
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quinoline based mono- and bis-(thio)carbohydrazones: synthesis, anticancer activity in 2D and 3D cancer and cancer stem cell models†
Aleksandra
Božić
a,
Aleksandar
Marinković
a,
Snežana
Bjelogrlić
 b,
Tamara R.
Todorović
c,
Ilija N.
Cvijetić
d,
Irena
Novaković
e,
Christian D.
Muller
f and
Nenad R.
Filipović
*g
b,
Tamara R.
Todorović
c,
Ilija N.
Cvijetić
d,
Irena
Novaković
e,
Christian D.
Muller
f and
Nenad R.
Filipović
*g
aFaculty of Technology and Metallurgy, University of Belgrade, Karnegijeva 4, Belgrade, Serbia
bNational Cancer Research Center of Serbia, Pasterova 14, Belgrade, Serbia
cFaculty of Chemistry, University of Belgrade, Studentski trg 12-16, Belgrade, Serbia
dInnovation Center of the Faculty of Chemistry, University of Belgrade, Studentski trg 12-16, Belgrade, Serbia
eInstitute of Chemistry, Technology and Metallurgy, University of Belgrade, Njegoševa 12, Belgrade, Serbia
fInstitut Pluridisciplinaire Hubert Curien, UMR 7178, CNRS, Université de Strasbourg, 67401 Illkirch, France. E-mail: cdmuller@unistra.fr
gFaculty of Agriculture, University of Belgrade, Nemanjina 6, Belgrade, Serbia. E-mail: nenadf.chem@gmail.com
First published on 27th October 2016
Abstract
A comparative study of antitumor activity of mono- and bis-quinoline based (thio)carbohydrazones was investigated by a series of tests on two human malignant cell lines: acute monocytic leukemia (THP-1) and pancreatic adenocarcinoma cancer stem cells (AsPC-1). Thiocarbohydrazones (TCHs) revealed superior pro-apoptotic activity over carbohydrazones (CHs) on both tested cell phenotypes, also displaying multi-target profile activities. Programmed cell death triggered by TCHs was partially caspase-dependent, mainly caspase-8 related. Activity against cancer stem cells (CSCs) was evaluated on 2D monolayers and 3D spheroidal models, where two out of three tested bis-TCHs successfully stimulated apoptosis accompanied by a reduction in size of treated spheres. Additionally, all bis-TCHs induced significant decrease in percentage of CD44-expressing AsPC-1 cells that indicate on their ability to induce reprogramming of CSC phenotype. Current results highly support further assessment of bis-TCHs in order to specify their specific targets in cancer cells and particularly in the CSCs subpopulation.
Introduction
Hydrazones are condensation products of hydrazine or its alkyl, acyl and aryl analogues with carbonyl compounds. They are known to possess a broad spectrum of biological activities such as antioxidant, analgesic, antiplatelet, antimicrobial, anticonvulsant, antidepressant, anti-inflammatory, anti-tubercular, cardio protective, anti-HIV, anthelmintic, anti-diabetic, trypanocidal and anticancer activities.1–6 Many of these compounds were patented in this respect.7 Monohydrazide derivatives of carbonic and thiocarbonic acid, semicarbazide and thiosemicarbazide, respectively react with N-heteroaromatic carbonyl compounds giving (thio)semicarbazones – molecules well described as tridentate NNO(S) chelators and ribonucleotid reductase (RR) inhibitory agents.8 RR plays a fundamental role in the critical early events involved in tumor promotion and activity of this enzyme is tightly linked to the neoplastic expression state and is currently one of the main targets for DNA inhibition by anticancer agents.9The most studied hydrazone is triapine (3-amino-2-formylpyridine thiosemicarbazone) since more than 30 clinical phase I and phase II trials has been done so far.10 Triapine was developed in 1994 as an RR inhibitor and its mechanism of action was thought to be iron chelation from RR active site.11 Later on its iron(II) complex was supposed to be active species which inhibits RR indirectly by generation of reactive oxygen species (ROS) which are formed in Fentone type reactions.12 Recent evidences proved that iron(II) complex inhibits RR, but probably via direct tyrosil radical quenching not involving ROS.13 There are also different theories were and how the active Fe(II)–triapine complex is formed in the body. For example, one study resulted in the model which proposed triapine binding for RR surface, followed by labilization of the diferric center and iron complex formation.14 On the other hand, there are evidences that interaction of triapine with iron occurs in the liver, which is known as the site of ferritin production.15 Unrevealing of triapine's mechanism of activity is still the topic of interest. Recently mitochondria16,17 and endoplasmatic reticulum18 were recognized as new triapine biological targets.
Carbohydrazide and thiocarbohydrazide are dihydrazide derivatives of carbonic and thiocarbonic acids. As their monohydrazide analogues (thio)semicarbazide, (thio)carbohydrazide also form corresponding monohydrazones. The presence of additional hydrazide group allows preparation of dihydrazones. Despite the fact that mono- and bis-(thio)carbohydrazides are also known to chelate iron,19–25 which make them potential RR inhibitors, there are only few investigations published where their anticancer activities were explored,25–33 with no study about their possible mechanism of action.
Quinoline is a privileged scaffold in anticancer drug discovery34 and various quinoline (thio)semicarbazones showed promising results when tested against cancer cell lines.35–40 Here, we present results of the first systematically organized investigation of antitumor activity of three mono-carbohydrazones and three mono-thiocarbohydrazones with their corresponding bis-structural counterparts. Despite the development of new therapeutics with increasing success in treating malignancies, recurrence of disease can develop years after efficient therapy. It was demonstrated that a small fraction of cells residing at the top of genetic hierarchy within solid tumors, declared as cancer stem cells (CSCs), are responsible for disease relapse.41 CSCs are also assigned culpable for tumor growth, progression, dissemination and resistance to therapy. Specified important role of CSCs in tumor growth, metastases and relapse, define that the major goal in effort to cure the cancer should be elimination or terminal differentiation of CSCs. Currently, there is no available treatment option proved as efficient against this subpopulation of cancer cells. Apart from having significantly enhanced DNA repairing mechanisms, CSCs hold a unique strategy to avoid the harmful effects of chemotherapeutic and radiation treatments which exclusively affect proliferating cells. There are several circumstances when CSCs resort the dormancy phase to gain genetic and epigenetic alterations that are necessary for neoplastic conversion, adaptation to new microenvironment and survival.42,43 Therapy-induced dormancy describes response of CSCs to repeated cycles of therapy that enter a dormant phase and thereafter relapse as a more aggressive phenotype.44 To eliminate CSCs, applied treatments should induce their apoptotic death, or to interferes with their hideaway in dormancy phase by stimulating mitotic division thus increasing their vulnerability to chemo and radiotherapy. Therefore, in order to investigate ability of our compounds to act as comprehensive anti-cancer agents with competency to eliminate both CSCs and non-CSCs, current biological study was organized on two diverse cell lines acute monocytic leukemia (THP-1) and pancreatic adenocarcinoma cancer stem cells (AsPC-1), where AsPC-1 cells are highly expressing CSC phenotype.45,46 Additionally, alongside standardly used 2D monolayer model, we here also used spheroidal 3D culture as more accurate screening platform for drug activity considering its architecture well mimics in vivo tumor mass.
Results and discussion
Chemistry
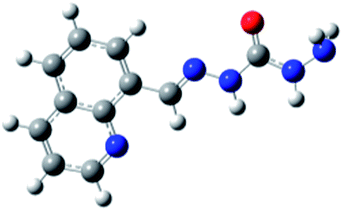
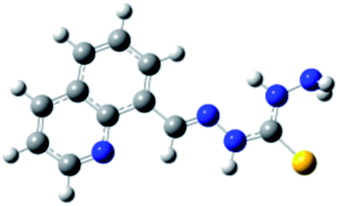
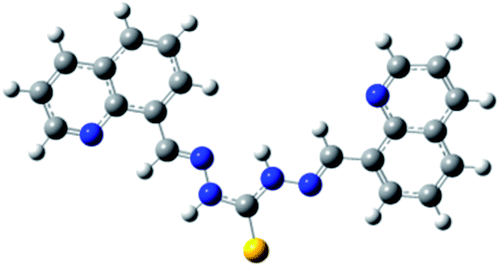
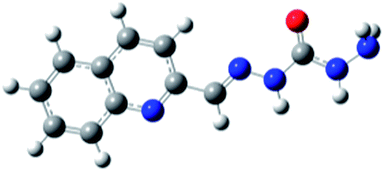
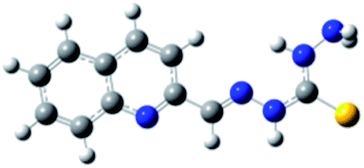
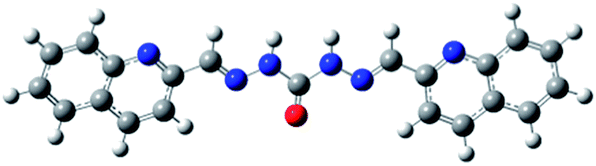
Carbohydrazones (CHs) O1–O6 and thiocarbohydrazones (TCHs) S1–S6 were prepared by the condensation reaction of carbonic acid dihydrazide (dhO) or its sulphur analogue thiocarbonic acid dihydrazide (dhS) and appropriate aldehyde (Scheme 1). The synthesis of O4 and S4 have been published earlier.47 The composition and purity of all compounds was verified by elemental analysis, while their structural characterization was done by IR and NMR spectroscopy.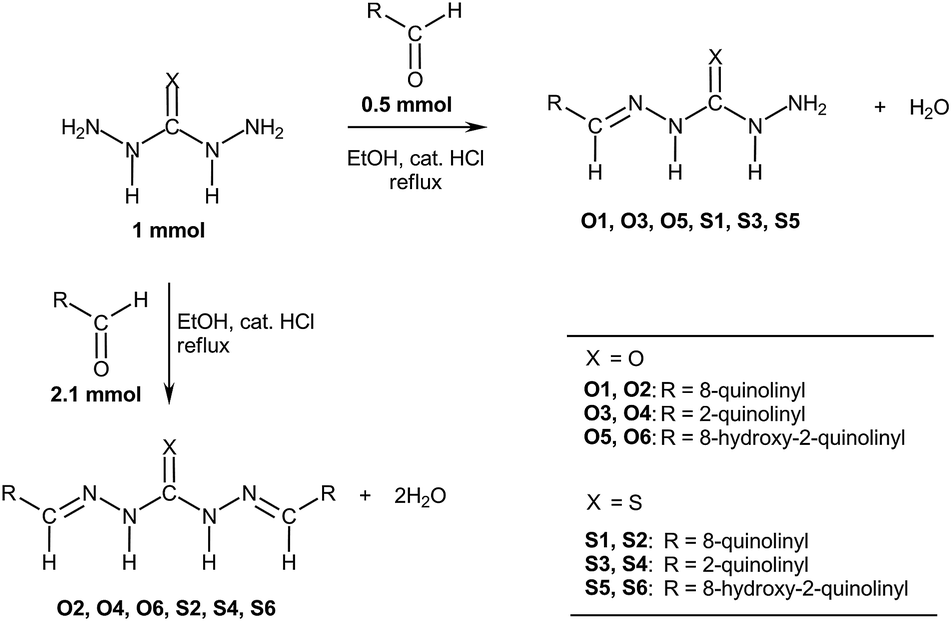 | ||
| Scheme 1 General procedure for the synthesis of carbohydrazones O1–O6 and thiocarbohydrazones S1–S6. | ||
In order to optimize geometries of the all compounds DFT calculations were performed. There are significant structural differences between optimized geometries of CHs and TCHs (Table 1). Analogue monohydrazones differ in the chalcogen atom orientation, while in the case of bis-hydrazones symmetrical conformations were obtained in the case of CHs and unsymmetrical ones in the case of TCHs. Optimized geometries of bis-hydrazones are in accordance with data obtained by NMR spectroscopy. Results of the GIAO/B3LYP calculations of the 1H and 13C chemical shifts of all compounds are shown in ESI (Tables S1–S5†). Namely, the main difference between 1H NMR spectra of oxygen based bis-hydrazones (bis-CHs) and sulphur based bis-hydrazones (bis-TCHs) is presence of two distinct signals for both, hydrazide and imine nitrogen protons in the case of sulphur compounds. The presence of two distinct signals for NH protons was already reported for bis-TCHs,48 and that phenomenon was explained by the presence of intermolecular hydrogen bonds between hydrazide hydrogen atom and imine nitrogen atom, while in the case of bis-CHs there is no such hydrogen bond.
| CHs | IC50 (mM) | TCHs | IC50 (mM) | ||
|---|---|---|---|---|---|
| O1 |
|
0.87 ± 0.11 | S1 |
|
0.14 ± 0.03 |
| O2 |
|
16.40 ± 1.02 | S2 |
|
0.16 ± 0.04 |
| O3 |
|
0.67 ± 0.06 | S3 |
|
0.13 ± 0.02 |
| O4 |
|
10.50 ± 0.46 | S4 |
|
0.11 ± 0.03 |
| O5 |
|
0.21 ± 0.05 | S5 |
|
0.11 ± 0.02 |
| O6 |
|
1.50 ± 0.09 | S6 |

|
0.09 ± 0.02 |
| Ascorbic acid | 0.08 ± 0.01 | ||||
All compounds obey Lipinski's rule of five,49 (Table S6, ESI†) so these compounds are drug-like, with promising bioavailability and pharmacokinetics. The proton donating ability of investigated quinoline based (thio)carbohydrazones O1–O6 and S1–S6 was assayed using a protocol for the determination of radical scavenging activity, the DPPH (1,1-diphenyl-2-picrylhydrazyl radical) method.50 Ascorbic acid was used as the reference compound (positive control). To the best of our knowledge there is no systematic comparative study of free-radical scavenging activity of CHs and TCHs. Our results indicate that TCHs were far more active in DPPH radical scavenging than their oxygen counterparts, with IC50 values close to that of vitamin C (Table 1). In both group of compounds, those which contain hydroxyl group are the most active, in the case of both mono- and bis-hydrazones. It seems that the main reason for pronounced antioxidant activity of TCHs is presence of thione (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S) group, since the obtained IC50 values are in the narrow range (0.09–0.16 mM), while S6 had almost the same activity as vitamin C. Other thione antioxidants such as ergoteine and thiosemicarbazones are well known for their free-radical scavenging activities.51,52 In the case of CHs, mono derivatives are 7–19 times more active than their bis-hydrazone analogues, which indicates a role of free –NH–NH2 group in the mechanism of their antioxidant activity. More detailed study of antioxidant activities of CHs and TCHs, which has a goal to decipher the mechanism of action, is currently in progress in our group.
S) group, since the obtained IC50 values are in the narrow range (0.09–0.16 mM), while S6 had almost the same activity as vitamin C. Other thione antioxidants such as ergoteine and thiosemicarbazones are well known for their free-radical scavenging activities.51,52 In the case of CHs, mono derivatives are 7–19 times more active than their bis-hydrazone analogues, which indicates a role of free –NH–NH2 group in the mechanism of their antioxidant activity. More detailed study of antioxidant activities of CHs and TCHs, which has a goal to decipher the mechanism of action, is currently in progress in our group.
Investigation of anticancer activities
| CHs | THP-1 | AsPC-1 | TCHs | THP-1 | AsPC-1 |
|---|---|---|---|---|---|
| a n.d. – not determined in the range of applied concentrations (1–100 μM). | |||||
| O1 | n.d.a | n.d. | S1 | 8.4 ± 0.3 | n.d. |
| O2 | 35.2 ± 0.7 | 64 ± 4 | S2 | 14 ± 5 | 75 ± 3 |
| O3 | n.d. | n.d. | S3 | 15.8 ± 0.5 | n.d. |
| O4 | n.d. | n.d. | S4 | 2.4 ± 0.2 | n.d. |
| O5 | n.d. | n.d. | S5 | 24 ± 9 | n.d. |
| O6 | n.d. | 57 ± 2 | S6 | 18 ± 6 | 74 ± 5 |
Apoptotic response in THP-1 cells treated with compound S4 was too drastic when added in the initial concentration range and impeded determination of the dose–response curves and computation of ED50 concentration. Therefore, concentrations of S4 had to be reduced to 0.5–30 μM (Fig. S1, ESI†). Compound S4, as well as S1, defined bell-shaped dose–response curves with the peak activities at 10 and 50 μM, respectively (Fig. S2A and B†). Considering the percentage of cells stained by Annexin V, the highest activity on THP-1 cells was achieved by O2 that caused for almost all treated cells to be labeled with both Annexin V and propidium iodide (PI) in samples subjected to 50, 75 and 100 μM, followed by a drastic decrease in cell death events at already 30 μM (Fig. S1, ESI†). Such activity profile for O2 was described with nearly vertical linear phase on dose–response curve typical for drugs with a tight therapeutic window, meaning that there is a narrow range of O2 concentrations that can achieve desired therapeutic effect while avoiding toxic threshold.
Contrary to the results obtained with THP-1 cells, only four compounds displayed pro-apoptotic activity against pancreatic AsPC-1 cells (Table 2 and Fig. S3, ESI†). Interestingly, those four compounds are structural siblings, where O2 and O6 are CHs, which analogically corresponds to TCHs S2 and S6, respectively. Both compounds S6 and particularly S2 were revealed as less efficient against AsPC-1 cells compared to THP-1 cells, a result not so surprising since AsPC-1 cells are known as highly resistant to common cytotoxic drugs as well as other cancer stem cell lines are.53,54 According to the percentage of Annexin V positive events, O2 displayed the highest activity, as previously seen on THP-1 cells, but on AsPC-1 cell line the peak activity starts only at 75 μM (Fig. S1 and S3, ESI†). While treatment of THP-1 cells with O6 resulted in a significant percentage of necrosis as noted above, on AsPC-1 cells it induced agglomeration of double-stained cells with a very small percentage of Annexin V single-stained events (Fig. S3, ESI†). A striking lack of gradual introducing of cells from early to late phases of apoptosis seen in AsPC-1 samples treated with O2 and O6, is highly indicative as double-stained events were actually belonging to necrotic cells. For this reason, compounds O2 and O6 were withdrawn from further investigation on relying mechanisms of apoptotic death in THP-1 and AsPC-1 cells.
In general, all evaluated compounds, at some concentration level, did induce accumulation of THP-1 cells in the G1-to-S transition point, while most of them caused concentration–dependent fluctuations in arrangement of cells within phases of mitotic division. As represented in Fig. 1A, treatment of THP-1 cells with S1 applied in a wide range (from 1 up to 100 μM) resulted in a concentration–dependent fluctuation in cell cycle distribution. In the samples treated with its lowest concentration, S1 stimulated cell accumulation at the G2/M phase. At 10 μM cells were arrested at the G1-to-S transition point accompanied with a significant incidence of apoptotic death, while starting from the sample subjected to the concentration of 30 μM, cells shifted to the G0/G1 block and sustained up to the highest 100 μM concentration. Although it might seem that the same mechanism of activity caused cell cycle arrest at the G0/G1 phase, percentage of apoptotic cells and concentration–response curve indicate otherwise. The highest percentage of cells at the G0/G1 phase was found in the samples treated with S1 at concentrations of 30 and 50 μM, which defined the peak of the bell-shaped curve (Fig. S2B†). In the samples subjected to 75 and 100 μM magnitude of the G0/G1 block slightly decreased, followed by a notably reduced frequency of apoptotic cells. This decrease in pro-apoptotic activity might indicate that S1 at higher concentration levels emerged additional mechanism of action that antagonized with the one dominating at concentrations between 30 and 50 μM.
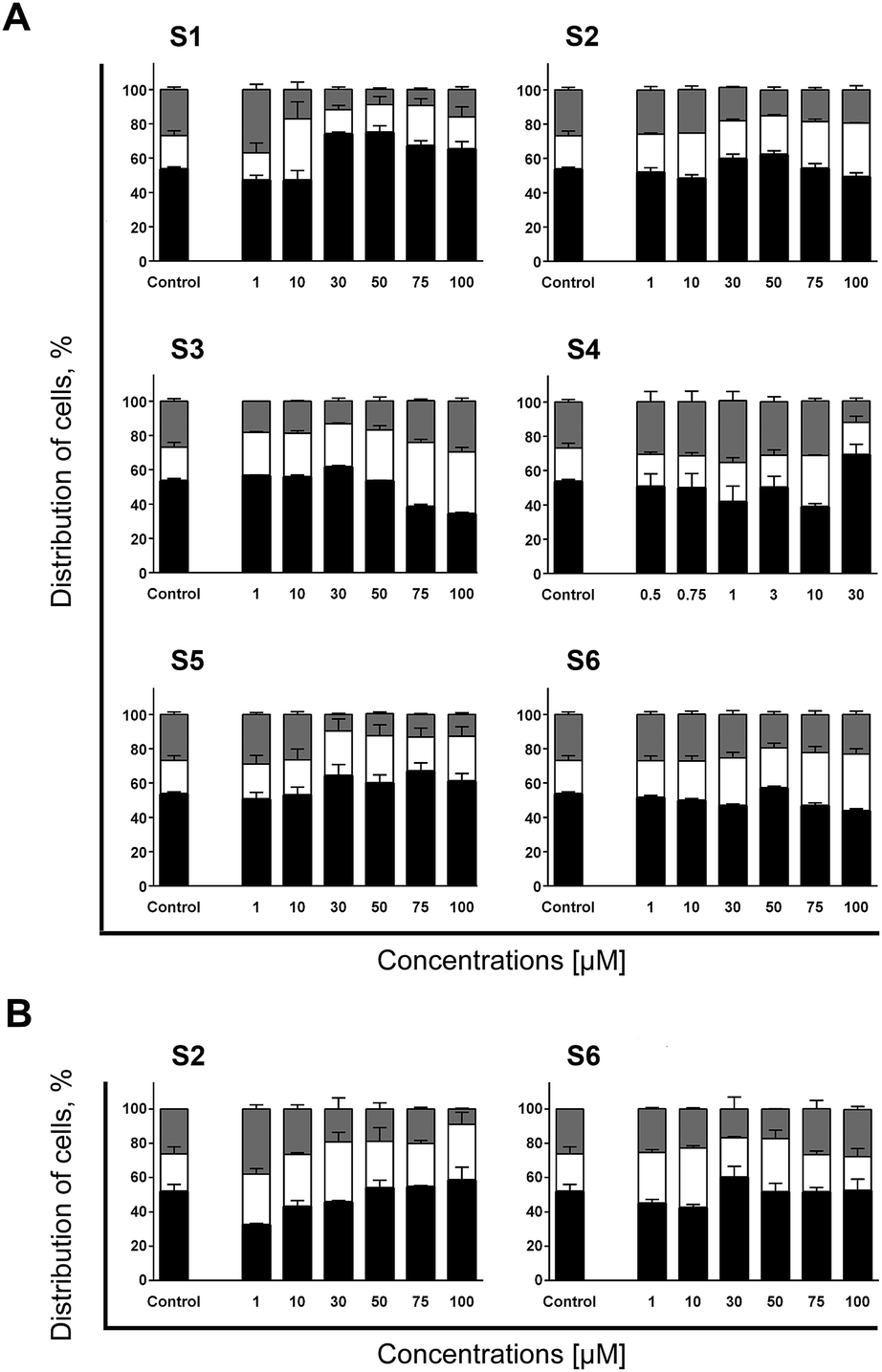 | ||
| Fig. 1 Distribution within phases of mitotic division of THP-1 cells (A) and AsPC-1 cells (B) determined after 24 h incubation with investigated compounds. Changes in cell cycle arrangement have been assessed in the same samples previously analyzed for Annexin V/PI (results represented in Fig. S1 and S2, ESI†). Incidences of cells found at the G0/G1 (black), S (white) and G2/M (gray) phases were determined according to non-treated control population. Results are represented as the mean ± SD of two replicates from independent experiments. | ||
Another compound that induced variations in the cell cycle distribution in concentration–dependent manner was S4 (Fig. 1A). Similarly to S1, S4 initially induced THP-1 cells to slightly accumulate at the G2/M phase that was modestly rising concentration-dependently within a range from 0.5 to 1 μM. The incidence of apoptotic death at those concentration levels was meaningless (Fig. S1, ESI†). However, starting from the sample treated with S4 at 3 μM, percentage of cells at the G2/M phase began to decrease followed by accumulation at the G1-to-S transition point and was accompanied with significant increase in incidence of apoptotic death. Finally, at the highest concentration (30 μM) a G0/G1 block was recorded together with almost halved percentage of apoptosis as compared to concentration of 10 μM. It is interesting to emphasize that S1 and S4 have more similarity in terms of transitive changes within cell cycles than their corresponding bis-TCH S2 and mono-TCH S3 analogues, respectively. Although S1 and S4 are not structurally correlated and differ in number of aromatic rings as well as in position of the side chain attached to the hetero-aromatic ring, those molecules share comparable transitive changes within cell cycle distribution. However, that was not the case with their structural counterparts, S2 and S3 (Fig. 1A). Namely, S2 and S3 at lower concentration levels induced accumulation at the G1-to-S transition point, then progress toward a gathering of cells at the S phase. In the case of the compounds with hydroxyl group (S5 and S6), a negligible variations in a cell cycle distribution were seen in the samples treated with concentrations that induced apoptosis in THP-1 cells (Fig. 1A and S1, ESI†). Both those treatments resulted in accumulation of cells at the G1-to-S transition point at concentration of 30 μM, which in samples subjected to S5 was getting intensified following the rise of applied concentration, whereas in cells treated with S6 a concentration–dependent accumulation of cells at the S phase was seen.
In AspC-1 cells concentration–dependent fluctuations in cell cycle distribution was seen in samples treated with S2 (Fig. 1B). Accumulation of treated cells at the G2/M phase seen in the samples treated with S2 at concentration of 1 μM was accompanied with low incidence of apoptosis (Fig. S3, ESI†). At next concentration levels of 10 and 30 μM, arrest at the S phase became dominant alteration followed by gradually reduced percentage of cells at the G2/M phase. This switch in cell cycle arrest was accompanied with increased occurrence of apoptotic death. However, rise in percentage of cells at the G0/G1 phase, which started at 50 μM, coincided with decreased percentage of apoptosis, especially in the sample treated with concentration of 75 μM. While treatment of THP-1 cells with S6 triggered accumulation at the S phase followed by an increased rate of apoptosis (Fig. 1A and S1, ESI†), in AsPC-1 cells, the same treatment induced the reversed outcome (Fig. 1B and S3, ESI†). At the concentration range from 10 to 50 μM S6 inducted a modest percentage of AsPC-1 cells into apoptosis, whereas at concentrations of 75 and 100 μM it resulted in the highest apoptotic response but did not cause any significant alteration in cell cycle distribution when compared to non-treated control.
The classical concept of concentration–response relationship indicates that the interaction between applied drug concentration and caused effect in treated biological model is assumed sigmoidal. Sigmoidal or threshold model of concentration–response curve implies that drugs at low concentrations do not have significant biological effect, while with rise of concentrations the effect linearly increases till the saturation is reached. Increasing number of evidences emerge that threshold model of concentration-related response does not adequately explain all aspects of drug activity. Instead, biphasic or hormetic concentration–response model has been shown as far more appropriate to describe pharmacodynamics features of many drugs used for the treatment of cancer.55–57
Despite a large base of published evidences that support biphasic model, to our best knowledge there are only few studies dealing with the underlying cause of non-sigmoidal response, and both had docetaxel as the subject of investigation.58,59 Hermandez-Vargas et al.59 were following changes in cell death, cell cycle, and gene transcription in MCF-7 and MDA-MB-231 breast cancer cell lines after treatment with docetaxel. Their results revealed that docetaxel targeted different phases of cell cycle according to drug concentration, whereas a biphasic growth–response curve was in consistence with the existence of at least two different mechanisms of docetaxel's activity. Later on, another group of authors concluded that biphasic dose–response gained after treatment of prostate cancer cell lines with docetaxel was occurred due to mitotic catastrophe at low concentrations, while apoptosis was the cause of cell death at concentrations higher than 10 μM.58 Additionally, Szomolay and Shahrezaei55 proposed a model where alterations in phosphorylation–dephosphorylation cycles due to different speeds of phosphatase and kinase activity can result in a variety of dose–response behaviors.
In the current study, we report overlapping and coincidental variations in cell cycle changes followed by altered intensity in apoptotic responses, characterized by biphasic concentration–response curves for compounds S1 and S4. Other investigated compounds displayed as well concentration–dependent rearrangements of cells within mitotic division, but those were not accompanied with oscillations in apoptotic response and consequential biphasic curve. At the present, we cannot rationally propose bio-molecular reasons for the described results, but we can undoubtedly state that our compounds targeted different phases of cell cycle depending on their concentration. According to those facts, our compounds can be indicated as those with polypharmacological profiles, which implies on a single drug that is acting on multiple cellular targets.60 This type of drugs offers several advantages over standard drug combination and the main of them include easer dose titration and evasion of serious drug interactions regarding their toxicological profiles, metabolism and excretion. Therefore, recognition of biological molecules targeted by current compounds will be the subject of our further investigations.
Evident correlation between structure and degree of caspase dependency of apoptotic process in THP-1 cells induced by the treatment with investigated compounds was found in two pairs of mono- and corresponding bis-TCHs (Fig. S4A, ESI†). There were almost the same percentages of inhibited apoptotic events by co-incubation with Z-VAD-fmk between counterparts S3 and S4 (25 ± 8% and 24 ± 8%, respectively), as well as between S5 and S6 (45 ± 12% and 42 ± 12%, respectively), together with strong compliance regarding proportions of inhibited early and late apoptotic events. However, while in the case of S5 and S6 similarity was evident also in relation of inhibited incidence of necrotic deaths (34 ± 4% and 42 ± 12%, respectively), Z-VAD-fmk in the samples subjected to S3 drastically increased the percentage of necrotic events compared to the samples treated with S4 (−221 ± 65% and −4.0 ± 0.6%, respectively). The only pair of mono- and bis-TCHs that did not show a strong equivalence regarding dependency in caspases activities and apoptosis is S1 and S2. While co-incubation with Z-VAD-fmk and S2 resulted with poorly reduced incidence of apoptotic cells (28 ± 14%), the addition of pan-caspase inhibitor almost completely inhibited apoptosis promoted by the treatment with S1 (81.2 ± 0.3%). Incompatibility regarding induction of caspases activation between these two compounds was also determined concerning influence of Z-VAD-fmk on the frequency of necrotic events. It is obvious that contrary to hydroxyl derivate S5, another two mono-TCHs, S1 and S3, have substantially challenged survival of THP-1 cells thus those had to end in necrosis, which was not the case with their bis-TCH counterparts.
Interestingly, apoptosis in AsPC-1 cells due to the treatment with S2 revealed as more caspase-dependent (43 ± 16%) compared to its effect on the THP-1 cells, with necrosis almost equivalently reduced in both cell lines by the treatment with Z-VAD-fmk (Fig. S4B, ESI†). Quite contrary, process of apoptotic death triggered by S6 in AsPC-1 cell line was less caspase-dependent (32 ± 4%) than in THP-1 cells, together with slightly stimulated necrotic events. These results indicate those two compounds most probably drove THP-1 and AsPC-1 cells to apoptotic death by different mechanisms, or in other words, their activities were phenotype specific.
Strong correlation in apoptosis caspase-dependency found between mono- and bis-TCHs has been repeated in the assay of caspase-8 and -9 activation. The most impressive is almost the same pattern in assortment of cells with activated calspase-8, -9, or both between samples treated with S3 and S4 (Fig. 2A). It is important to accentuate that these two compounds significantly suppressed caspase-9 activation, which strongly indicates that apoptosis induced in THP-1 cells by S3 and S4 has not been driven by intrinsic apoptotic pathway. Prominent similarity was clearly obvious for another pair of mono- and bis-TCHs. Namely, S1 and S2 were the only compounds that displayed diverse level of caspase-dependency on THP-1 cells (Fig. S4A, ESI†), while here both induced significant increase in percentage of cells with activated caspase-8 only and moderately reduced percentage of cells with single activated caspase-9 (Fig. 2A). The addition of hydroxyl group on aromatic ring resulted in a slight difference in caspases' activation between samples treated with mono- and bis-TCHs S5 and S6 respectively. Compound S5 was the only one not markedly increasing the percentage of cells with activated caspase-8, but rose proportion of cells with single activated caspase-9 although not significantly compared to non-treated control.
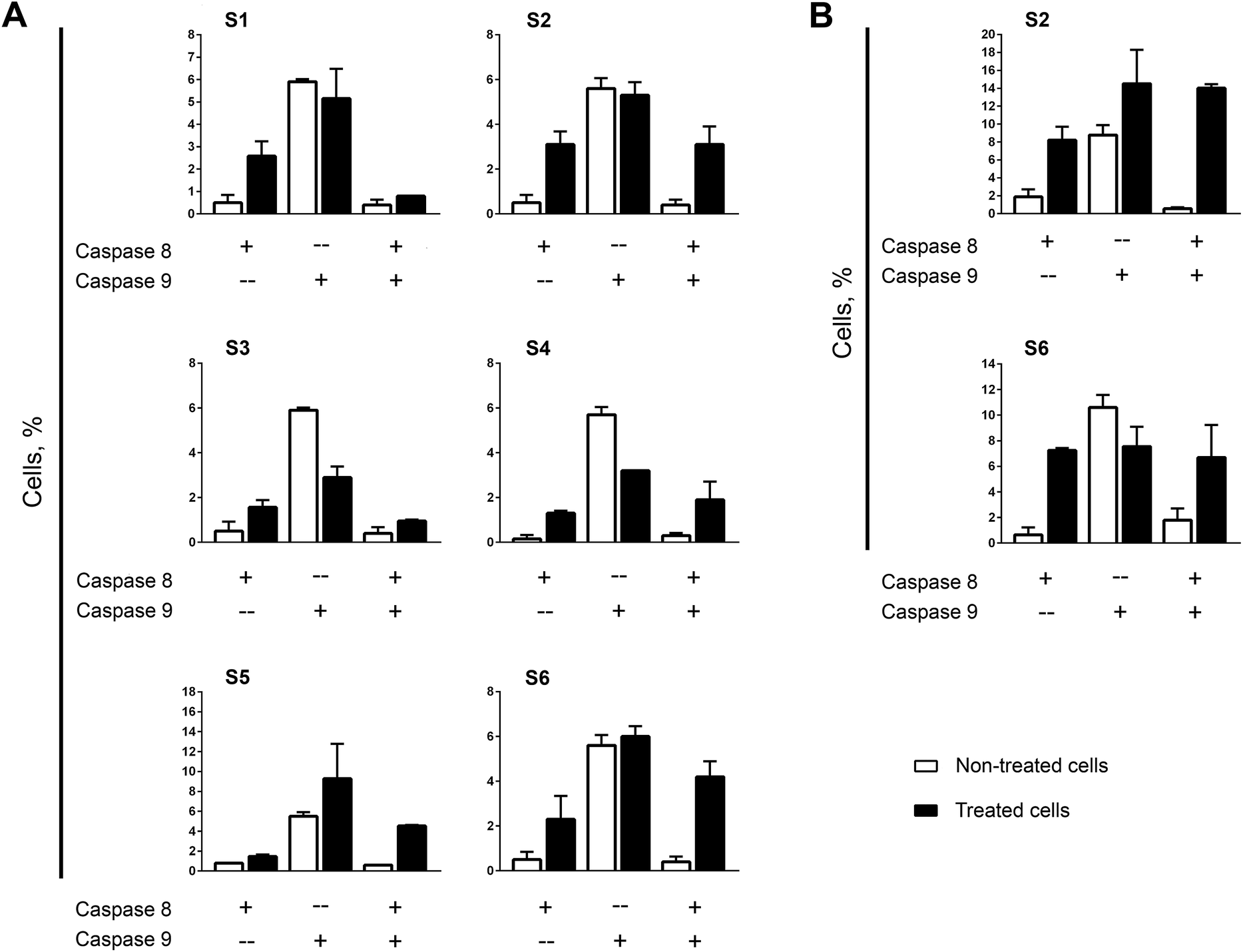 | ||
| Fig. 2 Induction of caspase-8 and -9 activation in THP-1 cells (A) and AsPC-1 cells (B), determined after 6 h incubation with investigated compounds applied at their ED50 concentrations. Results are presented as the mean ± SD percentage of cells that expressed activated caspase-8 only, caspase-9 only, and both caspases of two replicates from independent experiments. | ||
However, another repeating occurrence became evident when comparing the effects of mono- and bis-homologs: in all samples subjected to bis-TCHs, the percentages of cells expressing both caspase-8 and -9 were considerably higher compared to samples treated with their mono-counterparts (Fig. 2A). A cross talk that exists between extrinsic and intrinsic apoptotic pathways allows them to activate each other. Thus, active caspase-9 cleaves and further activates caspase-3 that subsequently in a feedback amplification loop activates caspase-8.63 Otherwise, activated caspase-8 via bid processing propels mitochondrial outer membrane permeabilization with cytochrome c release. The latter is involved in formation of apoptosome together with apoptotic protease activating factor 1 (APAF1), ATP and pro-caspase-9, which results in the activation of caspase-9.61 As was reviewed above, with exception of compound S5, all mono- and bis-TCHs primarily triggered activation of caspase-8, but the percentages of cells with both activated caspases in samples treated with S1 and S3 were on the level of non-treated control that makes quite possible these two ligands interfered with the cross-talk activation of caspase-9. On the other hand, in THP-1 cells treated with S5 and S6, almost the same percentages of cells with both active caspases were determined, while in cells subjected to S5 caspase-8 was activated subsequently to caspase-9.
Likewise assayed in THP-1 cells, activation of caspase-8 dominates in AsPC-1 samples treated by bis-TCHs S2 and S6 (Fig. 2B). However, S2 in AsPC-1 cells also initiated activation of caspase-9 independently of caspase-8, while S6 reduced caspase-9 activity compared to non-treated control. These results reinforce the assumption stated above on phenotype specific mechanisms of S2 and S6 activities.
Mitochondria are a significant source of ROS, together with endoplasmic reticulum and peroxisomes.70 Here, we have investigated pro-oxidant properties of all our compounds that induced apoptotic cell death in THP-1 and AsPC-1 cell lines, by following their ability to induce superoxide formation in mitochondria using MitoSOX Red fluorescent tracker. Although selectivity of MitoSOX Red toward superoxide radicals has been questioned by Zielonka and colleagues,71,72 other authors demonstrated its specificity and confirmed ability for site-specific detection of mitochondrial superoxides.73,74
Basal production of ROS was very similar in the both cell lines when expressed as percentage of ROS-producing cells, (5.4 ± 0.6% for THP-1 and 8.4 ± 0.3% for AsPC-1). In THP-1 cells, considerably higher pro-oxidative activity was obtained after mono-TCHs S1 and S3 treatments as compared to their bis-structural analogues S2 and S4 (Fig. 3A). Less striking discrepancy in ROS-generating ability was revealed for S5 and S6. Considering structural features, S1, S3 and S5 have highly reactive –NH–NH2 group that is probably in relation to their increased pro-oxidant capacity. On the other hand, –OH group attached to the quinoline ring of S6 contributed to a ROS-generating capacity in comparison to other bis-TCHs S2 and S4. However, two hydroxyl groups in S6 could not raise the ROS-producing potential to the level of mono-TCHs. Moreover, quite intriguing results were obtained with treatment of AsPC-1 cells by S2 and S6 as they were the only TCHs that revealed pro-apoptotic activity on this cell line (Fig. 2B). While there was almost no difference comparing percentages of ROS-positive cells in THP-1 and AsPC-1 samples treated with S6, treatment of AsPC-1 cells with S2 resulted in significantly higher amplitude of ROS production than in THP-1 cell line. This particular outcome with S2 cannot be put in relation to its structure, but only as another proof for phenotype-specific activity with no rational explanation that can be given so far.
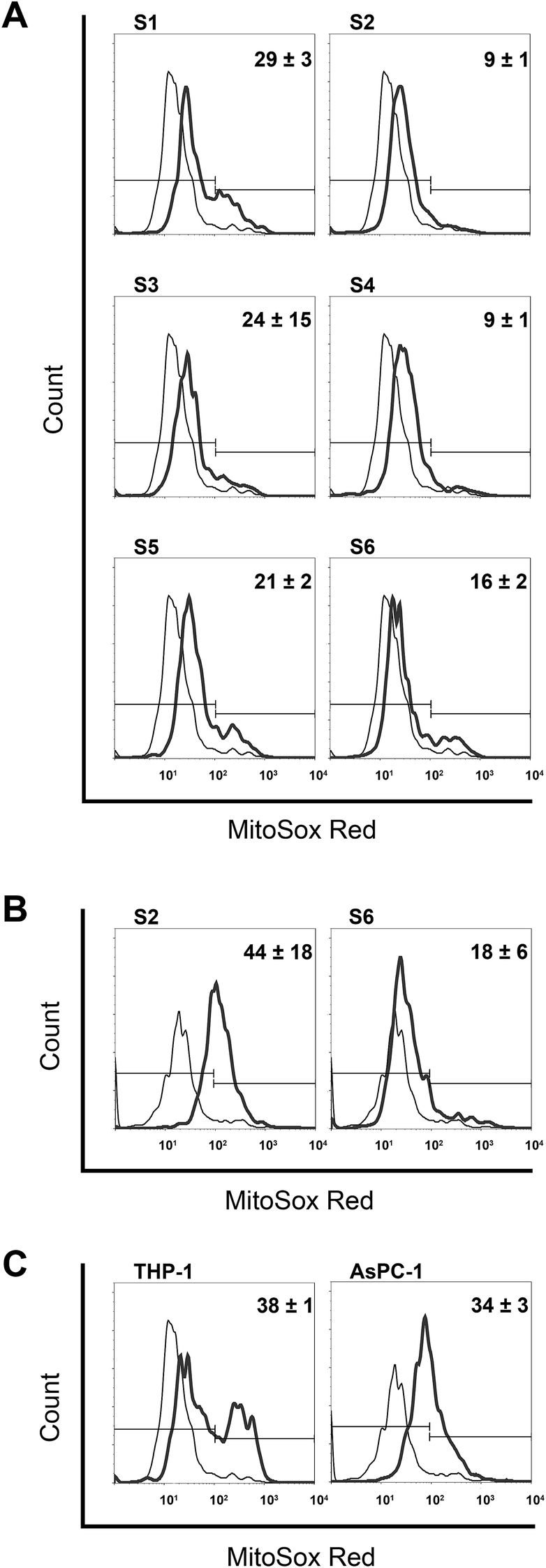 | ||
| Fig. 3 Mitochondrial superoxide radical production in THP-1 cells (A), AsPC-1 cells (B), and in both cell lines subjected to O2 (C). All investigated compounds were added in concentration of 50 μM during 6 h of incubation. Histograms present distribution of MitoSox Red positive cells in non-treated controls (thin line) and treated cells (thick line) with the mean ± SD percentage of cells positive for ROS in two treated replicates from independent experiments. | ||
Although O2 was excluded from detailed analysis on mechanisms involved in apoptotic death, we chose here to explore its pro-oxidant activity and compare these results with those of its structural analogue S2. In THP-1 cells O2 induced a four-fold higher ROS production than S2 (Fig. 3A and C). From the point of structure–activity relationship, oxygen placed in O2 instead of sulfur in S2 may be considered as the cause for increased pro-oxidant activity of O2. On the other hand, O2, as well as S6, gave the equivalent result on AsPC-1 and THP-1 cells (Fig. 3A–C). This outcome additionally underlines phenotype-specific pro-oxidant activity of S2, which deserves further detailed experimental analysis. Furthermore, by comparing mitochondrial ROS-generating activity and apoptotic responses in THP-1 and AsPC-1 cell lines (Fig. S1 and S3, ESI†), the lack of clear correlation between these two cellular events become obvious. Compounds S2, S4 and S6, which were strong pro-apoptotic inducers compared to their mono-TCHs counterparts, reveal here less pro-oxidant activity. Compound O2 inducing high percentage of ROS-positive cells on both cell lines triggers necrotic cell death and not apoptosis. Finally, significant discrepancy in mitochondrial ROS production found in THP-1 and AsPC-1 cells treated with S2, is in reverse proportion with incidence of apoptotic death. The only interdependency in relation of high ROS generation was seen in AsPC-1 cells treated with S2 that also displayed accelerated activation of caspase-9, previously confirmed as associated mechanisms by which oxidative stress initiates apoptotic death.75 Prevailing activation of caspase-8 in the majority of investigated samples (Fig. 2), together with the fact that treatment with S2 induced poor apoptotic response in AsPC-1 cells, strongly indicates that mitochondrial ROS formation did not play an important role in triggering apoptosis in THP-1 and AsPC-1 cells.
AsPC-1 cells were incubated over 72 h with low concentrations of the investigated compounds, and afterwards stained with anti-CD44s-FITC in order to follow possible changes in expression of this stemness CSCs surface marker.76,77 CD44 is a transmembrane glycoprotein that serves as a receptor for various components of extracellular matrix.78,79 Its most important roles include intermediation in cell–cell and cell–matrix interactions, as well as cancer cell migration. Thus, CD44 binding regulates CSCs survival, self-renewal, and chemoresistance.79,80 Our experimental results were analyzed regarding two parameters: percentage of positive CD44 cells, and the median fluorescent intensity (MFI) expressed in arbitrary units (AU) for CD44-positive cells subgroup. Thus computed MFI represents median incidence of CD44 expression per cell, irrespectively of size of CD44-positive population.
In non-treated AsPC-1 samples, 89 ± 3% of cells were expressing CD44 with a MFI of 3416 ± 43 AU. In cells subjected to S2 and S6 at concentration of 10 μM, a slight reduction of the number of CD44-positive subpopulation was recorded (Fig. S5A and B, ESI†). This effect was accompanied with decrease in MFI to 2918 ± 91 AU and 2396 ± 167 AU for the samples treated with S2 and S6, respectively. These changes in MFI signify that the applied treatments down-regulate the number of CD44 receptors per cell. At concentration of 1 μM, S2 and S6 significantly dropped the percentages of CD44-positive cells. Compound S6 accomplished the best response, whereas MFI values were still a bit higher (1876 ± 120 AU) compared to samples treated with S2 (1711 ± 42 AU). On the contrary, compound S4, the one that did not achieve apoptotic response in AsPC-1 cells, now revealed an ability to induce their differentiation with inverse concentration-dependency matching to the two other compounds (Fig. S5C, ESI†). It is interesting to note that S4 at 10 μM accomplished the greatest down-regulation of CD44 expression according to MFI values (1522 ± 48 AU). These results suggest that our compounds may have the ability to stimulate reprogramming of AsPC-1 cells independently of any pro-apoptotic activity, and thus should be further studied by following the expression of other CSC phenotype markers like Oct4, Nanog or Sox2.81
Activities of four compounds, which did induce cell death in the 2D AsPC-1 model irrespectively of apoptosis or necrosis, were evaluated (Fig. S3, ESI†). The 3D spheres were treated for eight days, and compounds were tested in three concentrations (1, 10 and 100 μM). Here are reviewed only results for concentrations that at the day 8 achieved a reduction of spheroid size when compared to day 0 (growth rate < 1, Fig. 4).
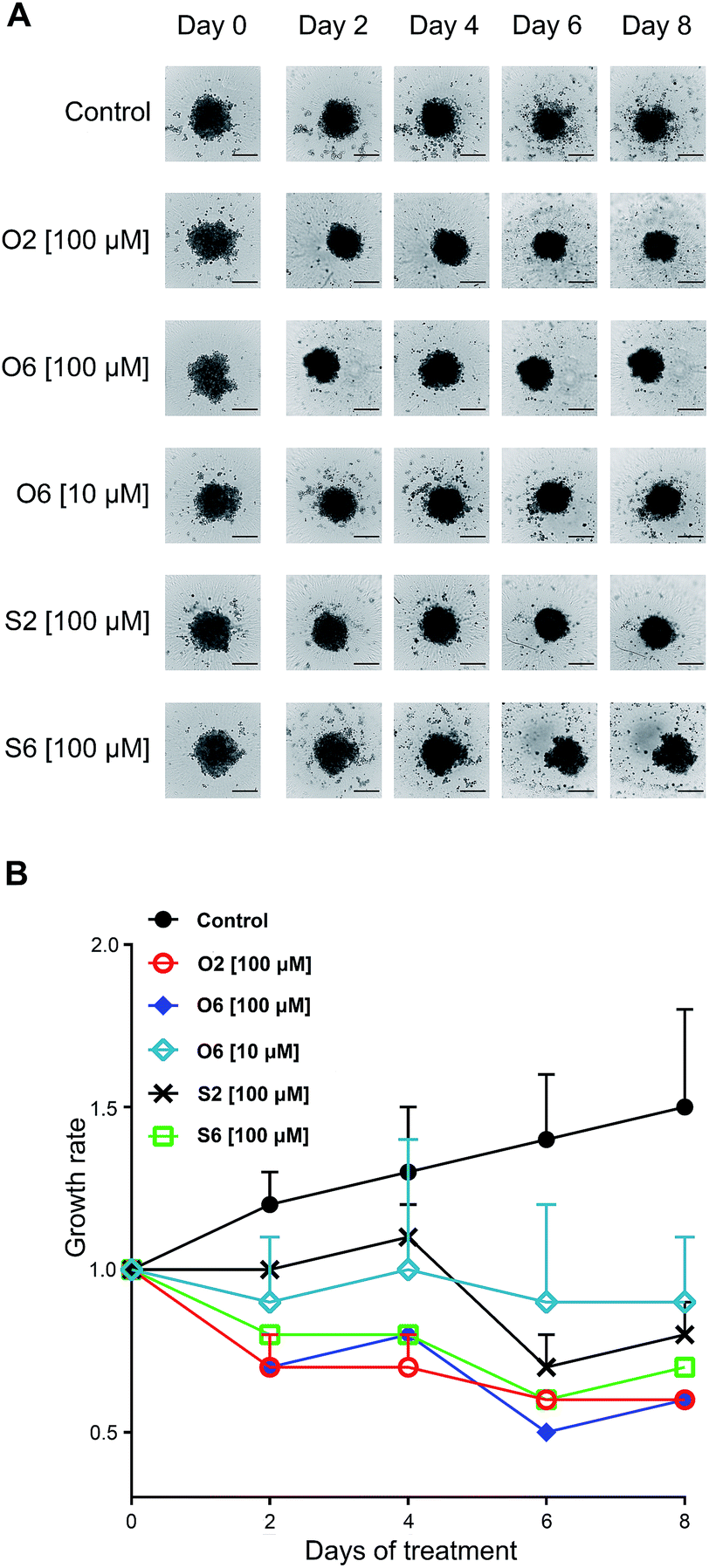 | ||
| Fig. 4 Changes in size and morphology of AsPC-1 3D culture over the 8 day treatment period (A). Images have been acquired every other day, starting from day 0 on the Celigo imaging cytometer using Celigo software. Scale bar: 500 μm. Growth rates of non-treated and treated AsPC-1 spheroids over the time of experiment (B). | ||
Growth of non-treated spheres recorded a linear rate, with a maximal increase of 1.5 ± 0.3 fold at day 8. Compound O6 was the only one effective in two evaluated concentrations. While at concentration of 10 μM O6 successfully inhibited growth of tumor mass, at 100 μM it reduced tumor mass to a half of its initial size after six days of incubation. The second most successful treatment was at 100 μM for O2 as the only compound which activity was described with a steadily downhill growth rate curve, with the tumor sizes at the day 8 equal to those subjected to O6 at 100 μM. It is important to notice that both O6 and O2 were withdrawn from further investigations due to results of Annexin V/PI staining, considering both of them induced necrotic death, a situation that in vivo induce inflammation thus not suitable for any putative drug. Furthermore, in 2D model samples treated with O2, at the same concentration, more than 89% of cells were double-stained, while O6 was relatively less efficient. Similarly to those treated with O2, spheres exposed to S6 were continually losing size until day 6. The most surprising was the activity of S2, a compound which on 2D model at 100 μM triggered apoptotic death in less than 25% of the population. On 3D culture, S2 displayed a delayed response, which was documented with slight gain in size of treated spheres during the first four days, that was followed by significant collapse of their masses undoubtedly caused by significant incidence of cell death. Therefore, for S2, according to results on 2D model, it might be proclaimed as compound of no interest regarding treatment of AsPC-1 cells. Here S2 revealed as almost equally effective as tumor growth inhibition of S6. This result is an excellent testimonial for the use of 3D cultures in drug screening as it is more accurate and closer to in vivo situation.
Interaction between S2 and human serum albumin (HSA)
HSA is well known for its ligand binding capacity and repository for an extraordinarily diverse range of molecules which makes it an important factor in the pharmacokinetic behavior of many drugs by affecting their efficiency and rate of delivery. Its feature to accumulate in solid tumors raises interest of medicinal chemists to explore ability of newly synthesized compounds to bind to and use HSA as a carrier, and yields the rationale for developing the albumin-based drug delivery systems for tumor targeting drugs.84 Here we explored performed experimental and docking analysis protocol to obtain information on selected TCH S2 interaction with HSA.Criterions used to choose the compound for evaluation of HSA binding were pro-apoptotic activity on both investigated cell lines and lipophilicity as one of the most important factors involved in the extent of drug binding.85 Although log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P was argued as not reliable physicochemical descriptor to distinguish strong and weak binders among diverse ligands,86 its higher value defined for S2 was the rational to investigate its binding properties rather than for S6 (Table S5, ESI†). Binding of S2 to HSA was studied by UV/Vis spectroscopy. After the addition of HSA to the solution of S2, blue shift of absorption maximum was observed (Fig. 5). More details about the HSA–S4 binding were obtained from fluorescence titration experiments and molecular docking.
P was argued as not reliable physicochemical descriptor to distinguish strong and weak binders among diverse ligands,86 its higher value defined for S2 was the rational to investigate its binding properties rather than for S6 (Table S5, ESI†). Binding of S2 to HSA was studied by UV/Vis spectroscopy. After the addition of HSA to the solution of S2, blue shift of absorption maximum was observed (Fig. 5). More details about the HSA–S4 binding were obtained from fluorescence titration experiments and molecular docking.
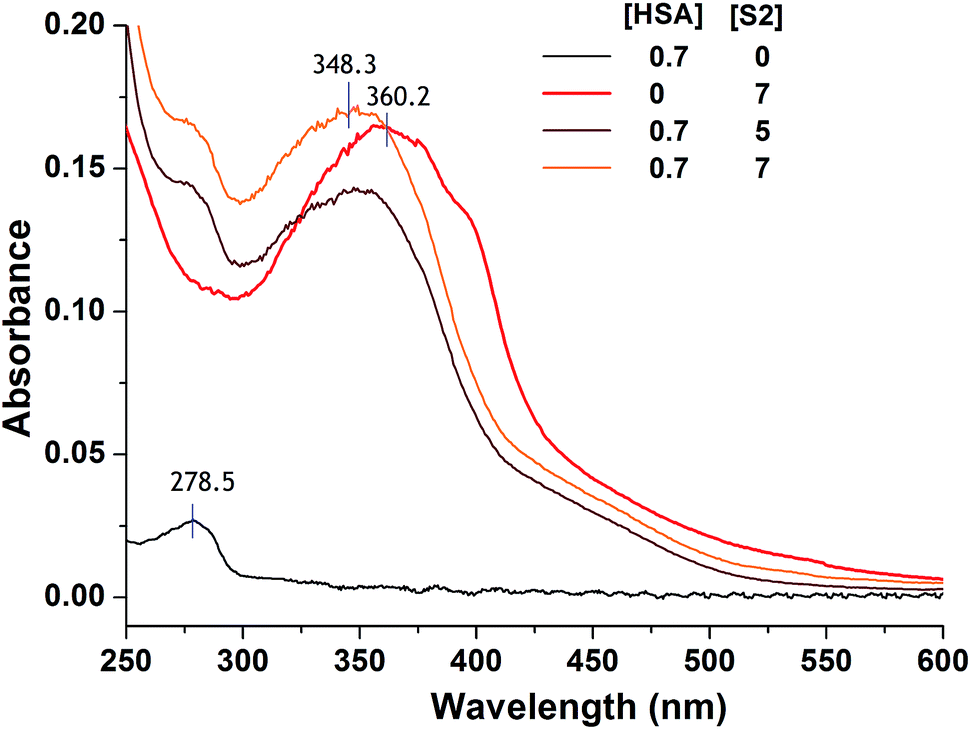 | ||
| Fig. 5 Changes in absorption spectrum of S2 after addition of HSA. All concentrations are given in μM. Spectra are recorded in 30 mM phosphate buffered saline (PBS), pH 7.36. | ||
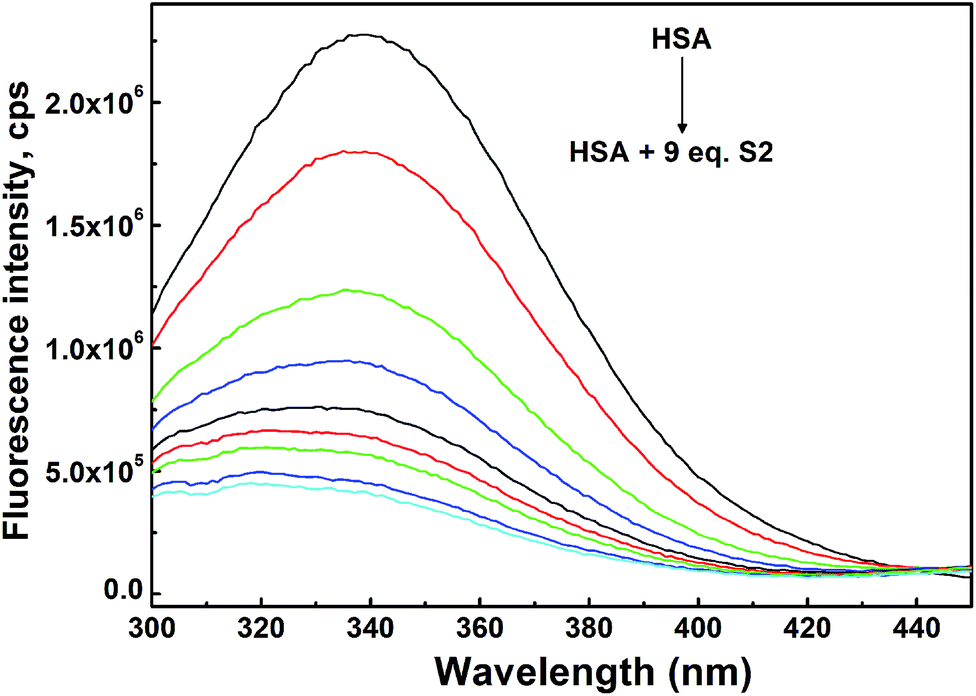 | ||
| Fig. 6 Changes in fluorescence emission spectra of HSA (c = 7 × 10−7 M) upon addition of S2 in following concentrations: (0.0; 0.35; 0.7; 1.4; 2.1; 2.8; 3.5; 4.9, 6.3) × 10−6 M; T = 310 K; 30 mM PBS, pH = 7.4. | ||
With the increasing amount of quencher, results may deviate from the linearity due to instrumental inner filter effect. This effect was removed applying Lakowicz procedure, eqn (1).
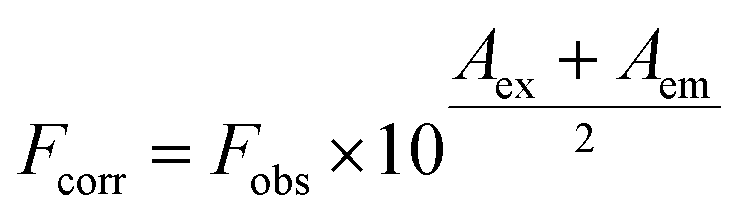 | (1) |
Fluorescence data were processed using double log plot, according to the eqn (2).
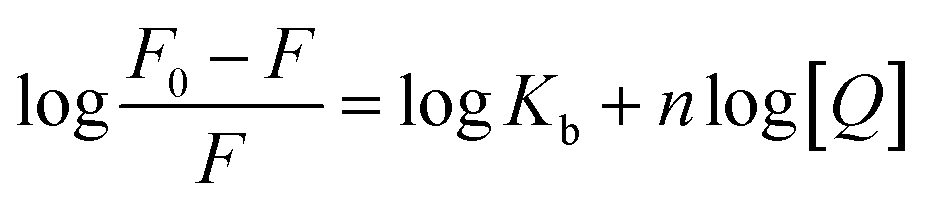 | (2) |
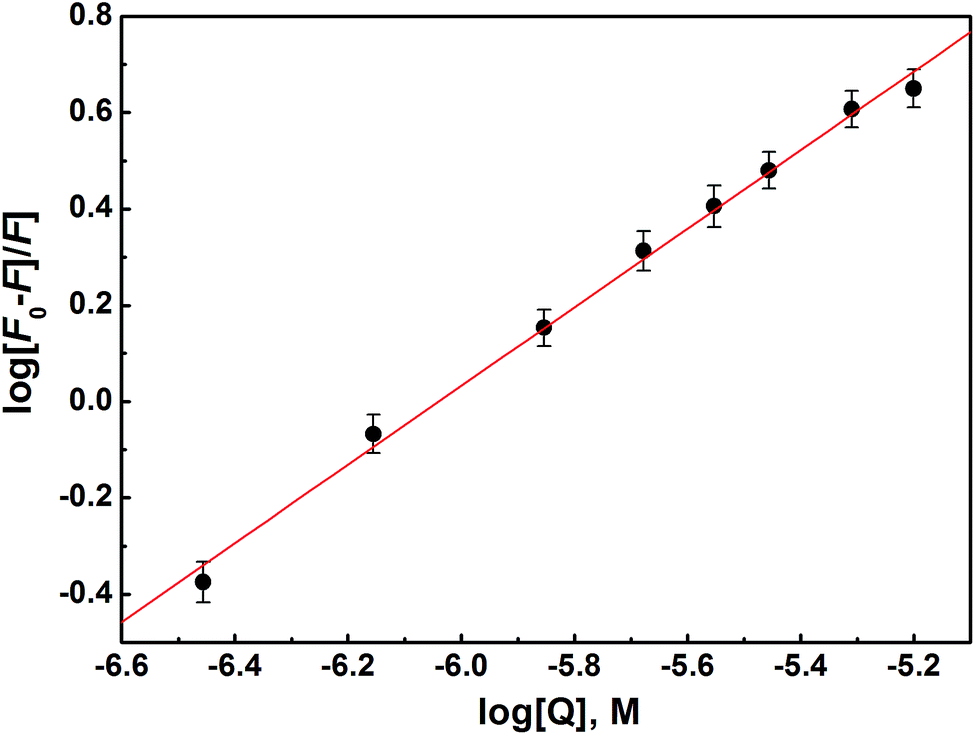 | ||
| Fig. 7 Double-log plot for determination of binding constant Kb, and the number of binding sites n for HSA–S2 interaction. | ||
| T (K) | K b (M−1) × 104 | logKb |
n | r 2 |
|---|---|---|---|---|
| 293 | 8.5 ± 2.2 | 4.9 ± 0.1 | 0.82 ± 0.02 | 0.995 |
| 301 | 6.1 ± 2.8 | 4.8 ± 0.2 | 0.78 ± 0.04 | 0.986 |
| 310 | 4.6 ± 0.8 | 4.7 ± 0.1 | 0.77 ± 0.01 | 0.998 |
Fluorescence quenching is classified as static or dynamic. Dynamic quenching is highly dependent upon diffusion. Higher temperatures result in faster diffusion and hence larger values for biomolecular quenching constant. On the other hand, higher temperatures will typically result in the dissociation of weakly bound complexes, and therefore decrease the biomolecular quenching constant in static process. The Kb value decreases with the increase of temperature, which is an indication of static quenching mechanism.
Compound S2 binds to approximately one binding site, as suggested with the value of n. The logKb is in the range optimal for biologically active compounds.88–90 These results suggest that S2 strongly binds to HSA, so it can be effectively carried and stored in the human body.
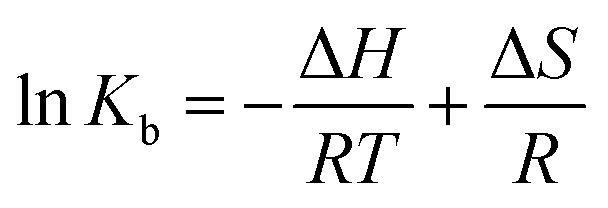 | (3) |
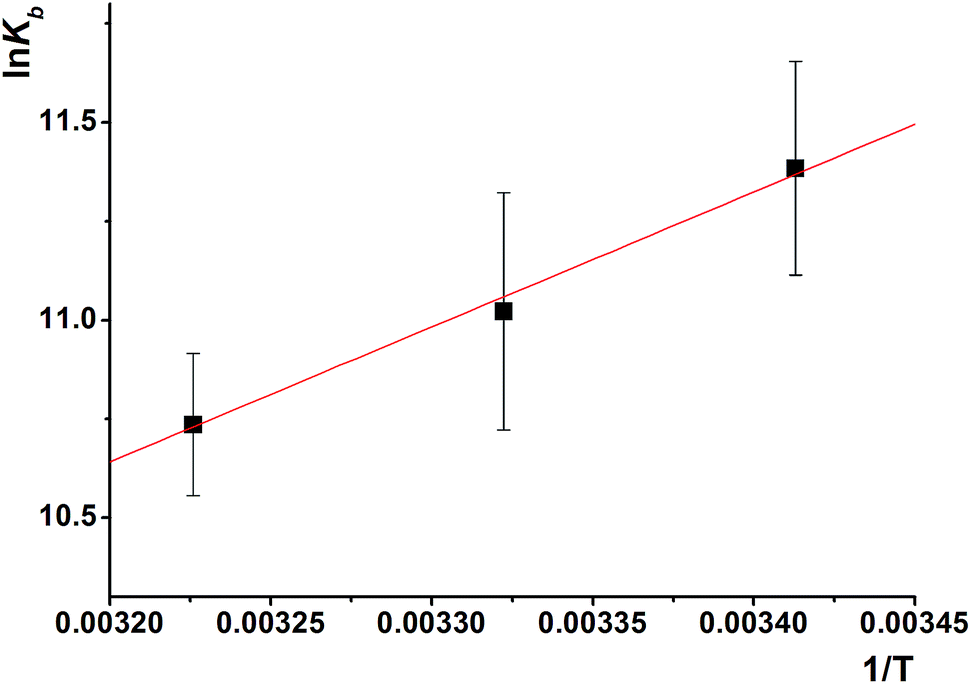 | ||
| Fig. 8 The plot of lnKbvs. 1/T for the interaction of S2 with HSA. | ||
| T (K) | lnKb (M−1) |
ΔG (kJ mol−1) | ΔH (kJ mol−1) | ΔS (J mol−1 K−1) |
|---|---|---|---|---|
| 293 | 11.38 | 27.731 | 28.731 | −3.55 |
| 301 | 11.02 | 27.584 | ||
| 310 | 10.74 | 27.670 |
According to Ross' view,91 the signs and magnitudes of thermodynamic parameters for protein reactions can account for the main forces contributing to protein stability. From the thermodynamic stand-point, ΔH > 0 and ΔS > 0 implies a hydrophobic interaction, ΔH < 0 and ΔS < 0 reflects the van der Waals force or hydrogen bond formation and ΔH < 0 and ΔS > 0 suggesting an electrostatic force.
The negative enthalpy and negative entropy indicate that the van der Waals forces or hydrogen bonding are dominant for HSA–S2 binding. Process is enthalpically driven, and hydrogen bonds between HSA and S2 are the most important intermolecular forces.
The most favorable docking solution for each binding site is shown in Fig. 9. S2 has many polar interactions within the site IIA of HSA. It makes hydrogen bonds with the residues His242 (at distance 3.3 Å), Arg222 (2.3–2.45 Å), Lys199 (2.8 Å) as well as Trp214 (3.2 Å). Hydrophobic and/or π–π stacking interactions between AB39 and Phe211 and Trp214 are also possible. Lesser number of polar interactions are observed for AB39 binding into the sites IB and IIIA.
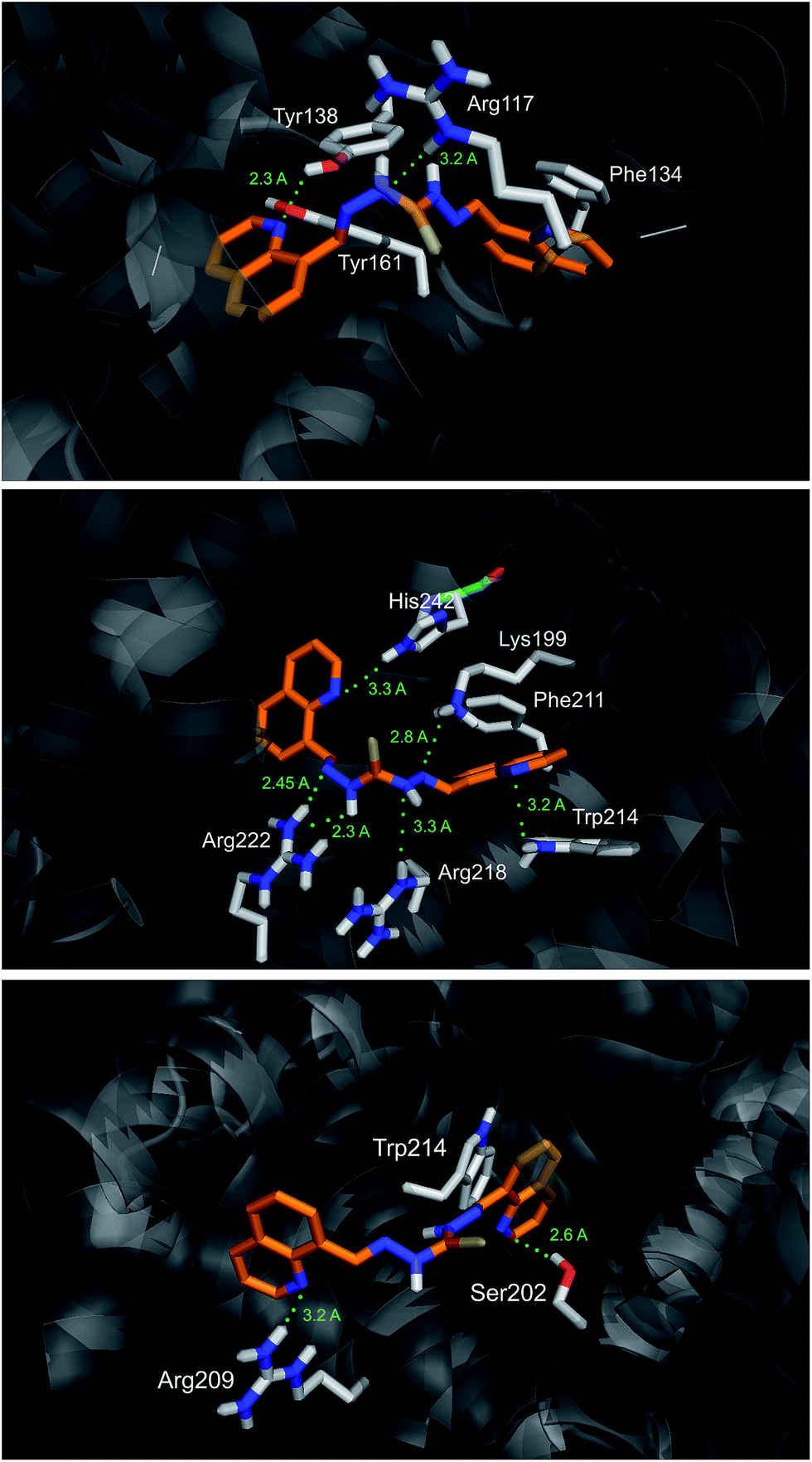 | ||
| Fig. 9 S2 (orange) docked into the site IB (top), IIA (middle), and IIIA (bottom) of HSA. Amino acid residues, which interact with the S2 are shown as sticks and colored by atom. The most important polar interactions are highlighted as green dots, together with corresponding distances. | ||
Binding mode of S2 into the site IIA of HSA confirms the dominant contribution of hydrogen bonding in overall binding energy.
Conclusion
Results of the current study validate superior anti-cancer activity of TCHs over CHs. Used set of biological tests provides preliminary information on mechanism of their activity. Particularly attractive is their ability to target different phases of mitotic division in concentration–dependent manner, which for some compounds was overlapping with alterations in intensity of apoptotic response of treated cells. That information strongly indicate on multi-target activity profile of investigated TCHs. Furthermore, apoptosis induced by these compounds was not exclusively caspase-dependent, while activation of caspase-8 was assayed as the principal trend signifying on endoplasmic reticulum stress or even activation of death receptor signaling as the triggering incident of apoptotic death. Induction of ROS genesis in mitochondria was shown to be trivial accident since there was no obvious correlation between potency in ROS production and intensity of apoptotic response. Among tested compounds, only two bis-TCHs also revealed pro-apoptotic activity on CSCs, while two bis-CHs induced necrotic death on this cell model. However, investigation on 3D spheroidal CSC model that is a superior platform for testing the drug activity compared to standardly used 2D monolayer, gave more accurate perspective on anti-tumor activity of evaluated drugs. According to those data, compound S6 as the only TCH that revealed strong pro-apoptotic activity on AsPC-1 2D model, showed on spheroids a sustained antitumor activity with significantly reduced size of treated spheres compared to their initial volume. Moreover, compound S6 drastically reduced percentage of CD44 positive CSCs over the 3 day incubation time, and therefore displayed its ability to act also as a pro-differentiation agent at low concentrations levels. Those results strongly encourage additional study of mono- and bis-TCHs, with mechanism of caspase-8 activation as the primary interest in any further investigations.Experimental
Reagents and instrumentation
2-Quinolinecarboxaldehyde (≥98%), thiocarbohydrazide (98%) and carbohydrazide (≥98%) were obtained from Sigma. 8-Quinolinecarboxaldehyde (98%) and 8-hydroxy-2-quinolinecarboxaldehyde (98%) were obtained from Acros Organics. All solvents (reagent grade) were obtained from commercial suppliers and used without further purification.Elemental analyses (C, H, N, S) were performed by the standard micromethods using the ELEMENTAR Vario EL III CHNS/O analyzer. Fourier-transform infrared (FTIR) spectra were obtained using FTIR BOMEM MB 100 in the form of KBr pellets. FTIR spectra (Fig. S5 and S6, ESI†) were recorded in the transmission mode between 400 and 4000 cm−1 with a resolution of 4 cm−1. Abbreviations used for IR spectra: vs, very strong; s, strong; m, medium; w, weak. All NMR spectral measurements were performed on a Bruker Avance III 500 spectrometer equipped with a broad-band direct probe. The spectra were recorded at room temperature in DMSO-d6. Chemical shifts are given on δ scale relative to tetramethylsilane (TMS), as internal standard for 1H and 13C. Coupling constants (J) were expressed in Hz. Abbreviations used for NMR spectra: s, singlet; dd, doublet of doublets; ddd, double double doublet. 1D (1H and 13C) and 2D (COSY, NOESY, 1H–13C HSQC and 1H–13C HMBC) spectra of all compounds are shown in Fig. S8–S79 (ESI†) together with the atom numbering scheme (Table S1, ESI†).
Synthesis of compounds
Example: 8-quinolinealdehyde carbohydrazone (O1). Solution of 8-quinolinecarboxaldehyde (0.0785 g, 0.5 mmol) in ethanol (10 mL) was added into hot solution of dhO (0.0901 g, 1 mmol) in ethanol (40 mL). After the catalytic amount of conc. hydrochloric acid was added, reaction mixture was stirred and refluxed for 3 h. The resulting precipitate was collected by filtration and washed successively with cold ethanol and diethyl ether. Yellow solid was recrystallized from absolute methanol. Yield: 0.0733 g (64%). Mp 185 °C. Elemental analysis calcd for C11H11N5O (Mw = 229.24 g mol−1): C, 57.63; H, 4.84; N, 30.55%; found: C, 57.71; H, 4.78; N, 30.62%. IR (KBr, cm−1) νmax: 3316s (NH2), 3200s (NH), 3096m (CHaryl), 1681vs (C
O), 1621m (CN); 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 4.12 (s, 2H, H–N5), 7.57 (dd, 1H, H–C3, 3J3,4 = 8.3 Hz, 3J3,2 = 4.1 Hz), 7.63 (t, 1H, H–C6, 3J6,5 = 3J6,7 = 7.4 Hz), 7.98 (dd, 1H, H–C5, 3J5,6 = 7.8 Hz, 4J5,7 = 1 Hz), 8.16 (s, 1H, H–N4), 8.39 (dd, 1H, H–C4, 3J4,3 = 8.3 Hz, 4J4,2 = 2.0 Hz), 8.58 (d, 1H, H–C7, 3J7,6 = 7.4 Hz), 8.94 (dd, 1H, H–C2, 3J2,3 = 4.1 Hz, 3J2,4 = 2.0 Hz), 9.14 (s, 1H, H–C9), 10.65 (s, 1H, H–N3). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 121.67 (C3), 125.61 (C7), 126.45 (C6), 127.94 (C4a), 128.90 (C5), 131.59 (C8), 136.55 (C4), 136.89 (C9), 145.01 (C8a), 150.08 (C2), 157.21 (C10).
2-Quinolinealdehyde carbohydrazone (O3). O3 was synthesized similarly to O1, starting from 2-quinolinecarboxaldehyde and dhO. White solid was recrystallized from absolute methanol. Yield: 56%. Mp 183 °C. Elemental analysis calcd for C11H11N5O (Mw = 229.24 g mol−1): C, 57.63; H, 4.84; N, 30.55%, found: C, 57.58; H, 4.62; N, 30.69%. IR (KBr, cm−1) νmax: 3297s (NH2), 3188s (NH), 3084m (CHaryl), 1679vs (C
O), 1638m (CN). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 4.15 (s, 2H, H–N5), 7.58 (ddd, 1H, H–C6, 3J6,7 = 8.2 Hz), 7.74 (ddd, 1H, H–C7, 3J7,6 = 8.2 Hz), 7.93–7.99 (br.m.ovlp., 2H, H–C5, H–C8), 8.03 (s, 1H, H–C9), 8.27 (d, 1H, H–C4, 3J4,3 = 8.4 Hz), 8.34–8.46 (br.m.ovlp., 2H, H–C3, H–N4, 3J3,4 = 8.4 Hz), 10.84 (s, 1H, H–N3). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 118.03 (C3), 126.84 (C6), 127.66 (4a), 127.92 (C5), 128.69 (C8), 129.82 (C7), 136.19 (C4), 140.64 (C9), 147.26 (C8a), 154.34 (C2), 156.76 (C10).
8-Hydroxy-2-quinolinealdehyde carbohydrazone (O5). O5 was synthesized similarly to O1, starting from 8-hydroxy-2-quinolinecarboxaldehyde and dhO. Yellow solid was recrystallized from absolute methanol. Yield: 72%. Mp 214–215 °C. Elemental analysis calcd for C11H11N5O2 (Mw = 245.24 g mol−1): C, 53.83; H, 4.525; N, 28.56%; found: C, 53.66; H, 4.68; N, 28.74%. IR (KBr, cm−1) νmax: 3371s (OH), 3335s (NH2), 3198s (NH), 3049m (CHaryl), 1696vs (C
O), 1600m (CN). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 4.14 (s, 2H, H–N5), 7.08 (dd, 1H, H–C7, 4J7,5 = 1.4 Hz), 7.36 (dd, 1H, H–C5, 4J5,7 = 1.4 Hz), 7.41 (m, 1H, H–C6), 8.09 (s, 1H, H–C9), 8.24 (d, 1H, H–C4, 3J4,3 = 8.55 Hz), 8.30–8.50 (br.m.ovlp., 2H, H–C3, H–N4, 3J3,4 = 8.55 Hz), 9.71 (s, 1H, OH), 10.88 (s, 1H, H–N3). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 111.59 (C7), 117.74 (C5), 118.35 (C3), 127.73 (C6), 128.52 (C4a), 136.06 (C4), 137.93 (C8a), 140.50 (C9), 152.25 (C2), 153.24 (C8), 156.83 (C10).
8-Quinolinealdehyde thiocarbohydrazone (S1). S1 was synthesized similarly to O1, starting from dhS instead of dhO. Yellow solid was recrystallized from water/ethanol mixture (1
:4, v/v). Yield 75%. Mp 163–164 °C. Elemental analysis calcd for C11H11N5S (Mw = 245.30 g mol−1): C, 53.86; H, 4.52; N, 28.55; S, 13.07%; found: C, 53.75; H, 4.64; N, 28.40; S, 13.22%. IR (KBr, cm−1) νmax: 3438s (NH), 3249w (NH2), 2921m (CHaryl), 1500vs (amide II); 1237m (CS). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 4.91 (s, 2H, H–N5), 7.59 (dd, 1H, H–C3, 3J3,2 = 4.15 Hz, 3J3,4 = 8.35 Hz), 7.65 (t, 1H, H–C6, 3J6,5 = 7.9 Hz, 3J6,7 = 7.25 Hz), 8.03 (dd, 1H, H–C5, 3J5,6 = 7.9 Hz), 8.41 (dd, 1H, H–C4, 3J4,3 = 8.35 Hz), 8.73 (d, 1H, H–C7, 3J7,6 = 7.25 Hz), 8.96 (dd, 1H, H–C2, 3J2,3 = 4.15 Hz), 9.30 (s, 1H, H–C9), 9.95 (s, 1H, H–N4), 11.67 (s, 1H, H–N3). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 121.75 (C3), 126.33 (C7), 126.40 (C6), 127.94 (C4a), 129.59 (C5), 131.21 (C8), 136.59 (C4), 138.97 (C9), 145.30 (C8a), 150.29 (C2), 175.96 (C10).
2-Quinolinealdehyde thiocarbohydrazone (S3). S3 was synthesized similarly to O1, starting from 2-quinolinecarboxaldehyde and dhS. Yellow solid was recrystallized from absolute ethanol. Yield: 56%. Mp 183 °C. Elemental analysis, calcd for C11H11N5S (Mw = 245.30 g mol−1): C, 53.86; H, 4.52; N, 28.55; S, 13.07%; found: C, 53.66; H, 4.48; N, 28.32; S, 13.17%. IR (KBr, cm−1) νmax: 3425m (NH), 3267s (NH2), 2962w (CHaryl), 1500vs (amide II); 1230s (C
S). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 4.97 (s, 2H, H–N5), 7.59 (ddd, 1H, H–C6, 3J6,7 = 8.2 Hz), 7.75 (ddd, 1H, H–C7, 3J7,6 = 8.2 Hz), 7.97 (dd, 2H, H–C5, H–C8), 8.18 (s, 1H, H–C9), 8.34 (d, 1H, H–C4, 3J4,3 = 8.7 Hz), 8.52 (d, 1H, H–C3,3J3,4 = 8.7 Hz), 10.18 (s, 1H, H–N4), 11.78 (s, 1H, H–N3). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 118.00 (C3), 126.75 (C6), 127.46 (4a), 127.57 (C5), 128.40 (C8), 129.57 (C7), 135.89 (C4), 141.98 (C9), 146.97 (C8a), 153.68 (C2), 175.37 (C10).
8-Hydroxy-2-quinolinealdehyde thiocarbonohydrazone (S5). S5 was synthesized similarly to O1, starting from 8-hydroxy-2-quinolinecarboxaldehyde and dhS. Yellow solid was recrystallized from DMF/methanol mixture (1
:9, v/v). Yield 83%. Mp 208–209 °C. Elemental analysis calcd for C11H11N5OS (Mw = 261.30 g mol−1): C, 50.56; H, 4.24; N, 26.80; S, 12.27%; found: C, 50.42; H, 4.34; N, 28.77; S, 12.42%. IR (KBr, cm−1) νmax: 3450s (OH), 3424s (NH), 3165m (NH), 3015m (CHaryl), 1503vs (amide II); 1228s (CS). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 4.96 (s, 2H, H–N5), 7.09 (dd, 1H, H–C7, 3J7,6 = 7.65 Hz, 4J7,5 = 1.1 Hz), 7.37 (dd, 1H, H–C5, 3J5,6 = 7.9 Hz, 4J5,7 = 1.1 Hz), 7.42 (t, 1H, H–C6, 3J6,5 = 7.9 Hz, 3J6,7 = 7.6 Hz), 8.23 (s, 1H, H–C9), 8.27 (d, 1H, H–C4, 3J4,3 = 8.7 Hz), 8.52 (d, 1H, H–C3, 3J3,4 = 8.7 Hz), 9.80 (s, 1H, OH), 10.17 (s, 1H, H–N4), 11.84 (s, 1H, H–N3). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 112.00 (C7), 117.71 (C5), 118.65 (C3), 127.97 (C6), 128.71 (C4a), 136.03 (C4), 138.10 (C8a), 142.16 (C9), 151.95 (C2), 153.36 (C8), 175.72 (C10).
Example: bis-(8-quinolinealdehyde) carbohydrazone (O2). Solution of 8-quinolinecarboxaldehyde (0.33 g, 2.1 mmol) in ethanol (10 mL) was added into hot solution of dhO (0.0901 g, 1 mmol) in ethanol (40 mL). After the catalytic amount of conc. hydrochloric acid was added, reaction mixture was stirred and refluxed for 3 h. The resulting precipitate was collected by filtration and washed successively with cold ethanol and diethyl ether. Yellow solid was recrystallized from absolute methanol. Yield: 0.199 g (54%). Mp 219–220 °C. Elemental analysis calcd for C21H16N6O (Mw = 368.14 g mol−1): C, 68.47; H, 4.38; N, 22.81%; found: C, 68.32; H, 4.91; N, 22.73%. IR (KBr, cm−1) νmax: 3331s (NH), 3055m (CHaryl), 1707vs (C
O), 1614w (CN). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 7.61 (dd, 2H, H–C3H–C17, 3J3,2 = 3J17,18 = 4.1 Hz, 3J3,4 = 3J17,16 = 8.3 Hz); 7.72 (t, 2H, H–C6H–C14, 3J6,5 = 3J14,15 = 7.9 Hz); 8.04 (dd, 2H, H–C5H–C15, 3J5,6 = 3J15,14 = 7.9 Hz); 8.43 (dd, 2H, H–C4H–C16, 3J4,3 = 3J16,17 = 8.3 Hz, 4J4,2 = 4J16,18 = 1.75 Hz); 8.60 (d, 2H, H–C7H–C13); 8.99 (dd, 2H, H–C2H–C18, 3J2,3 = 3J18,17 = 4.1 Hz, 4J2,4 = 4J18,16 = 1.75 Hz); 9.50 (s, 2H, H–C9H–C11); 11.09 (s, 2H, H–N3, H–N4). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 121.77 (C3C17); 125.72 (C7C13); 126.49 (C6C14); 128.02 (C4aC15a); 129.28 (C5C15); 131.65 (C8C12); 136.62 (C4C16); 139.89 (C9C11); 145.19 (C8aC12a), 150.16 (C2C18); 152.28 (C10).
Bis-(2-quinolinealdehyde) carbohydrazone (O4). O4 was synthesized similarly to O2, starting from 2-quinolinecarboxaldehyde and dhO, with a slight modification of previously published procedure.47 White solid was recrystallized from DMF/methanol mixture (1
:9, v/v). Yield 78%. Mp 162–164 °C. Elemental analysis calcd for C21H16N6O (Mw = 368.14 g mol−1): C, 68.47; H, 4.38; N, 22.81%; found: C, 68.81; H, 4.80; N, 22.56%. IR (KBr, cm−1) νmax: 3392s (NH), 3096m (CHaryl), 1708vs (CO), 1630w (CN). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 7.63 (ddd, 2H, H–C6H–C16, 3J6,7 = 3J16,17 = 6.8 Hz, 3J6,5 = 3J16,15 = 8.2 Hz), 7.79 (ddd, 2H, H–C7H–C17, 3J7,6 = 3J17,16 = 6.8 Hz), 8.02 (ddd, 4H, H–C5H–C15, H–C8H–C18, 3J5,6 = 3J15,16 = 8.2 Hz), 8.31 (s, 2H, H–C9H–C11), 8.38–8.60 (br.m.ovlp., 4H, H–C3H–C13, H–C4H–C14), 11.31 (s, 2H, H–N3H–N4). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 117.78 (C3C13), 127.11 (C7C17), 127.79 (C6C16), 127.99 (C4aC14a), 128.84 (C5C15), 130.00 (C8C18), 136.47 (C4C14), 144.06 (C9C11), 147.35 (C8aC18a), 151.67 (C2C12), 153.99 (C10).
Bis-(8-hydroxy-2-quinolinealdehyde) carbohydrazone (O6). O6 was synthesized similarly to O2, starting from 8-hydroxy-2-quinolinecarboxaldehyde and dhO. Yellow solid was recrystallized from DMF/methanol mixture (1
:9, v/v). Yield: 66%. Mp 248–249 °C. Elemental analysis calcd for C21H16N6O3 (Mw = 400.39 g mol−1): C, 62.99; H, 4.03; N, 20.99, %; found: C, 62.84; H, 4.11; N, 21.22%. IR (KBr, cm−1) νmax: 3408s (OH), 3230m (NH2), 3116m (NH), 2925m (CHaryl), 1684vs (CO), 1601w (CN). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 7.12 (dd, 2H, H–C7H–C17, 3J7,6 = 3J17,16 = 7.4 Hz, 4J7,5 = 4J17,15 = 1.5 Hz), 7.41 (dd, 2H, H–C5H–C15, 4J5,7 = 4J15,17 = 1.5 Hz), 7.45 (t, 2H, H–C6H–C16, 3J6,7 = 3J16,17 =7.4 Hz), 8.17–8.39 (br.m.ovlp., 4H, H–C3H–C13, H–C4H–C14), 8.48 (s, 2H, H–C9H–C11), 9.80 (s, 2H, H–O1H–O2), 11.34 (s, 2H, H–N3H–N4). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 112.13 (C7C17), 117.90 (C5C15), 118.12 (C3C13), 128.13 (C6C16), 128.75 (C4aC14a), 136.40 (C4C14), 138.13 (C8aC18a), 144.02 (C9C11), 151.92 (C2C12), 153.37 (C8C18), 162.45 (C10).
Bis-(8-quinolinealdehyde) thiocarbohydrazone (S2). S2 was synthesized similarly to O2, starting from dhS instead of dhO. Yellow solid was recrystallized from DMF/ethanol mixture (1
:4, v/v). Yield 75%. Mp 192–193 °C (decomp.). Elemental analysis calcd for C21H16N6S (Mw = 384.12 g mol−1): C, 65.61; H, 4.19; N, 21.86; S, 8.34%; found: C, 65.44; H, 4.03; N, 21.77; S, 8.21%. IR (KBr, cm−1) νmax: 3441m (NH), 3119w (CHaryl), 1519vs (amide II); 1231s (CS). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 7.62 (dd, 2H, H–C3H–C17, 3J3,4 = 3J17,18 = 4.1 Hz), 7.75 (t, 2H, H–C6H–C14, 3J6,5 = 3J14,15 = 7.95 Hz), 8.09 (dd, 2H, H–C5H–C15, 3J5,6 = 3J15,14 = 7.95 Hz), 8.35–8.64 (br.m.ovrl. 3H, H–C4H–16, H–C13), 8.88 (s, H–C7), 9.00 (dd, 2H, H–C2H–C18, 3J2,3 = 3J18,17 = 4.1 Hz), 9.48 (s, 1H, H–C11), 9.91 (s, 1H, H–C9), 12.14 (d, 2H, H–N3H–N4). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 121.85 (C3C17), 125.98 (C13), 126.48 (C6C14), 126.70 (C7), 128.01 (C4aC15a), 129.99 (C5C15), 130.97 (C12), 131.46 (C8), 136.65 (C4C16), 140.36 (C8aC12a), 140.48 (C11), 145.40 (C9), 150.34 (C2C18), 175.21 (C10).
Bis-(2-quinolinealdehyde) thiocarbohydrazone (S4). S4 was synthesized similarly to O2, starting from 2-quinolinecarboxaldehyde and dhS, with a slight modification of previously published procedure.47 Yellow solid was recrystallized from DMSO/methanol mixture (9
:1, v/v). Yield 68%. Mp 184 °C. Elemental analysis calcd for C21H16N6S (Mw = 384.12 g mol−1): C, 65.61; H, 4.19; N, 21.86; S, 8.34%; found: C, 65.78; H, 4.01; N, 21.56; S, 8.12%. IR (KBr, cm−1) νmax: 3424s (NH), 2923m (CHaryl), 1488vs (amide II); 1229s (CS). 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 7.67 (ddd, 2H, H–C6H–C16, 3J6,7 = 3J16,17 = 7.05 Hz, 3J6,5 = 3J16,15 = 7.95 Hz), 7.82 (ddd, 2H, H–C7H–C17, 3J7,6 = 3J17,16 = 7.05 Hz), 8.06 (ddd, 4H, H–C5H–C15, H–C8H–C18, 3J5,6 = 3J15,16 = 7.95 Hz), 8.20 (s, 1H, H–C11), 8.40 (s, 1H, H–C9), 8.46–8.63 (br.m.ovlp., 4H, H–C3H–C13, H–C4H–C14), 12.16 (s, 1H, H–N4), 12.51 (s, 1H, H–N3). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 118.41 (C13), 118.72 (C3), 127.75 (C6C16), 128.23 (C4aC14a), 127.37 (C5C15), 130.47 (C7C17), 130.47 (C8C18), 136.98 (C4C14), 144.46 (C11), 149.59 (C9), 147.70 (C8aC18a), 152.03 (C12), 153.96 (C2), 175.97 (C10).
Bis-(8-hydroxy-2-quinolinealdehyde) thiocarbohydrazone (S6). S6 was synthesized similarly to O2, starting from 8-hydroxy-2-quinolinecarboxaldehyde and dhS. Yellow solid was recrystallized from DMF/methanol mixture (1
:9, v/v). Yield 74%; mp 240–241 °C. Elemental analysis calcd for C21H16N6O2S (Mw = 416.46 g mol−1): C, 60.56; H, 3.87; N, 20.18; S, 7.70%; found: C, 60.68; H, 4.03; N, 20.07; S, 7.52%. IR (KBr, cm−1) νmax: 3479s (OH), 3428m (NH), 2923m (CHaryl), 1500vs (amide II); 1231s (CS); 1H NMR (500.26 MHz, DMSO-d6) δ (ppm): 7.14 (d, 2H, H–C7H–C17, 3J7,6 = 3J17,16 = 7.4 Hz), 7.43 (m, 4H, H–C5H–C15, H–6H–16), 8.05–8.41 (br.m.ovlp., 4H, H–C3H–C13, H–C4H–C14), 8.53 (s, 1H, H–C11), 8.89 (s, 1H, H–C9), 9.89 (s, 2H, O1HO2H), 12.18 (s, 1H, H–N4), 12.52 (s, 1H, H–N3). 13C NMR (126.0 MHz, DMSO-d6) δ (ppm): 112.24 (C7C17), 117.87 (C5C15), 118.62 (C3C13), 128.38 (C6C16), 128.90 (C4aC14a), 136.48 (C4C14), 138.20 (C8aC18a), 143.85 (C11), 149.17 (C9), 151.60 (C2C12), 153.44 (C8C18), 175.61 (C10).
DFT theoretical calculations, NMR and lipophilicity predictions
Initial conformations of compounds O1–O6 and S1–S6 were generated in OMEGA 2.4.3,90 using MMFF94s force field.92 The structures were additionally optimized using ωB97X-D functional,93 and 6-311G(d,p) basis set. The solvation effect was simulated with polarized continuum model (PCM),94 of acetonitrile. In order to confirm that the obtained geometry represents the global minimum, vibrational frequency analysis was performed afterward (Opt + Freq keywords). No imaginary frequencies were found, so stationary point reached do represent global minimum. Different tautomeric forms and geometric isomers around azomethine (CN) bond(s) were modeled, and the lowest energy form of each compound was (thio)keto, with E configuration on every CN bond. Starting from global minimum found, NMR chemical shifts are calculated on ωB97X-D/6-311G(d,p) level using GIAO approximation,95 applying PCM model of DMSO. Chemical shifts are scaled to 1H and 13C chemical shifts of TMS. Lipophilicity of compounds was predicted starting from DFT-optimized geometries, calculating virtual logP derived from molecular lipophilicity potential.96
Free radical scavenging antioxidant assay
The proton donating ability was assayed using a protocol for the determination of radical scavenging activity.50 Compounds were dissolved in DMSO and were diluted into ten different concentrations. Commercially available DPPH radical was dissolved in methanol at a concentration of 6.58 × 10−5 M. Into a 96-well microplate, 140 μL of DPPH solution was loaded and 10 μL DMSO solution of the tested compounds was added, or pure DMSO (10 μL) as the control. The microplate was incubated for 30 min at 298 K in the dark and the absorbance was measured at 517 nm using a Thermo Scientific Appliskan. All the measurements were carried out in triplicate. The scavenging activity of the compounds was calculated using the eqn (4):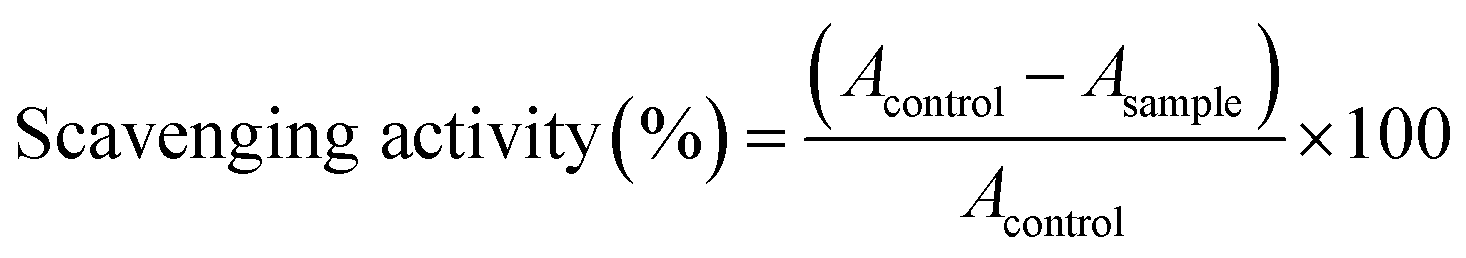 | (4) |
Cell cultures
The two ATCC cell lines were all purchased from LGC Standards (Molsheim, France). Human pancreatic adenocarcinoma (AsPC-1, ATCC® CRL-1682) cell line was maintained in DMEM high glucose medium (Dominique Dutscher, 67172 Brumath cedex, France, Cat. no. L0102-500), while human acute monocytic leukemia cell line (THP-1, ATCC® TIB-202) was maintained in RPMI-1640 (Life Technologies, Paisley, UK, Cat. no. 11875-093), supplemented with 10% (v/v) heat inactivated fetal bovine serum (FBS, Life Technologies, Paisley, UK, Cat. no. 10270-106) and 1% (v/v) penicillin–streptomycin (10000 units per mL and 10000 μg mL−1, Life Technologies, Paisley, UK, Cat. no. 15140-122). Cells were kept at 37 °C in humidified atmosphere containing 5% (v/v) CO2 during their exponential growing phase and in the course of incubation with investigated compounds.
Investigated compounds were initially dissolved in DMSO to the stock concentration of 20 mM. Further dilutions to the experimental concentrations applied on the cells have been done with RPMI-1640 or DMEM media immediately before each experiment, thus the final concentration of DMSO on cells treated with the highest applied concentration of investigated compound was 0.5% (v/v).
Annexin V and PI staining
Cells were seeded in 96 flat bottom well plates (Corning® Costar®, Cat. no. CLS3596) in a volume 0.1 mL, at a density of 10000 per well. AsPC-1 cells were left overnight to settle, while treatment of THP-1 cells started 2 h after seeding. Investigated compounds were added in a range of six concentrations. As controls, non-treated cells, cells treated with 0.5% DMSO, and cells treated with celastrol (Enzo Life Sciences, Cat. no. ALX-350-332-M025) at 50 μM concentration were used.
After 24 h of treatment, supernatant medium with non-adherent cells were removed from plate with AsPC-1 cells and placed into another 96 well plate. Fresh PBS was added to remaining adherent cells afterwards the plate was centrifuged at 450 g for 10 min. Supernatant was discarded and 200 μL of trypsin–EDTA (BioWest, Nuaille, France, Cat. no. L0930-100) was added to each well. Cells were detached in about 15 minutes of incubation at 37 °C. Trypsin–EDTA was removed after additional spinning cycle, afterwards previously removed supernatant with non-adherent cells were added to trypsinized cells and stained with Annexin V-FITC (Immuno Tools, Friesoythe, Germany, Cat. no. 31490013) and PI (Miltenyl Biotec Inc, Auburn, USA, Cat. no. 130-093-233) in a volumes of 3 μL. In the case of THP-1 cells, Annexin V-FITC and PI were added to the wells with cells right after the incubation time was over.
Plates were analyzed on Guava® easyCyte 12HT Benchtop flow microcapillary cytometer (Millipore, Merck, Darmstadt, Germany) using the dedicated InCyte® software package. Cells were classified according to Annexin V-FITC (green fluorescence) and PI (red fluorescence) labeling on viable (double negative), pre-apoptotic cells (Annexin V-FITC single-stained cells), necrotic cells (PI single-stained cells), and cells in advanced phases of apoptosis (double-stained cells). Described trypsinization protocol was applied each time AsPC-1 cells were prepared for flow cytometry analyses, unless is stated otherwise.
Calculation of ED50 concentration
Percentages of Annexin V single-stained and double-stained cells were summarized for each concentration of investigated compound. The computed percentages were plotted against corresponding concentrations on a concentration–response graph. ED50 concentration was calculated as the one that corresponds to half-way of the sigmoidal concentration–dependent curve using asymmetric five-parameter logistic equation (GraphPad Prism 6 software).Cell cycle analysis
Distribution of cells within phases of mitotic division has been evaluated on remaining cells after Annexin V/PI analysis, which right after the read out was finished were fixed in ethanol overnight at 4 °C. Before reading, plates were centrifuged on 450 g for 10 min, ethanol was discarded and PBS added in a volume of 100 μL per well. Cells were stained with 50 μL of FxCycle™ PI/RNAse staining solution (Molecular Probes, Cat. no. F10797), and incubated at 37 °C for 30 min. Plates were analysed on Guava® easyCyte 12HT Benchtop flow microcapillary cytometer using the dedicated InCyte® software package.Inhibition of caspase activity
Cells were treated with investigated compound at ED50 concentration for 6 h with or without pan-caspase inhibitor Z-VAD-fmk (Promega, Madison, USA, Cat. no. G7232). Z-VAD-fmk was added in a concentration that was previously tested and confirmed as non-toxic to cells over 6 h incubation (10 μM and 20 μM for THP-1 and AsPC-1 cell line, respectively). As controls, non-treated cells, cells treated with Z-VAD-fmk only, and cells treated with ED50 concentration only were used. After incubation period was ended, treated cells were carried out for Annexin V/PI staining as described above, and analyzed on Guava® easyCyte 12HT Benchtop flow microcapillary cytometer using the dedicated InCyte® software package. The percent of apoptosis inhibited by Z-VAD-fmk co-treatment was determined using eqn (5):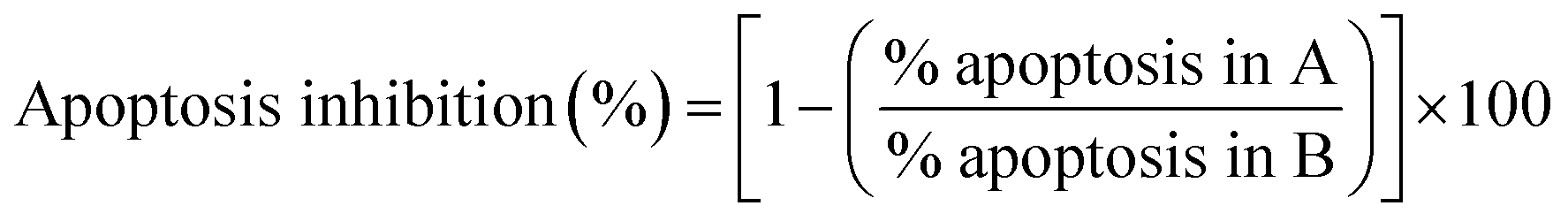 | (5) |
Caspase-8 and caspase-9 activities
Cells were treated with investigated compound at ED50 concentration for 6 h afterwards activity of caspase-8 and -9 were assayed by means of Guava Caspase 9 SR and Caspase 8 FAM kit (EMD Millipore, Cat. no. 4500-0640), following manufacturer's instructions. Cells were analysed on Guava® easyCyte 12HT Benchtop flow microcapillary cytometer using the dedicated InCyte® software package. In acquired data cells were discriminated according to expression of caspase-8 (Grn-B fluorescence), caspase-9 (Yel-B fluorescence), and 7-AAD (Red-B fluorescence).Generation of radical oxygen species in mitochondria
Cells were treated over 6 h with investigated compounds in a concentration of 50 μM, afterwards were stained with MitoSox Red (Molecular Probes, Cat. no. M36008) according to manufacturer's recommendations. Analysis was performed on Guava® easyCyte 12HT Benchtop flow microcapillary cytometer using the dedicated InCyte® software package.Investigation of pro-differentiation activity
AsPC-1 cells were seeded in a volume 0.1 mL, at density 5000 per well of 96 well flat bottom plates. After 24 h, investigated compounds were added in a volume of 0.2 mL to the final concentrations of 1 and 10 μM per well. Wells with control cells were supplemented with medium up to the volume of 0.3 mL. All cells, treated and non-treated controls, were incubated for another 72 h, afterwards evaluation of changes in expression of cell surface marker of differentiation has been performed. When incubation was ended, cells were trypsinized as described above. Anti-CD44-FITC antibody (Miltenyi Biotec, Cat. no. 130-095-195) was added in a volume of 1 μM to each well. Non-treated controls were labeled with anti-CD44-FITC or anti-IgG-FITC (Miltenyl Biotec, Cat. no. 130-093-192). Plate was incubated at 37 °C for 30 min, when additional spinning cycle (450 g for 10 min) with PBS removal was performed to remove the excess of antibodies. Finally, fresh PBS was added to the wells and cells were analyzed on Guava® easyCyte 12HT Benchtop flow microcapillary cytometer using the dedicated InCyte® software package.Growth inhibition of 3D tumor models
Three-dimensional AsPC-1 tumor models were made in 96 well plates (Corning, Sigma-Aldrich, St. Louis, Mo, USA, Cat. no. 4515). Tumors were left to grow for additional four days, afterwards investigated compounds were added in concentrations of 100, 10 and 1 μM. Evaluation has been maintained during 8 day incubation period, with media exchanged on the day 4. Changes in the tumors sizes have been assessed on Celigo® imaging cytometer (Cyntellect, Brooks Life Science Systems, Poway, CA, USA) using Celigo software. Growth rates of non-treated and treated spheroids were computed for each day during 8 day incubation by dividing the area on the day-n with the area on the day 0.HSA binding experiments
Fatty acid free HSA (<0.007% fatty acids, Mw = 66478 Da) was purchased from Sigma, as well as potassium dihydrogen phosphate, disodium hydrogen phosphate, sodium chloride and potassium chloride used for PBS preparation. Stock solution of HSA (c = 1.91 × 10−4 M) was prepared by dissolving accurate weighted mass of commercially available lyophilized HSA in freshly prepared 1 × PBS (pH 7.36), and stock solution was kept in freezer in 100 μL portions. The stock solution of S2 (c = 6.87 × 10−4 M) was prepared by dissolving proper amount of substance in DMSO, because of its low solubility in buffer. For HSA–S2 interaction studies, HSA solution was freshly prepared from the stock by dilution with a buffer (HSA concentration was kept constant, c = 7 × 10−7 M), and titrated with S2 stock solution to avoid large sample dilution. Substance was added in 0.5, 1, 2, 3, 4, 5, 7 and 9 molar equivalents. After each aliquot addition, system was stirred and left to equilibrate for 15 min before UV-Vis absorption and fluorescence emission spectra recording. After addition of nine equivalents of substance, final volume of DMSO did not exceeded 2% (v/v). It was shown that addition of 15% of DMSO did not induce structural changes in BSA, protein structurally similar to HSA. Therefore, it is unlikely that the conformation of HSA will be changed with the level of DMSO used in this study.97 Milipore water was used in the preparation of buffer solution.
Fluorescence measurements were performed on spectrofluorometer Fluoromax-4 Jobin Yvon (Horiba Scientific, Japan), equipped with Peltier element for temperature control and magnetic stirrer for cuvette, using quartz cell with 1 cm path length and 4 mL volume. Before recording of fluorescence spectrum, diluted HSA solution was ultrafiltrated using filters with 0.23 μm pore size. An excitation wavelength was 280 nm, with 5 nm slits; emission spectra were recorded in 300–450 nm wavelength range, with 5 nm slits, and 0.1 s integration time. Background PBS signal was subtracted from each spectrum.
UV-Vis spectra were recorded on a GBC Cintra6 UV/Vis spectrophotometer, using quartz cell with 1 cm path length and 4 mL volume. All spectra were recorded against the corresponding blank (PBS) in the 250–450 nm wavelength range at room temperature.
Molecular docking studies
For exploring the binding of S2 to site IB of HSA, crystal structure of HSA with idarubicin dimer (PDB code 4LB2) was used.98 For site IIA, PDB structure of indol amide derivative (PDB code 3LU7) was used.99 For site IIIA, structure of HSA–diflunisal complex (PDB code 2BXE) was used.88 To define the receptor site, the residues in sphere of 12 Å around co-crystallized ligand were used. Ligands and water molecules were removed prior to receptor preparation in AutoDock Tools 1.5.6.89 Initial conformation of S2 was generated in OMEGA 2.4.3,90 using MMFF94s force field.92 The geometry of lowest energy conformer was further optimized using semiempirical PM6 method implemented in MOPAC 2016.100 Docking was carried in Autodock Vina 1.1.2 software.101 VEGAZZ 3.0.3 was used as a GUI.102Acknowledgements
The authors acknowledge networking support by the COST Action CM1106 StemChem – “Chemical Approaches to Targeting Drug Resistance in Cancer Stem Cells”. The work was funded by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grants 172055 and 172013).References
- S. Rollas and Ş. G. Küçükgüzel, Molecules, 2007, 12, 1910–1939 CrossRef CAS PubMed.
- A. Kajal, S. Bala, N. Sharma, S. Kamboj and V. Saini, Int. J. Med. Chem., 2014, 2014, 1–11 Search PubMed.
- G. Verma, A. Marella, M. Shaquiquzzaman, M. Akhtar, M. R. Ali and M. M. Alam, J. Pharm. BioAllied Sci., 2014, 6, 69–80 CrossRef PubMed.
- K. Padmini, P. J. Preethi, M. Divya, P. Rohini, M. Lohita, K. Swetha and P. Kaladar, Int. J. Pharma Res. Rev., 2013, 2, 43–58 Search PubMed.
- M. Asif and A. Husain, J. Appl. Chem., 2013, 2013, 1–7 CrossRef.
- V. J. Negi, Int. J. Pharm. Chem., 2013, 2, 12–13 CrossRef.
- R. d. C. Maia, R. Tesch and C. A. M. Fraga, Expert Opin. Ther. Pat., 2014, 24, 1161–1170 CrossRef CAS PubMed.
- K. C. Agrawal, B. A. Booth, R. L. Michaud, E. C. Moore and A. C. Sartorelli, Biochem. Pharmacol., 1974, 23, 2421–2429 CrossRef CAS PubMed.
- N. S. H. N. Moorthy, N. M. F. S. A. Cerqueira, M. J. Ramos and P. A. Fernandes, Mini-Rev. Med. Chem., 2013, 13, 1862–1872 CrossRef CAS PubMed.
- B. Fischer, K. Kryeziu, S. Kallus, P. Heffeter, W. Berger, C. R. Kowol and B. K. Keppler, RSC Adv., 2016, 6, 55848–55859 RSC.
- M. Liu, T. Lin and A. C. Sartorelli, Biochem. Pharmacol., 1994, 48, 335–344 CrossRef.
- J. M. Shao, B. S. Zhou, a. J. Di Bilio, L. J. Zhu, T. L. Wang, C. Qi, J. Shih and Y. Yen, Mol. Cancer Ther., 2006, 5, 586–592 CrossRef CAS PubMed.
- Y. Aye, M. J. C. Long and J. Stubbe, J. Biol. Chem., 2012, 287, 35768–35778 CrossRef CAS PubMed.
- A. Popović-Bijelić, C. R. Kowol, M. E. S. Lind, J. Luo, F. Himo, É. A. Enyedy, V. B. Arion and A. Gräslund, J. Inorg. Biochem., 2011, 105, 1422–1431 CrossRef PubMed.
- K. Pelivan, W. Miklos, S. van Schoonhoven, G. Koellensperger, L. Gille, W. Berger, P. Heffeter, C. R. Kowol and B. K. Keppler, J. Inorg. Biochem., 2015, 160, 61–69 CrossRef PubMed.
- C. R. Myers, Free Radical Biol. Med., 2016, 91, 81–92 CrossRef CAS PubMed.
- J. M. Myers, W. E. Antholine, J. Zielonka and C. R. Myers, Toxicol. Lett., 2011, 201, 130–136 CrossRef CAS PubMed.
- R. Trondl, L. S. Flocke, C. R. Kowol, P. Heffeter, U. Jungwirth, G. E. Mair, R. Steinborn, E. A. Enyedy, M. A. Jakupec, W. Berger and B. K. Keppler, Mol. Pharmacol., 2014, 85, 451–459 CrossRef PubMed.
- S. Hill, S. Datta, J. Liu, R. Inglis, C. J. Milios, P. L. Feng, J. J. Henderson, E. del Barco, E. K. Brechin and D. N. Hendrickson, Dalton Trans., 2010, 39, 4693–4707 RSC.
- H. Gang, G. Dong, D. Chun-Ying, M. Hong and M. Qing-jin, New J. Chem., 2002, 26, 1371–1377 RSC.
- Y. Zhao, D. Guo, Y. Liu, C. He and C. Duan, Chem. Commun., 2008, 2, 5725–5727 RSC.
- L. Zhang, J. J. Wang and G. C. Xu, Inorg. Chem. Commun., 2014, 39, 66–69 CrossRef CAS.
- A. Bacchi, M. Carcelli, P. Pelagatti, C. Pelizzi and G. Pelizzi, J. Inorg. Biochem., 1999, 75, 123–133 CrossRef CAS PubMed.
- A. A. A. Abu-Hussen and A. A. A. Emara, J. Coord. Chem., 2004, 57, 973–987 CrossRef CAS.
- D. Y. Wu, O. Sato, Y. Einaga and C. Y. Duan, Angew. Chem., Int. Ed., 2009, 48, 1475–1478 CrossRef CAS PubMed.
- G. M. Abu El-Reash, O. A. El-Gammal and A. H. Radwan, Spectrochim. Acta, Part A, 2014, 121, 259–267 CrossRef CAS PubMed.
- J. R. Dimmock, S. N. Pandeya, J. W. Quail, U. Pugazhenthi, T. M. Allen, G. Y. Kao, J. Balzarini and E. DeClercq, Eur. J. Med. Chem., 1995, 30, 303–314 CrossRef CAS.
- F. A. French and E. J. Blanz, J. Med. Chem., 1966, 9, 585–589 CrossRef CAS PubMed.
- O. A. El-Gammal, G. M. Abu El-Reash, S. E. Ghazy and A. H. Radwan, J. Mol. Struct., 2012, 1020, 6–15 CrossRef CAS.
- S. Rhee, J. Pharm. Soc. Korea, 1972, 16, 162–175 CAS.
- K. Gangarapu, S. Manda, S. Thota, R. Yerra, S. S. Karki, J. Balzarini, E. De Clercq and H. Tokuda, Lett. Drug Des. Discovery, 2012, 9, 934–941 CrossRef CAS.
- M. P. Sathisha, U. N. Shetti, V. K. Revankar and K. S. R. Pai, Eur. J. Med. Chem., 2008, 43, 2338–2346 CrossRef CAS PubMed.
- J. M. Pérez, V. Cerrillo, A. I. Matesanz, J. M. Millán, P. Navarro, C. Alonso and P. Souza, ChemBioChem, 2001, 2, 119–123 CrossRef.
- V. R. Solomon and H. Lee, Curr. Med. Chem., 2011, 18, 1488–1508 CrossRef CAS PubMed.
- S. Adsule, V. Barve, D. Chen, F. Ahmed, Q. P. Dou, S. Padhye and F. H. Sarkar, J. Med. Chem., 2006, 49, 7242–7246 CrossRef CAS PubMed.
- Q. Ba, M. Hao, H. Huang, J. Hou, S. Ge, Z. Zhang, J. Yin, R. Chu, H. Jiang, F. Wang, K. Chen, H. Liu and H. Wang, Clin. Cancer Res., 2011, 17, 7625–7633 CrossRef CAS PubMed.
- M. Serda, D. S. Kalinowski, A. Mrozek-Wilczkiewicz, R. Musiol, A. Szurko, A. Ratuszna, N. Pantarat, Z. Kovacevic, A. M. Merlot, D. R. Richardson and J. Polanski, Bioorg. Med. Chem. Lett., 2012, 22, 5527–5531 CrossRef CAS PubMed.
- H. Huang, Q. Chen, X. Ku, L. Meng, L. Lin, X. Wang, C. Zhu, Y. Wang, Z. Chen, M. Li, H. Jiang, K. Chen, J. Ding and H. Liu, J. Med. Chem., 2010, 53, 3048–3064 CrossRef CAS PubMed.
- F. Bisceglie, A. Musiari, S. Pinelli, R. Alinovi, I. Menozzi, E. Polverini, P. Tarasconi, M. Tavone and G. Pelosi, J. Inorg. Biochem., 2015, 152, 10–19 CrossRef CAS PubMed.
- F. A. French, E. J. Blanz, S. C. Shaddix and R. W. Brockman, J. Med. Chem., 1974, 17, 172–181 CrossRef CAS PubMed.
- B. Beck and C. Blanpain, Nat. Rev. Cancer, 2013, 13, 727–738 CrossRef CAS PubMed.
- M. S. Sosa, P. Bragado and J. A. Aguirre-Ghiso, Nat. Rev. Cancer, 2014, 14, 611–622 CrossRef CAS PubMed.
- R. P. Galvao, A. Kasina, R. S. McNeill, J. E. Harbin, O. Foreman, R. G. W. Verhaak, A. Nishiyama, C. R. Miller and H. Zong, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 4214–4223 CrossRef PubMed.
- J. W. Uhr and K. Pantel, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12396–12400 CrossRef CAS PubMed.
- M. Yang, H. Wang, Y. Hou, H. Tung, T. Chiu and Y. Shan, Mol. Cancer, 2015, 1–17 Search PubMed.
- P. Longati, X. Jia, J. Eimer, A. Wagman, M.-R. Witt, S. Rehnmark, C. Verbeke, R. Toftgård, M. Löhr and R. L. Heuchel, BMC Cancer, 2013, 13, 95–108 CrossRef CAS PubMed.
- E. Manoj, M. R. P. Kurup and E. Suresh, J. Chem. Crystallogr., 2008, 38, 157–161 CrossRef CAS.
- K. H. M. Ebrahim Tehrani, F. Kobarfard, P. Azerang, M. Mehravar, Z. Soleimani, G. Ghavami and S. Sardari, Iran. J. Pharm. Res., 2013, 12, 331–346 Search PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- R. L. Prior, X. Wu and K. Schaich, J. Agric. Food Chem., 2005, 53, 4290–4302 CrossRef CAS PubMed.
- M. T. Zimmerman, C. A. Bayse, R. R. Ramoutar and J. L. Brumaghim, J. Inorg. Biochem., 2015, 145, 30–40 CrossRef CAS PubMed.
- V. Calcatierra, Ó. López, J. G. Fernández-Bolaños, G. B. Plata and J. M. Padrón, Eur. J. Med. Chem., 2015, 94, 63–72 CrossRef CAS PubMed.
- B. Beck and C. Blanpain, Nat. Rev. Cancer, 2013, 13, 727–738 CrossRef CAS PubMed.
- I. Murtaza, V. M. Adhami, B. Bin Hafeez, M. Saleem and H. Mukhtar, Int. J. Cancer, 2009, 125, 2465–2473 CrossRef CAS PubMed.
- B. Szomolay and V. Shahrezaei, BMC Syst. Biol., 2012, 6, 26 CrossRef CAS PubMed.
- E. J. Calabrese and M. P. Mattson, Journal of Cell Communication and Signaling, 2011, 5, 25–38 CrossRef PubMed.
- A. R. Reynolds, Dose-Response, 2010, 8, 253–284 CrossRef CAS PubMed.
- E. Tsakalozou, A. M. Eckman and Y. Bae, Biochem. Res. Int., 2012, 2012, 1–10 CrossRef PubMed.
- H. Hernández-Vargas, J. Palacios and G. Moreno-Bueno, Oncogene, 2007, 26, 2902–2913 CrossRef PubMed.
- A. S. Reddy and S. Zhang, Expert Rev. Clin. Pharmacol., 2013, 6, 41–47 CrossRef CAS PubMed.
- D. R. McIlwain, T. Berger and T. W. Mak, Cold Spring Harbor Perspect. Biol., 2015, 7, a026716 CrossRef PubMed.
- A. Jimbo, E. Fujita, Y. Kouroku, J. Ohnishi, N. Inohara, K. Kuida, K. Sakamaki, S. Yonehara and T. Momoi, Exp. Cell Res., 2003, 283, 156–166 CrossRef CAS PubMed.
- R. S. Hotchkiss and D. W. Nicholson, Nat. Rev. Immunol., 2006, 6, 813–822 CrossRef CAS PubMed.
- J. Alexandre, F. Batteux, C. Nicco, C. Chéreau, A. Laurent, L. Guillevin, B. Weill and F. Goldwasser, Int. J. Cancer, 2006, 119, 41–48 CrossRef CAS PubMed.
- D. Llobet, N. Eritja, M. Encinas, A. Sorolla, A. Yeramian, J. A. Schoenenberger, A. Llombart-Cussac, R. M. Marti, X. Matias-Guiu and X. Dolcet, Anti-Cancer Drugs, 2008, 19, 115–124 CrossRef CAS PubMed.
- I. Bairati, F. Meyer, M. Gelinas, A. Fortin, A. Nabid, F. Brochet, J.-P. Mercier, B. Tetu, F. Harel, B. Abdous, E. Vigneault, S. Vass, P. del Vecchio and J. Roy, J. Clin. Oncol., 2005, 23, 5805–5813 CrossRef CAS PubMed.
- G. Bjelakovic, D. Nikolova, L. L. Gluud, R. G. Simonetti and C. Gluud, JAMA, 2007, 297, 842–857 CrossRef CAS PubMed.
- D. Trachootham, Y. Zhou, H. Zhang, Y. Demizu, Z. Chen, H. Pelicano, P. J. Chiao, G. Achanta, R. B. Arlinghaus, J. Liu and P. Huang, Cancer Cell, 2006, 10, 241–252 CrossRef CAS PubMed.
- E. A. Bey, M. S. Bentle, K. E. Reinicke, Y. Dong, C.-R. Yang, L. Girard, J. D. Minna, W. G. Bornmann, J. Gao and D. A. Boothman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11832–11837 CrossRef CAS PubMed.
- G. C. Brown and V. Borutaite, Mitochondrion, 2012, 12, 1–4 CrossRef CAS PubMed.
- J. Zielonka and B. Kalyanaraman, Free Radical Biol. Med., 2010, 48, 983–1001 CrossRef CAS PubMed.
- J. Kang and S. Pervaiz, Biochem. Res. Int., 2012, 2012, 1–14 CrossRef PubMed.
- L. Papa, M. Hahn, E. L. Marsh, B. S. Evans and D. Germain, J. Biol. Chem., 2014, 289, 5412–5416 CrossRef CAS PubMed.
- A. E. Dikalova, A. T. Bikineyeva, K. Budzyn, R. R. Nazarewicz, L. McCann, W. Lewis, D. G. Harrison and S. I. Dikalov, Circ. Res., 2010, 107, 106–116 CrossRef CAS PubMed.
- Y. Zuo, B. Xiang, J. Yang, X. Sun, Y. Wang, H. Cang and J. Yi, Cell Res., 2009, 19, 449–457 CrossRef CAS PubMed.
- A. Jaggupilli and E. Elkord, Clin. Dev. Immunol., 2012, 2012, 1–11 CrossRef PubMed.
- Y. Yan, X. Zuo and D. Wei, Stem Cells Transl. Med., 2015, 4, 1033–1043 CrossRef CAS PubMed.
- K. Williams, K. Motiani, P. V. Giridhar and S. Kasper, Exp. Biol. Med., 2013, 238, 324–338 CrossRef CAS PubMed.
- D. Naor, S. B. Wallach-Dayan, M. A. Zahalka and R. V. Sionov, Semin. Cancer Biol., 2008, 18, 260–267 CrossRef CAS PubMed.
- L. Li, X. Hao, J. Qin, W. Tang, F. He, A. Smith, M. Zhang, D. M. Simeone, X. T. Qiao, Z.-N. Chen, T. S. Lawrence and L. Xu, Gastroenterology, 2014, 146, 1108–1118 CrossRef CAS PubMed.
- F. Emhemmed, S. Ali Azouaou, F. Thuaud, V. Schini-Kerth, L. Désaubry, C. D. Muller and G. Fuhrmann, Biochem. Pharmacol., 2014, 89, 185–196 CrossRef CAS PubMed.
- D. R. Grimes, C. Kelly, K. Bloch and M. Partridge, J. R. Soc., Interface, 2014, 11, 20131124 CrossRef PubMed.
- R.-Z. Lin and H.-Y. Chang, Biotechnol. J., 2008, 3, 1172–1184 CrossRef CAS PubMed.
- N. R. Filipović, S. Bjelogrlić, A. Marinković, T. Ž. Verbić, I. N. Cvijetić, M. Senćanski, M. Rodić, M. Vujčić, D. Sladić, Z. Striković, T. R. Todorović and C. D. Muller, RSC Adv., 2015, 5, 95191–95211 RSC.
- L. Deschamps-Labat, F. Péhourcq, M. Jagou and B. Bannwarth, J. Pharm. Biomed. Anal., 1997, 16, 223–229 CrossRef CAS PubMed.
- K. W. Lexa, E. Dolghih and M. P. Jacobson, PLoS One, 2014, 9, e93323 Search PubMed.
- Principles of Fluorescence Spectroscopy, ed. J. R. Lakowicz, Springer Science Business Media, New York, USA, 3rd edn, 2006, p. 11 Search PubMed.
- J. Ghuman, P. A. Zunszain, I. Petitpas, A. A. Bhattacharya, M. Otagiri and S. Curry, J. Mol. Biol., 2005, 353, 38–52 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- P. C. D. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, J. Chem. Inf. Model., 2010, 50, 572–584 CrossRef CAS PubMed.
- P. D. Ross and S. Subramanian, Biochemistry, 1981, 20, 3096–3102 CrossRef CAS PubMed.
- T. A. Halgren, J. Comput. Chem., 1999, 20, 720–729 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
- P. Gaillard, P. A. Carrupt, B. Testa and A. Boudon, J. Comput.-Aided Mol. Des., 1994, 8, 83–96 CrossRef CAS PubMed.
- B. Ojha and G. Das, J. Phys. Chem. B, 2010, 114, 3979–3986 CrossRef CAS PubMed.
- Z. Wang, J. X. Ho, J. R. Ruble, J. Rose, F. Rüker, M. Ellenburg, R. Murphy, J. Click, E. Soistman, L. Wilkerson and D. C. Carter, Biochim. Biophys. Acta, Gen. Subj., 2013, 1830, 5356–5374 CrossRef CAS PubMed.
- D. Buttar, N. Colclough, S. Gerhardt, P. A. MacFaul, S. D. Phillips, A. Plowright, P. Whittamore, K. Tam, K. Maskos, S. Steinbacher and H. Steuber, Bioorg. Med. Chem., 2010, 18, 7486–7496 CrossRef CAS PubMed.
- J. J. P. Stewart, MOPAC2016, Stewart Computational Chemistry, Colorado Springs, CO, USA, 2016 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- A. Pedretti, L. Villa and G. Vistoli, J. Comput.-Aided Mol. Des., 2004, 18, 167–173 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23940d |
| This journal is © The Royal Society of Chemistry 2016 |