Ps lifetimes in the octahedral sites our data suggests that the voids expected within the octahedral sites are occupied by excess ligand or solvent molecules trapped during film preparation.
Acknowledgements
The authors would like to thank Dr Thomas Strunskus and Dr Oleksandr Polonskyi for acquiring and analyzing XPS spectra of the samples. Mr Andreas Kornowski, Mr Stefan Werner and Mr Robert Schön are acknowledged for measuring TEM and SEM images. We thank Ms Katrin Rehmke and Mr Stefan Bleck (Institute for Technical and Macromolecular Chemistry, University of Hamburg) for TGA measurements. Mr Mateusz Olichwer is acknowledged for supporting the preparation of graphical artwork.References
- J. Langer, S. M. Novikov and L. M. Liz-Marzán, Nanotechnology, 2015, 26, 322001 CrossRef PubMed.
- A. Zabet-Khosousi and A.-A. Dhirani, Chem. Rev., 2008, 108, 4072–4124 CrossRef CAS PubMed.
- M. Gauvin, Y. Wan, I. Arfaoui and M.-P. Pileni, J. Phys. Chem. C, 2014, 118, 5005–5012 CAS.
- H. Schlicke, E. W. Leib, A. Petrov, J. H. Schröder and T. Vossmeyer, J. Phys. Chem. C, 2014, 118, 4386–4395 CAS.
- J. Herrmann, K.-H. Müller, T. Reda, G. R. Baxter, B. Raguse, G. J. J. B. de Groot, R. Chai, M. Roberts and L. Wieczorek, Appl. Phys. Lett., 2007, 91, 183105 CrossRef.
- T. Vossmeyer, C. Stolte, M. Ijeh, A. Kornowski and H. Weller, Adv. Funct. Mater., 2008, 18, 1611–1616 CrossRef CAS.
- F. J. Ibañez and F. P. Zamborini, Small, 2012, 8, 174–202 CrossRef PubMed.
- L. Minati, A. Chiappini, F. Benetti, G. Speranza, D. Zonta, A. Piotrowska, M. Marciniak, A. Vaccari and M. Ferrari, Colloids Surf., A, 2015, 482, 431–437 CrossRef CAS.
- F.-Y. Chen, W.-C. Chang, R.-S. Jian and C.-J. Lu, Anal. Chem., 2014, 86, 5257–5264 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- H. Schlicke, D. Battista, S. Kunze, C. J. Schröter, M. Eich and T. Vossmeyer, ACS Appl. Mater. Interfaces, 2015, 7, 15123–15128 CAS.
- H. Schlicke, C. J. Schröter and T. Vossmeyer, Nanoscale, 2016, 8, 15880–15887 RSC.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- N. Goubet, J. Richardi, P.-A. Albouy and M.-P. Pileni, Adv. Funct. Mater., 2011, 21, 2693–2704 CrossRef CAS.
- A. Courty, J. Richardi, P.-A. Albouy and M.-P. Pileni, Chem. Mater., 2011, 23, 4186–4192 CrossRef CAS.
- J. J. Choi, C. R. Bealing, K. Bian, K. J. Hughes, W. Zhang, D.-M. Smilgies, R. G. Hennig, J. R. Engstrom and T. Hanrath, J. Am. Chem. Soc., 2011, 133, 3131–3138 CrossRef CAS PubMed.
- K. Bian, J. J. Choi, A. Kaushik, P. Clancy, D.-M. Smilgies and T. Hanrath, ACS Nano, 2011, 5, 2815–2823 CrossRef CAS PubMed.
- S. I. Stoeva, B. L. V. Prasad, S. Uma, P. K. Stoimenov, V. Zaikovski, C. M. Sorensen and K. J. Klabunde, J. Phys. Chem. B, 2003, 107, 7441–7448 CrossRef CAS.
- B. W. Goodfellow and B. A. Korgel, ACS Nano, 2011, 5, 2419–2424 CrossRef CAS PubMed.
- B. W. Goodfellow, M. R. Rasch, C. M. Hessel, R. N. Patel, D.-M. Smilgies and B. A. Korgel, Nano Lett., 2013, 13, 5710–5714 CrossRef CAS PubMed.
- Y. Yu, A. Jain, A. Guillaussier, V. R. Voggu, T. M. Truskett, D.-M. Smilgies and B. A. Korgel, Faraday Discuss., 2015, 181, 181–192 RSC.
- X. P. Liu, Y. Ni and L. H. He, Nanotechnology, 2016, 27, 135707 CrossRef CAS PubMed.
- U. Landman and W. D. Luedtke, Faraday Discuss., 2004, 125, 1–22 RSC.
- W. D. Luedtke and U. Landman, J. Phys. Chem., 1996, 100, 13323–13329 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- M. J. Hostetler, J. J. Stokes and R. W. Murray, Langmuir, 1996, 12, 3604–3612 CrossRef CAS.
- K. M. Salerno, D. S. Bolintineanu, J. M. D. Lane and G. S. Grest, Phys. Rev. E, 2015, 91, 62403 CrossRef PubMed.
- Z. L. Wang, Adv. Mater., 1998, 10, 13–30 CrossRef CAS.
- Z. L. Wang, S. A. Harfenist, R. L. Whetten, J. Bentley and N. D. Evans, J. Phys. Chem. B, 1998, 102, 3068–3072 CrossRef CAS.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Chem. Soc. Rev., 2010, 39, 1805–1834 RSC.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1169 CrossRef CAS PubMed.
- M. G. Panthani, C. M. Hessel, D. Reid, G. Casillas, M. Jose-Yacaman and B. A. Korgel, J. Phys. Chem. C, 2012, 116, 22463–22468 CAS.
- Z. Lee, K.-J. Jeon, A. Dato, R. Erni, T. J. Richardson, M. Frenklach and V. Radmilovic, Nano Lett., 2009, 9, 3365–3369 CrossRef CAS PubMed.
- A. A. Gunawan, B. D. Chernomordik, D. S. Plemmons, D. D. Deng, E. S. Aydil and K. A. Mkhoyan, Chem. Mater., 2014, 26, 3328–3333 CrossRef CAS.
- J. He, X.-M. Lin, H. Chan, L. Vuković, P. Král and H. M. Jaeger, Nano Lett., 2011, 11, 2430–2435 CrossRef CAS PubMed.
- N. Olichwer, A. Meyer, M. Yesilmen and T. Vossmeyer, J. Mater. Chem. C, 2016, 4, 8214–8225 RSC.
- N. Olichwer, E. W. Leib, A. H. Halfar, A. Petrov and T. Vossmeyer, ACS Appl. Mater. Interfaces, 2012, 4, 6151–6161 CAS.
- E. Dovgolevsky, U. Tisch and H. Haick, Small, 2009, 5, 1158–1161 CAS.
- M. Segev-Bar, G. Shuster and H. Haick, J. Phys. Chem. C, 2012, 116, 15361–15368 CAS.
- E. Chow, B. Raguse, K. H. Müller, L. Wieczorek, A. Bendavid, J. S. Cooper, L. J. Hubble and M. S. Webster, Electroanalysis, 2013, 25, 2313–2320 CAS.
- M. J. Puska and R. M. Nieminen, Rev. Mod. Phys., 1994, 66, 841–897 CrossRef CAS.
- A. Seeger and F. Banhart, Helv. Phys. Acta, 1990, 63, 403–428 CAS.
- O. E. Mogensen, in Springer Series in Chemical Physics, ed. V. I. Goldanskii, Springer-Verlag, Berlin, Heidelberg, 1995, vol. 58 Search PubMed.
- Y. C. Jean, P. E. Mallon and D. M. Schrader, Principles and Applications of Positron and Positronium Chemistry, World Scientific, New Jersey London Singapore Hong Kong, 2003 Search PubMed.
- K. Raetzke, M. Q. Shaikh, F. Faupel and P.-L. M. Noeske, Int. J. Adhes. Adhes., 2010, 30, 105–110 CrossRef CAS.
- R. Konietzny, T. Koschine, K. Rätzke and C. Staudt, Sep. Purif. Technol., 2014, 123, 175–182 CrossRef CAS.
- H. B. Tanh Jeazet, T. Koschine, C. Staudt, K. Raetzke and C. Janiak, Membranes, 2013, 3, 331–353 CrossRef PubMed.
- S. Harms, K. Rätzke, V. Zaporojtchenko, F. Faupel, W. Egger and L. Ravelli, Polymer, 2011, 52, 505–509 CrossRef CAS.
- S. Harms, K. Rätzke, F. Faupel, N. Chaukura, P. M. Budd, W. Egger and L. Ravelli, J. Adhes., 2012, 88, 608–619 CrossRef CAS.
- L. Chai, W. Al-Sawai, Y. Gao, A. J. Houtepen, P. E. Mijnarends, B. Barbiellini, H. Schut, L. C. van Schaarenburg, M. A. van Huis, L. Ravelli, W. Egger, S. Kaprzyk, A. Bansil and S. W. H. Eijt, APL Mater., 2013, 1, 22111 CrossRef.
- L. Ravelli, Phd thesis, Universität der Bundeswehr München, 2014.
- W. Egger, in Physics with Many Positrons, ed. A. Dupasquier, A. P. Mills Jr and R. S. Brusa, IOS Press, Amsterdam, 2010, pp. 419–449 Search PubMed.
- N. Zheng, J. Fan and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 6550–6551 CrossRef CAS PubMed.
- W. Bauer-Kugelmann, PhD thesis, Universität der Bundeswehr München, 2000.
- N. Goubet, H. Portalès, C. Yan, I. Arfaoui, P.-A. Albouy, A. Mermet and M.-P. Pileni, J. Am. Chem. Soc., 2012, 134, 3714–3719 CrossRef CAS PubMed.
- R. Lazzari, J. Appl. Crystallogr., 2002, 35, 406–421 CrossRef CAS.
- S. Förster, L. Apostol and W. Bras, J. Appl. Crystallogr., 2010, 43, 639–646 CrossRef.
- S. Förster, A. Timmann, M. Konrad, C. Schellbach, A. Meyer, S. S. Funari, P. Mulvaney and R. Knott, J. Phys. Chem. B, 2005, 109, 1347–1360 CrossRef PubMed.
- C. D. Bain, E. B. Troughton, Y.-T. Tao, J. Evall, G. M. Whitesides and R. G. Nuzzo, J. Am. Chem. Soc., 1989, 111, 321–335 CrossRef CAS.
- Z. Kajcsos, I. Dézsi and D. Horváth, Appl. Phys., 1974, 5, 53–56 CAS.
- H. Nakanishi, S. J. Wang and Y. C. Jean, International Symposium on Positron Annihilation Studies of Fluids, World Scientific, Singapore, 1988 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS spectra and SEM images of the GNP superlattice films, details on calculations. See DOI: 10.1039/c6ra24241c |
| ‡ Current address: Centre for Surface Chemistry and Catalysis, KU Leuven, Celestijnenlaan 200f, 3001 Leuven, Belgium. |
| This journal is © The Royal Society of Chemistry 2016 |