
Luminescence, chemical sensing and mechanical properties of crystalline materials based on lanthanide–sulfonate coordination polymers†
Richard F. D'Vries*a,
German E. Gomez *b,
Diego F. Lionellob,
M. Cecilia Fuertesb,
Galo J. A. A. Soler-Illiac and
Javier Ellenaa
*b,
Diego F. Lionellob,
M. Cecilia Fuertesb,
Galo J. A. A. Soler-Illiac and
Javier Ellenaa
aInstituto de Física de São Carlos, Universidade de São Paulo, CP. 369, 13560-970, São Carlos, SP, Brazil. E-mail: ridvries@ifsc.usp.br; Fax: +55 16 3373-9758
bGerencia de Química, Centro Atómico Constituyentes, Comisión Nacional de Energía Atómica (CAC-CNEA), Av. Gral. Paz 1499, 1650 San Martin, Buenos Aires, Argentina. E-mail: gegomez@unsl.edu.ar
cInstituto de Nanosistemas, Universidad Nacional de San Martín (INS-UNSAM), Av. 25 de Mayo 1021, San Martín, Buenos Aires, Argentina
First published on 10th November 2016
Abstract
Lanthanide-coordination polymers (Ln-CPs) constitute relevant compounds for the design of multifunctional materials. Nevertheless, studies devoted to an understanding of the relationships of combined structural–optical–mechanical properties have scarcely been reported. In this work, an exhaustive study of a series of CPs obtained from lanthanide metals, 3-hydroxinaphthalene-2,7-disulfonate (3-OHNDS) and 1,10-phenanthroline (Phen) as ligands is presented. Two crystalline phases were identified with the general formulae [Eu(3-OHNDS)(Phen)(H2O)]·3H2O (phase 1-Eu), and [Ln2(3-OHNDS)2(Phen)2(H2O)]·3H2O (phase 2-Ln), where Ln3+ = Tb, Dy, Ho, Er and Yb. Both phases were characterized by powder and single crystal X-ray diffraction, vibrational and thermal analysis and scanning electron microscopy. Moreover, nanoindentation analysis was performed in order to find the relationships between these structural features and the mechanical properties of the crystalline materials. The photoluminescence (PL) properties of the reported phases were also explored, involving excitation–emission experiments and quantification of color emission. Finally, one compound was selected as a chemical sensor model, exhibiting different optical behaviour in the presence of aromatic molecules, principally towards naphtalene molecules. These results make these compounds promising materials for the elaboration of selective chemical sensors.
Introduction
In the exploration of synthesis methodologies to obtain new crystalline materials such as coordination polymers (CPs) and metal–organic frameworks (MOFs), different procedures applying the principles of crystal engineering have been developed.1,2 However, it is hard to find systematic paths addressed to elucidating the synthetic mechanism and related structural features with physical, chemical or electronic properties.From the synthetic point of view, the use of polytopic linkers increases the possibility of obtaining multiple structural phases. It provides an excellent scenario to study synthesis, structure and its properties.
In this context, sulfonate linkers represent new opportunities to explore the synthesis of CPs, due to their versatility in forming covalent bonds and hydrogen interactions.3 Recent works have demonstrated that under hydro or solvothermal conditions the sulfonate group increases its coordination ability, improving its capacity to form compounds with different architectures, dimensionalities and topologies.4,5 These different structural features have a high impact on such resulting properties as catalysis,6,7 gas absorption,8,9 luminescence,4,10,11 magnetism and conductivity.12,13 Our research is focused on an understanding of sulfonate group behavior under hydrothermal conditions as well as on the formation equilibrium involved in the reaction.14,15 In this work we choose two aromatic π-conjugated linkers (3-OHNDS and 1,10-phenanthroline) and lanthanides as building blocks for the construction of optically active platforms. It is known that lanthanide ions present rich optical features derived from intra-configurational 4f–4f transitions, with some of them being of hypersensitive nature.16 This key property is important for sensing applications,17 which is a field in exponential growth.
On the other hand, nanoindentation has been widely adopted to analyze the mechanical properties of thin films, single crystals and monoliths.18 Thanks to pioneering work by Cheetham et al.,19 nanoindentation analysis has been applied to demonstrate correlations between the internal arrangement of crystal packing and anisotropic mechanical properties (density, stability and elasticity) in MOF-type materials.
The present approach involves a complete and systematic study regarding the synthesis strategies, photophysical characterization, chemical sensing and, for the first time, analysis of anisotropic mechanical properties of mixed ligand (naphtalenedisulfonate, phenanthroline) Ln-CPs.
Experimental section
Synthesis
All reagents and solvents employed were commercially available and used as supplied with no further purification: 3-hydroxynaphthalene-2,7-disulfonic acid (95%, Sigma-Aldrich); 1,10-phenanthroline (99%, Sigma-Aldrich); Ln(NO3)3·6H2O where Ln = Eu, Tb, Dy, Ho and Yb (99%, Sigma-Aldrich).Several temperatures and reaction times were tested. The molar composition of the initial reaction mixture in 3-OHNDS3−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ln3+:2Phen:1474H2O was set. The optimized synthesis procedure was the following:
:Ln3+:2Phen:1474H2O was set. The optimized synthesis procedure was the following:
[Eu(3-OHNDS)(Phen)(H2O)]·3H2O (phase 1-Eu) was obtained by the addition of 3-OHNDS (0.04 g, 0.116 mmol) and 1,10-phenanthroline (0.042 g, 0.232 mmol) to a solution of Eu(NO3)3·6H2O (0.05 g, 0.116 mmol) in 6 mL of distilled water under constant stirring at room temperature for 30 minutes. Finally, the reaction mixture was placed in a Teflon-lined stainless steel autoclave to react at 170 °C for 18 hours. After cooling at room temperature, the product mixture was filtered and washed with water and ethanol. A few small crystals were obtained and manually isolated from the product mixture.
A similar procedure was employed to obtain the compounds [Ln2(3-OHNDS)2(Phen)2(H2O)]·3H2O (phase 2-Ln), where Ln3+ = Tb, Dy, Ho, Er and Yb. [Ho2(3-OHNDS)2(Phen)2(H2O)]·3H2O compound was obtained by the addition of 3-OHNDS (0.04 g, 0.115 mmol) and 1,10-phenanthroline (0.041 g, 0.23 mmol) to a solution of Ho(NO3)3·6H2O (0.05 g, 0.115 mmol) in 6 mL of distilled water under constant stirring at room temperature for 30 minutes. Finally, the reaction mixture was placed in a Teflon-lined stainless steel autoclave for reaction under hydrothermal conditions at 200 °C for 24 hours. After cooling to room temperature, the crystalline products (yield of 71.2%) were filtered and washed with water and ethanol. Elemental analysis calculated (%) for [Ho2(3-OHNDS)2(Phen)2(H2O)]·3H2O: C, 38.72; N, 4.10; H, 2.51; S, 9.4; found: C, 38.94; N, 3.64; H, 2.83; S, 9.08.
Material characterization
The structures were solved by SHELXS-2013 software and then refined by SHELXL-2013,23 included in WinGX24 and Olex2.25 Non-hydrogen atoms of the molecules were clearly resolved and their full-matrix least-square refinement was conducted using anisotropic thermal parameters. All hydrogen atoms were stereochemically positioned and refined by the riding model.23 Hydrogen atoms of the water molecules were localized and fixed (with Uiso(H) = 1.5 U eq.) on the density map. ORTEP diagrams were prepared with diamond.26 TOPOS,27 mercury28 and diamond26 programs were used in the preparation of the artwork of the polyhedral and topological representations.
Results and discussion
Synthetic study
The choice of hydrothermal or solvothermal methodologies as the synthetic route of CPs and MOFs often involves several experiments to find the optimal conditions (temperature, time, solvents, stoichiometry, pH, etc.) in order to obtain pure crystalline materials.30 The variables explored in this work include temperature, reaction time and solvothermal methodology, always under a fixed stoichiometry (see Exp. section).For the study of the hydrothermal reaction conditions, holmium-derived phase 2-Ln was arbitrarily used for tests. An impure mixed product phase 2 (phase 2 ≫ impurity) was obtained as a result of a low temperature test, where some peaks of the impurity coincide with phase 1, as shown in ESI S6a.† These results indicate that both phases could be involved in a formation equilibrium where phase 1 is formed and quickly transformed in the most stable, phase 2.
As was observed in previous cases,7,31,32 the electronic configuration and the lanthanide contraction impacted on the thermodynamics and kinetics of the reaction which enables it to form different structural phases. For instance, two structural phases were obtained, depending on the region of the lanthanide series. It was possible to find phase 1 (La–Sm)4 and phase 2 (Tb–Yb) in the “light rare-earth” region (LREE)33 and “heavy rare-earth” region (HREE),33 respectively.
Moreover, in order to compare the results employing solvothermal methodology under pre-defined synthesis conditions (200 °C, 24 h), different experiments were performed: (i) Using methanol or ethanol, phase 2 was obtained as a unique reaction product. (ii) An unidentified phase and a mixture of decomposed products were obtained with acetonitrile and toluene, respectively (ESI S8†). In fact, it was possible to isolate and characterize the compound [Ho2(C7O2H5)4(Phen)(NO3)2] from the toluene reaction synthesis (ESI S1†). The formation of this compound indicates that it takes place in in situ toluene oxidation under solvothermal conditions. A structural analysis of the isostructural Pr3+ compound was previously reported.34
Structural description of Ln-CPs
Details of the data collection as well as refinement and crystallographic data for the compounds are summarized in Table 1. The ORTEP diagrams for [Eu(3-OHNDS)(Phen)(H2O)]·3H2O, and [Tb2(3-OHNDS)2(Phen)2(H2O)]·3H2O compounds are shown in Fig. 1.| Compound | Eu | Tb | Ho | Er | Yb | Dy |
| Emp. formula | C22H15N2O8S2Eu, 3(H2O) | C44H28N4O15S4Tb2, 3(H2O) | C44H28N4O15S4Ho2, 3(H2O) | C44H28N4O15S4Er2, 3(H2O) | C44H28N4O15S4Yb2, 3(H2O) | C22H15N2O8S2Dy, 3(H2O) |
| FW (g mol−1) | 704.48 | 1352.83 | 1364.85 | 1369.51 | 1381.07 | 1356.45 |
| Temp. (K) | 296 | 296 | 296 | 296 | 296 | 296 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | P21/n | P21/n | P21/n | P21/n |
| Unit cell | ||||||
| a (Å) | 9.2352(6) | 14.2636(6) | 14.2341(3) | 14.2425(8) | 14.2136(4) | 14.2586(6) |
| b (Å) | 11.8774(7) | 17.0354(7) | 17.0149(4) | 17.0358(9) | 17.0112(5) | 17.0438(7) |
| c (Å) | 12.6755(8) | 18.9491(7) | 18.8852(4) | 18.8857(10) | 18.8325(5) | 18.9287(7) |
| α (°) | 106.681(3) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 110.149(3) | 99.088(2) | 98.991(1) | 99.030(2) | 99.014(1) | 99.071(1) |
| γ (°) | 99.171(3) | 90 | 90 | 90 | 90 | 90 |
| Volume (Å3) | 1197.7(1) | 4546.6(3) | 4517.6(2) | 4525.5(4) | 4497.3(2) | 4542.5(3) |
| Z | 2 | 4 | 4 | 4 | 4 | 4 |
| ρ calcd (mg m−3) | 1.863 | 1.976 | 2.007 | 2.010 | 2.040 | 1.984 |
| Abs. coeff (mm−1) | 1.935 | 3.354 | 3.747 | 3.953 | 4.404 | 3.532 |
| F(000) | 691 | 2656 | 2672 | 2680 | 2696 | 2653 |
| θ range (°) | 1.8 to 26.5 | 1.6 to 26.5 | 1.6 to 26.4 | 1.6 to 26.4 | 1.6 to 26.4 | 1.6 to 26.4 |
| Reflections collected/Unique [R(int)] | 21024/4955 [0.059] |
140387/9363 [0.104] |
32602/9231 [0.043] |
129298/9276 [0.051] |
60860/9216 [0.060] |
89917/9287 [0.090] |
| Completeness (%) | 99.8 | 99.4 | 99.5 | 99.9 | 99.8 | 99.8 |
| Data/restraints/parameters | 4955/0/341 | 9363/0/647 | 9231/0/647 | 9276/0/647 | 9216/0/647 | 9287/0/655 |
| Gof on F2 | 1.05 | 1.12 | 1.02 | 1.09 | 1.03 | 1.03 |
| R1 [I > 2σ(I)] | 0.0354 | 0.0365 | 0.0305 | 0.0242 | 0.0276 | 0.0361 |
| wR2 [I > 2σ(I)] | 0.0894 | 0.0927 | 0.0658 | 0.0588 | 0.0576 | 0.0870 |
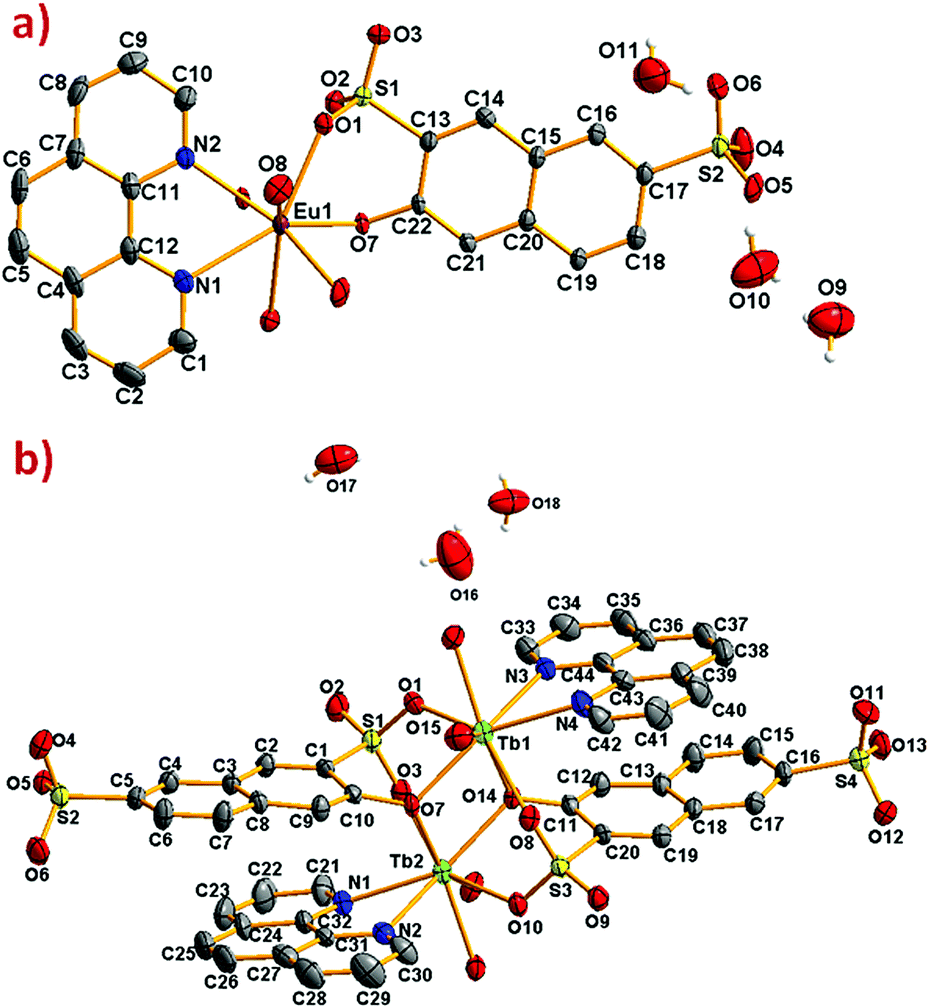 | ||
| Fig. 1 ORTEP diagram showing 50% of probability ellipsoids (a) [Eu(3-OHNDS)(Phen)(H2O)]·3H2O and (b) [Tb2(3-OHNDS)2(Phen)2(H2O)]·3H2O compounds. Hydrogen atoms were omitted for clarity. | ||
The compound [Eu(3-OHNDS)(Phen)(H2O)]·3H2O was obtained within a mixture of reaction products. Small crystals were manually isolated and structurally characterized by single crystal X-ray diffraction. The compound crystallizes in the triclinic P space group (named here as phase 1) belonging to a previously reported family of [Ln(3-OHNDS)(Phen)(H2O)]·3H2O compounds,4 (where Ln3+ = La, Pr, Nd and Sm). The asymmetric unit is formed by one crystallographically independent metal cation with the coordination of one 3-OHNDS ligand, one 1,10-phenanthroline and two water molecules.4 The structure also presents three non-coordinated water molecules in the asymmetric unit. The particular coordination modes of the ligand give rise to ladder-shaped chains as extended structures.
In phase 2-Ln, [Ln2(3-OHNDS)2(Phen)2(H2O)]·3H2O (where, Ln3+ = Tb, Dy, Ho, Er and Yb) crystallizes in the monoclinic space group P21/n, as a pure phase. The asymmetric unit is formed by two crystallographically independent metallic centers with an 8-coordinate environment [Ln1 = Ln2 = square antiprism polyhedron (SAPR-8)]35 (Fig. 2). Furthermore, while defining the primary building units (PBU), the Ln1 is coordinated to: (i) two nitrogen atoms of the 1,10-phenanthroline molecule, (ii) three oxygen atoms of the sulfonate group, (iii) two oxygen atoms of the phenyl group and (iv) one oxygen atom of the water molecule. Whereas the Ln2 coordination environment is formed by: (i) two nitrogen atoms of the 1,10-phenanthroline molecule, (ii) four oxygen atoms of the sulfonate group and (iii) two oxygen atoms of the phenolate group. Dimeric shared-edge secondary building units (SBUs) are formed by the junction of the metallic centers via syn–syn μη2 and μη2 modes of two sulfonates and two phenolate groups, respectively. SBUs are linked along the [101] crystallographic direction by the ligand through the sulfonate group giving rise to a 1D ladder-shaped coordination polymer. According to the classification proposed by Cheetham et al.,36 these coordination polymers can be classified as inorganic hybrid chains I1O0; where I1 means that the inorganic connectivity is 1D and O0 implies that the organic one is zero-dimensional.
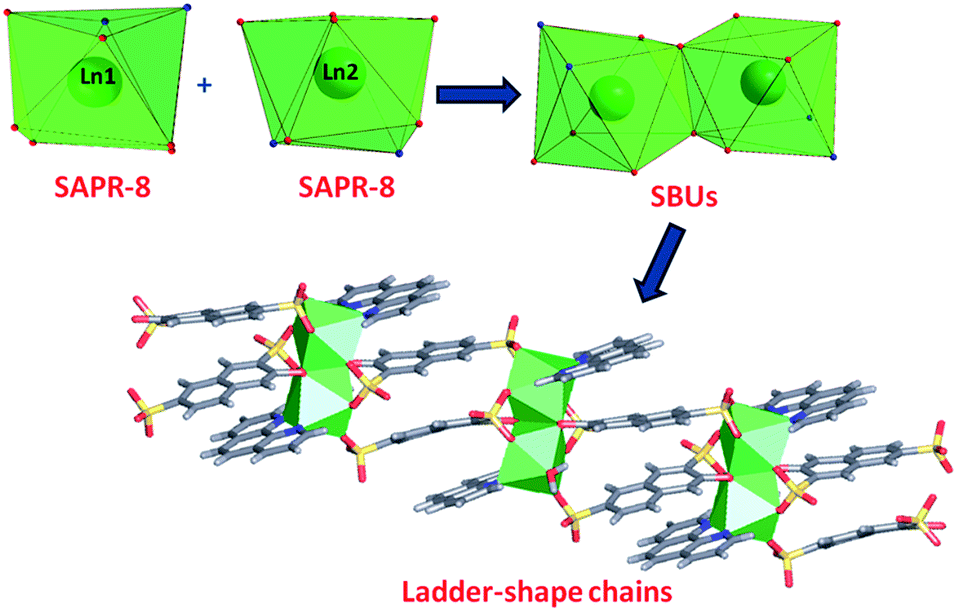 | ||
| Fig. 2 Coordination polyhedron, SBU and chains formed by the [Ln2(3-OHNDS)2(Phen)2(H2O)]·3H2O compounds. | ||
The ladder-shaped chains are joined together for the interstitial water molecules by strong hydrogen bonds. One water molecule links two chains along the [100] direction with distances O18–H18B⋯O5 of 3.000(6) and O18–H18A⋯O11 of 2.814(7) Å, respectively. Furthermore, hydrogen (O17–H17B⋯O8 = 3.231(6), O16–H16B⋯O6 = 3.083(7) Å) and π–π stacking interactions between the 1,10-phenanthroline rings along [100] were noticed. An intercentroid distance 3.6002(1) Å was also observed, to complete the formation of the 3D supramolecular crystal packing.
Structural comparison
Clear differences in the crystal packing of both phases are observed. The polymeric chains in phase 1 are formed along the [010] direction, while in phase 2 the same chains grow along the [101] direction (Fig. 3).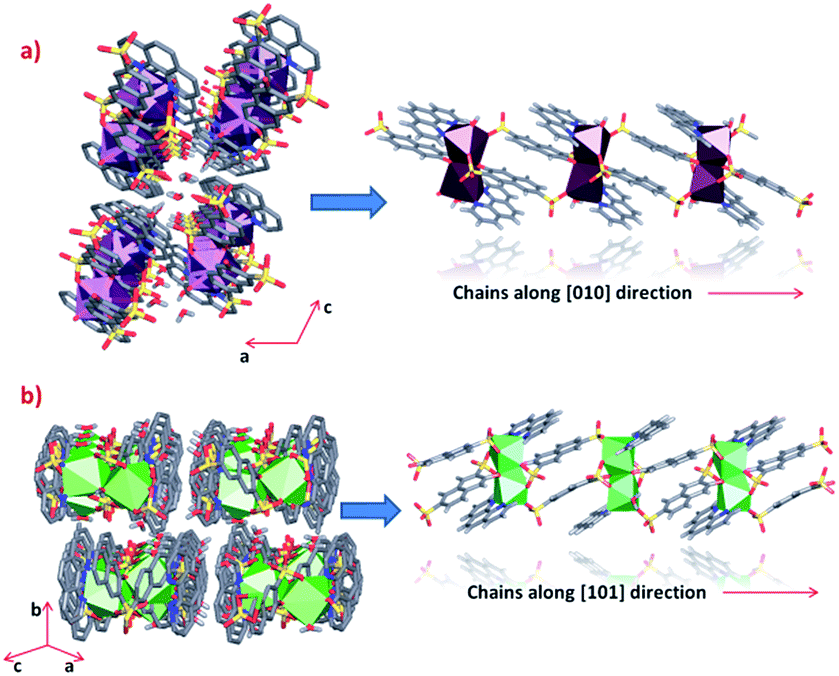 | ||
| Fig. 3 Crystal packing and polymeric chains formation of (a) phase 1 and (b) phase 2 (some hydrogen atoms from linkers were omitted for clarity). | ||
Through an analysis of the lanthanide coordination spheres, both phases are correlated with the loss of one coordination water molecule. This process provokes the coordination of one oxygen atom from the sulfonate group, generating the re-orientation of the chains. Hence, this results in the formation of two different crystal packings. Fig. 4 shows the formation of the thermodynamically stable syn–syn μη2 mode in phase 2, accompanied by a ligand rotation of almost 180°.
 | ||
| Fig. 4 Structural changes observed between phase 1 and phase 2. | ||
The differences in the crystal packing and the intermolecular interactions between the phases generate dissimilarities in the net topologies associated with such crystal structures. For this reason, the TOPOS16 program was employed to obtain the corresponding net simplifications. In both cases, the supramolecular packing is governed by multiple hydrogen bonds and weak hydrogen interactions. Considering the simplification of the supramolecular network due to stronger hydrogen bonds, it is possible to obtain a 2D supramolecular arrangement in both cases. Layers in phase 1 are formed by O18–H18A⋯O1 (2.935 Å) interactions which link the chains along the [100] direction. The 3-OHNDS ligand acts as a ditopic connector joining two SBUs, while the dimeric SBU is linked to four connectors where both of them are hydrogen bonds (Fig. 5a). This strong interaction gives rise to 2D layers in the (110) plane with a sql/Shubnikov tetragonal plane topology and a [44·62] point symbol.27
 | ||
| Fig. 5 Topological simplification of (a) phase 1 and (b) phase 2. | ||
In phase 2, chains interact via O5⋯H18B–O18–H18A⋯O11 bonds along the [100] direction, giving rise to 2D supramolecular layers in the plane (101) with topology 3,5L52 and [3·52][32·53·64·7] point symbol.27 The 3-OHNDS ligand acts as a 3-connected node linking the 5-connected dimeric SBUs (Fig. 5b).
Nanomechanical properties
To elucidate the structural–mechanical properties and relationships underlying the 3D framework, NI experiments were performed on selected single crystals of phase 2-Yb as a representative model. Phase 2-Yb was chosen as it was the largest crystal of all the series, Fig. 6. The crystal faces were identified via single-crystal X-ray diffraction (see Fig. 6a). The crystal indexing was carried out using the Mercury program (Fig. 6b).28 The orientations (010), (1 0−1) and (001) of the crystal faces were indented at a maximum penetration depth of 1200 nm (Fig. 6c). To avoid ambiguities, NI experiments on the acrylic substrate were made without crystals (see P–h curves in Fig. 7), with H and E values of 0.18 ± 0.01 and 4.5 ± 0.2 GPa. | ||
| Fig. 6 (a) Experimental procedure to determine the different facets of a phase 2-Yb single crystal. (b) Crystal model and (c) micrograph of single crystals of phase 2-Yb in different exposed hkl-oriented facets. In the same figures the Berkovich indents made to a maximum depth of 1200 nm are indicated. | ||
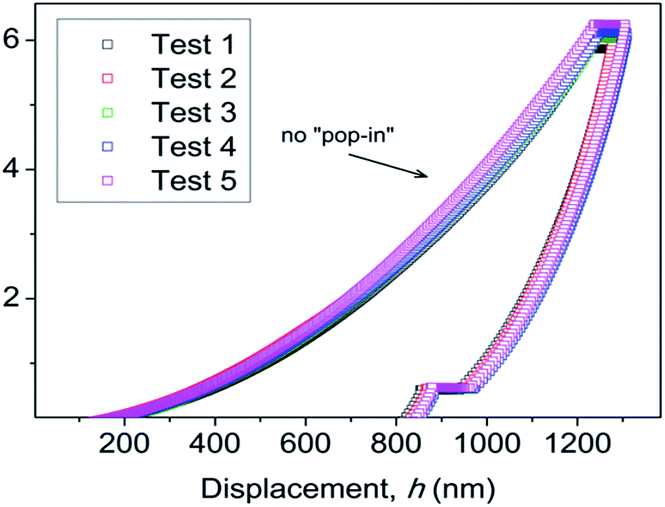 | ||
| Fig. 7 P–h profiles of the acrylic without crystals. | ||
Load-displacement (P–h) curves obtained on the natural facets of single crystals of phase 2-Yb are shown in Fig. 8. The average values of hardness (H) and Young's modulus (E), extracted from P–h curves of the three main faces are: (i) 0.32 ± 0.03 and 7.8 ± 0.5 GPa, (ii) 0.22 ± 0.02 and 5.6 ± 0.3 GPa and (iii) 0.05 ± 0.02 and 3.2 ± 0.6 GPa, assigned to the (010), (001) and (10−1) faces, respectively. This assignation was based on structural features.
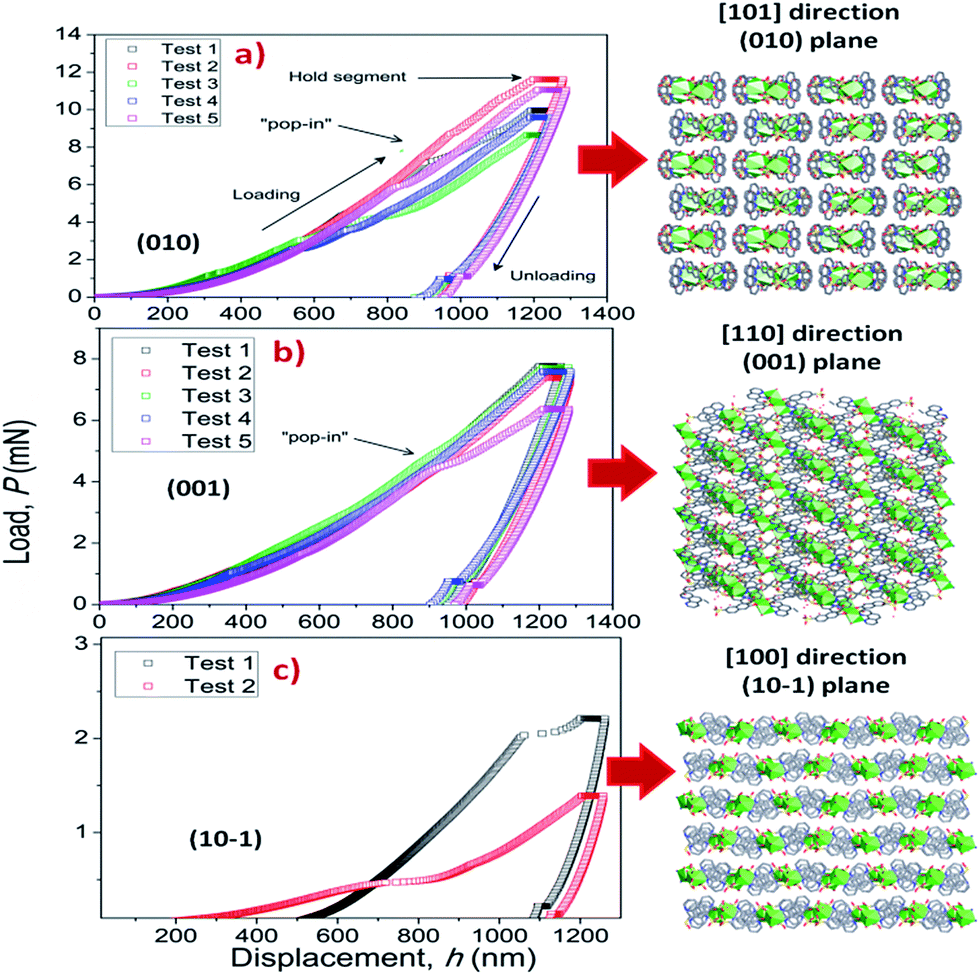 | ||
| Fig. 8 P–h profiles of the phase 2-Yb crystals in comparison with the crystal packing along (a) [101], (b) [110] and (c) [100] directions. | ||
Phase 2-Yb NI analysis shows high modulus values in the (010) and (001) faces. The indentation along [101] confers the highest stiffness in the crystal structure. It is due to the indenter axis, which in this case aligns parallel to the chain's growth direction. However, along the [110] direction (the stacking chain's direction, Fig. 8b), the modulus decreases by 28.2% with respect to the previous value.
Moreover, a decrease in H and E values should be expected when the indenter is perpendicular to the (101) plane. They could be explained in terms of the weak interactions exhibited in that plane (hydrogen bonds and π–π interactions, Fig. 8c). The same behavior was previously reported by Tan et al., where the [Cu3(H2O)2(O3PCH2CO2)2] compound is formed by layers parallel to the (010) plane. The authors reported the highest modulus values in the direction of the layer formation [011] and [110] (61.2 ± 2.2 and 55.2 ± 1.1 GPa, respectively) and a low value in the stacking layer direction (34.5 ± 0.9 GPa).37
It is important to note the presence of “pop-in” or displacement events detected on all crystal faces (see Fig. 8), which are an intrinsic characteristic of the material. This phenomenon could be explained by the breakages of weaker bonds and chain deformation (see the crystallographic section), being more prominent in the (010) plane. The same displacements have been described in low-dimensional MOFs such as [Cu3(H2O)2(O3PCH2CO2)2]37 and [Mn(2,2-dimethylsuccinate)].38 Tan et al.19 associated the presence of “pop-in” events with plastic deformations in the (001) plane of [Ce(oxalate)(formiate)]. The absence of “pop-in” steps when NI experiments were carried out on acrylic without phase 2-Yb crystals (Fig. 7) is noteworthy. To put these results in context, it is necessary to make comparisons with related compounds. As far as we know, this work is the first to report the mechanical properties of coordination polymers based on a sulfonate linker. According to the E vs. H map (Fig. 9), it was possible to locate phase 2-Yb between organic polymers and ceramics. For comparison, the E values found for phase 2-Yb are close to those reported for ZIFs (zeolitic imidazole frameworks) materials (3–9 GPa).39
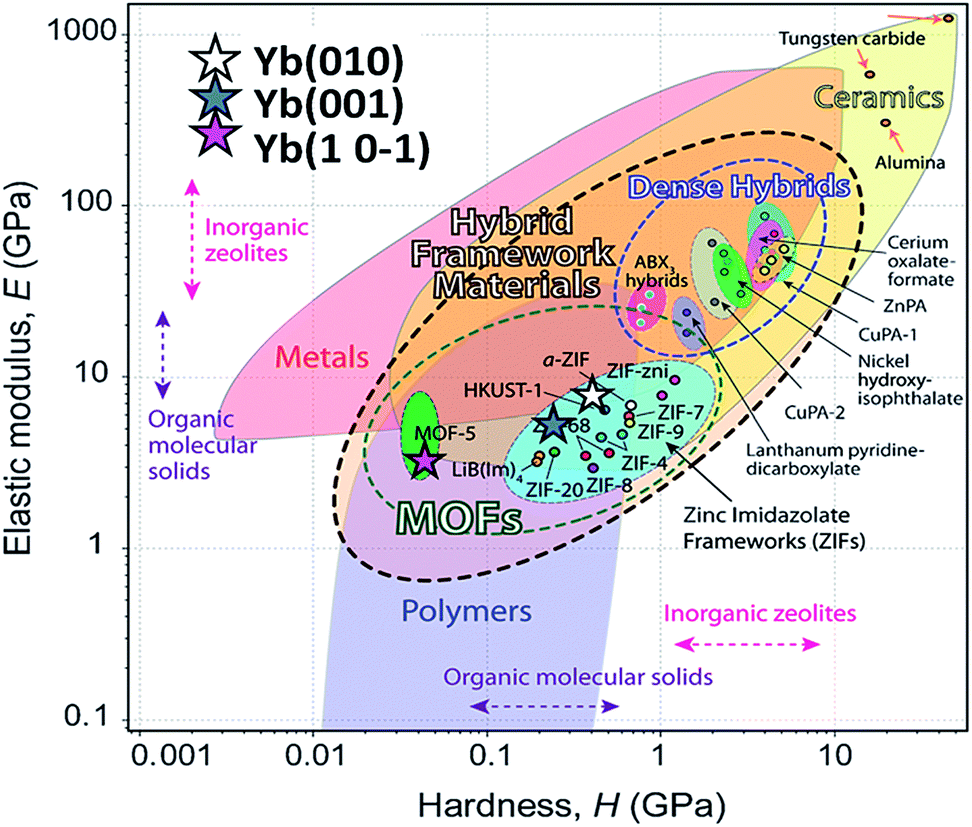 | ||
| Fig. 9 Young's modulus vs. hardness for different materials (from Tan et al.).29 The E and H values of different crystal facets of phase 2-Yb are included (stars) in comparison with other MOF-type materials. | ||
Luminescence properties
In order to make a photophysical characterization, a pre-treatment liquid ultrasonication (LU) was performed (from now on named for its metal component to simplify the notation, e.g. phase 2-Yb = Yb). LU has been employed as a “top-down” route to exfoliate low-dimensional MOFs materials.38,40 As can be seen in Fig. 10a, the as-synthesized Ln-CPs compounds (bulk phase) are obtained with a high degree of crystal condensation, forming “double-headed broom-like” structures. With LU, the big structures are separated in “needle”-shaped crystals with lengths between 60 and 100 μm and widths of ∼8 μm (Fig. 10b). We have recently demonstrated the impact of crystal condensation on the PL properties of 2D Eu-MOFs, showing that miniaturization–exfoliation procedures decreased the non-radiative pathways.41 Fig. 10d shows the system containing the ultrasonicated Yb crystals in ethanol (density 0.3 mg mL−1), illuminated with a red laser. As a consequence, the ethanolic suspension containing the needle-shaped crystals shows the characteristic Tyndall effect associated with colloidal systems. The nature of the ultrasonicated samples was studied by PXRD and FTIR techniques (see ESI S10–S12†). The PXRD pattern of ultrasonicated crystals showed the preferred orientation of the (020) crystallographic plane (ESI S11†). This is expected because LU takes places along that direction in which hydrogen bonds ensure crystalline packing. In accordance with the vibrational study (ESI 11†), the band located at 1668 cm−1, associated with the bending mode of water molecules, is affected by LU. Meanwhile the bands at 2854 and 2925 cm−1 related to νC–H aromatic modes are more intense after exfoliation, indicating that the LU occurs in the mentioned direction. These results confirmed the presence of a unique phase consistent with the bulk one. | ||
| Fig. 10 (a) SEM micrographs of “double-head broom-like” crystals, and (b) ultrasonicated needle-shape crystals of Yb. Demonstration of the Tyndall effect in colloidal suspensions containing ultrasonicated Yb crystals. (c) The path of the red laser beam is invisible in ethanol (ignoring reflections by the glass surfaces). (d) The laser beam is clearly visible through the colloidal ethanolic suspension of 0.3 mg mL−1 of Yb crystals. | ||
Due to the intense increase in investigations of luminescent MOFs,42,43 it was possible to identify basic steps in order to explore the PL properties. These studies include the collection of PL spectra, quantum yields (QYs) and lifetime values (τobs). In this sense, the quantification of the emitting light color is relevant to elaborate optical devices as chemical17 and physical sensors.44,45 In this work, the room temperature PL properties for Eu, Tb, Dy (“early” Ln3+), Ho, Er and Yb (“late” Ln3+) are reported and compared to analogous Ln3+/3-OHNDS coordination polymers (Ln = La, Pr, Nd, Sm).4
After monitoring the excitations at specific wavelengths [excitation spectrum of Eu monitored at 611 nm (5D0 → 7F2), Tb at 545 nm (5D4 → 7F5), Dy at 573 nm (7F9/2 → 6H13/2), and Ho, Er and Yb at 430 nm, a predominance of aromatic π → π*/n → n* transitions bands (300–400 nm range) was observed46 (see Fig. 11). Some compounds exhibited both ligands and 4f transitions (Eu, Tb, Dy). So, the selected excitation wavelengths (λexc) were 391 (25575 cm−1), 375 (26667 cm−1), 385 (25974 cm−1), 374 (26738 cm−1), 345 (28986 cm−1) and 338 nm (29586 cm−1) for Eu, Tb, Dy, Ho, Er and Yb, respectively. This fact indicates a rather antenna effect, as was seen in analogous Ln-CPs.4
 | ||
| Fig. 11 Excitation spectra of Ln-CPs. The corresponding assignations of labelled transitions can be found in ESI S9.† | ||
From the excitation spectrum for Eu (Fig. 11) two maximum peaks related to ligand (label a, 362 nm) and 5L6 → 7F0 Eu3+ (label b, 391 nm) transitions were located. It worth mentioning that the 5L6 → 7F0 transition is the highest peak in the excitation spectrum of carboxylate Eu-MOFs.47,48 In order to evaluate possible transfer phenomena, two experiments were carried out, employing two different excitation wavelengths, as shown in Fig. 12a. The resulting emission spectra show strong ligand centered luminescence (LCL) accompanied by a weak signal corresponding to the 5D0 → 7F2 transition (labelled as f in Eu-362 and Eu-391). This 4f transition is more intense when the sample is excited at 362 nm (27624 cm−1), evidence of possible energy migrations not only from ligands, but also from the 5D0 europium emissive level (19020 cm−1). These facts indicate a more efficient emission process when an excitation wavelength is selected close to the ligand absorption edge. The direct excitation into 4f levels (7F6 → 5H7) in Tb exhibits intense 5D4 → 7FJ (J = 5, 4, 3, 2 and 0) transitions (Tb-425), (labelled as e–h see ESI S9†). These are the most intense bands belonging to the organic molecules (labelled as c–d). The excitation at 375 nm (spectrum Tb-375) led to emissions dominated by ligand bands (labelled as c–f) accompanied by the Tb3+ ion's shell (labelled as g–j). In Tb-375 and Tb-425 spectra, the most intense 4f peak was the 5D4 → 7F5 transition. In the literature, this signal has been used to study the chemical sensor performance of many Tb-MOFs.49,50 The optical behavior of Dy was also evaluated, by exciting the sample at two different wavelengths. At λexc = 385 nm (25974 cm−1) the emission is dominated mainly by ligand bands with a poor contribution of 4f Dy3+ transitions (spectrum Dy-385); meanwhile at λexc = 446 nm (22422 cm−1) bands of aromatic linkers and some 4f transitions were identified. In Dy-MOFs,51,52 the most intense transition is 7F9/2 → 6H13/2 (see spectrum of Dy-446, Fig. 12c). On the other hand, Ho, Er and Yb compounds showed LCL characterized by π* → π/n* → n transitions in the visible range (see Fig. 12d), without 4f transitions.
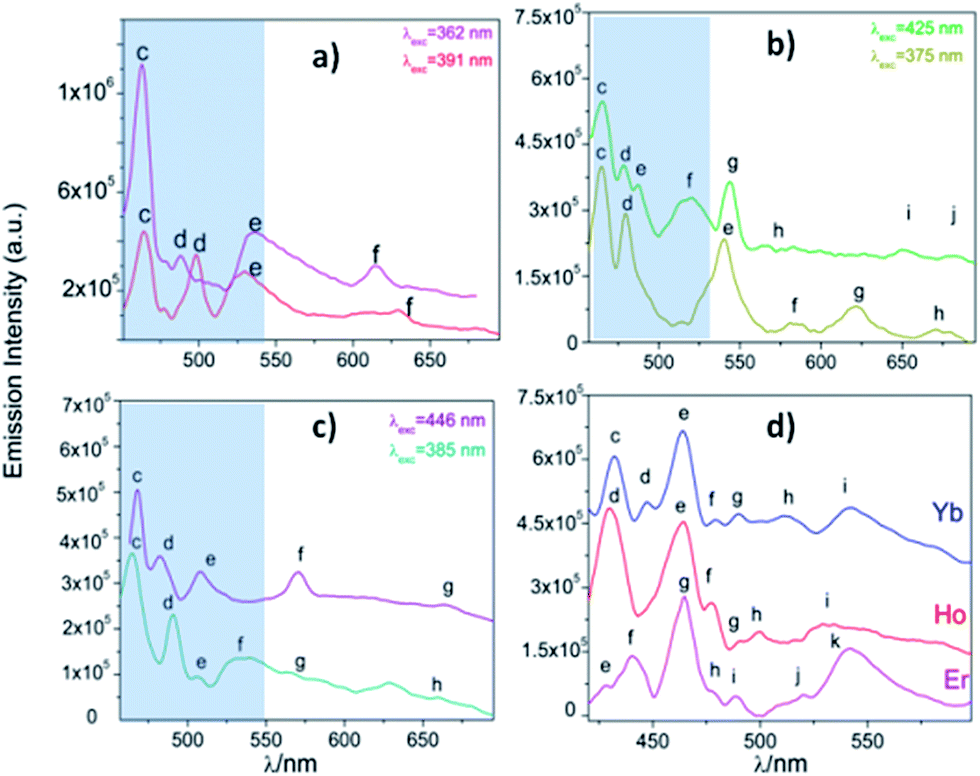 | ||
| Fig. 12 (a) Emission spectra of Eu (a), Tb (b), Dy (c) and Ho, Er and Yb (d). The corresponding labels are shown in ESI S9.† The marked blue zone denotes LCL. | ||
The behavior of strong quenching of Eu3+ emission ions and the low dominance of Tb3+ transitions could be explained by the multiphonon relaxation mechanism, previously reported for Eu and Tb-MOFs.53 The presence of two coordination (one in Tb and Dy) and three hydration water molecules per metal center in Eu, contributes to non-radiative pathways. Secondly, the concentration quenching of luminescence (condensation of emitting centers) is attributed to 4f emission attenuation. For instance, the proximities between the Ln-polyhedra (3.759 and 3.678 Å, for Tb and Yb, respectively) in Ln/3-OHNDS are shorter compared to the Ln-2,3-dimethylsuccinate (4.3001(1) Å),54 whose decrease in 4f intensity transitions was explained by this mechanism.
Quantification of luminescence: analysis of CIE x, y chromaticities
The quantification of the color (QC) emission in luminescent materials enables an accurate comparison between different materials, with their light-emitting performance. In this sense, the color coordinates are usually calculated by employing the CIE (Commision International de L'Éclairage) system, proposed in 1931.55 The QC is relevant for lanthanide device fabrication for many purposes.56,57 In this context, the color emissions of Eu, Tb, Dy, Ho, Er and Yb were quantified in terms of x, y color coordinates and compared to analogous phases. Eu, Tb and Dy compounds have different light-emitting behaviors (see Fig. 13a–c). The predominance of a blue color in this new set of compounds is explained by the influence of the 3-OHNDS ligand. Intrinsically, the 3-OHNDS ligand emits blue colour4 (x, y = 0.14, 0.1). Similar features could be found in analogous phases,4 where the majority of the samples presented x, y coordinates belonging to the blue zone of the CIE diagram, except [Sm(3-OHNDS)(H2O)2] and [Sm(3-OHNDS)(Phen)(H2O)]·3H2O, which emitted pink and white light. In addition, the character of white-light emission from Dy-446 is remarkable, exhibiting x, y coordinates 0.32, 0.37 close to dysprosium-based white emitters (0.33, 0.33).58,59 This fact is a highlighted feature of solid-state lighting, full color display and backlights60–62 ahead of other co-doped materials, because it is not necessary to add any other lanthanide into the structure to produce white light. As was described before, Ho, Er and Yb showed basically LCL, exhibiting x, y coordinates into the blue zone of the CIE diagram (see Fig. 13). The Table in the ESI S13† summarizes the QC data of so far reported coordination polymers based on lanthanides and 3-OHNDS. | ||
| Fig. 13 CIE diagrams for the studied systems. Corresponding x, y coordinates are displayed in ESI S13.† | ||
Sensing studies
There are many examples reporting sensing of vapor, liquid and substances in solution by fluorescence, quenching and enhancement processes. Lanthanide and metal transition MOFs have been employed as platforms to sense organic solvents,63 VOCs (volatile organic compounds),64 water65 and ions.50 Besides this, the use of these compounds in the detection of hazardous materials such as explosives66 is a significant issue with regard to security and safety measures. It is worth mentioning that studies of CPs or MOFs formed from naphthalene-linkers with applications as sensor platforms are really scarce. One relevant example is the work of Takashima et al.67 In this report they obtained a porous [Zn(BDC)2(dpNDI)] MOF (BDC = 1,4-benzenedicarboxylate; dpNDI = N,N′-di(4-pyridyl)-1,4,5,8-naphthalenediimide), with PL properties depending on the guest aromatic molecules.Generally, authors quantify the quenching effect by calculating the Quenching Efficiency (QE) defined as (I0 – I)/I0 × 100%, where I0 and I represent the intensity values before and after exposure, respectively.
To study the sensor capability of this new set of CPs, Tb was selected as a sensor model. According to the photophysical characterization, Tb showed dual luminescence centered at 464 nm (LCL) and at 541 nm (LnCL and 5D4 → 7F5 transition). Two VOCs (toluene and anisole) and two PAHs (polycyclic aromatic hydrocarbons) (anthracene and naphthalene) were chosen to study the sensor performance of Tb towards the intensity emission as signal transduction. Fluorescence measurements were carried out in a 1 cm quartz cuvette, containing some milligrams of Tb sample suspended in ethanol solutions of aromatics (for more details see the Experimental section). Fig. 14a and b show the emission spectra of Tb-nap, Tb-tol, Tb-ani and Tb-ant (λexc = 375 nm) in the LCL and LnCL region (Fig. 15).
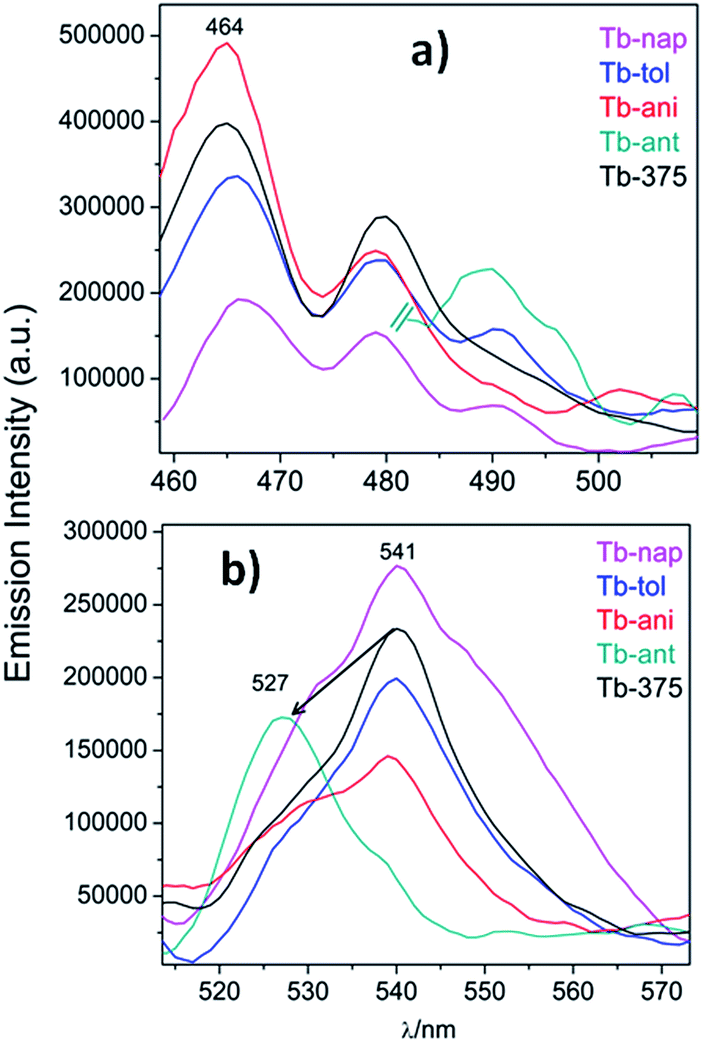 | ||
| Fig. 14 (a) Emission spectra of Tb-nap, Tb-tol, Tb-ani and Tb-ant zoomed into the LCL and (b) 5D4 → 7F5 emission zone. | ||
 | ||
| Fig. 15 (a) Variations of the relative intensity centered at 541 nm and (b) intensities ratio (I541/I464). | ||
Fig. 14a shows that the LCL of Tb is incremented in anisole (23.6%) and anthracene but quenched in naphthalene (QE = 51.6%). The LCL zone of Tb-ant is not complete because its signal is high enough that it saturated the fluorometer detector. In Fig. 14a, only one portion of the Tb-ant signal is shown. Furthermore, the 4f quenching effect is seen in the presence of anisole and anthracene (QE = 37.4% and 26.1% respectively). However, it was enhanced by 18.4% when Tb interacted with naphthalene (Fig. 14b). The spectrum of Tb-tol did not show any drastic changes in intensity. As can be observed in Fig. 14b, the 4f signal of Tb-ant was quenched and suffered a blue shift (Δλ = 14 nm). To the extent of our knowledge, this is the first example of hypsochromic shift in a 4f–4f emission transition in Tb-MOFs materials (Fig. 16).
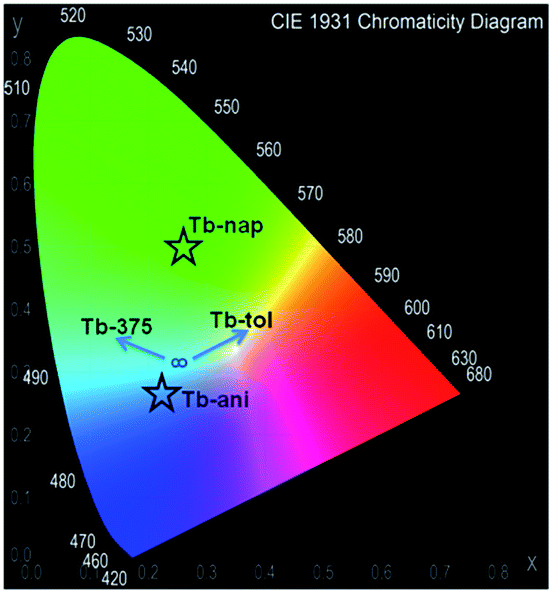 | ||
| Fig. 16 Chromaticity diagram of Tb-nap, Tb-tol, Tb-ani and Tb-ant in comparison with Tb-375. | ||
The CP-substrate interactions directly impacted on the intensity ratio I541/I464 (see Fig. 15b) to a major degree, when naphthalene molecules interacted with the emissive Tb3+ centers. This differential optical activity could be explained in terms of the photoelectron transfer (PET) process between aromatic molecules with different emissive sites. Finally, a QC was carried out (Fig. 16). This analysis reflected differences in the colorimetric response of Tb under different chemical environments around the emissive material: green luminescence in naphthalene (x, y = 0.26, 0.5), blue in anisole (x, y = 0.22, 0.26) and blue-green in toluene solution (x, y = 0.26, 0.31). It is also observed that the sensing did not affect the vibrational behavior of Tb (see ESI S14†).
Conclusions
As a product of hydrothermal synthesis of the series of lanthanide metals with 3-OHNDS and 1,10-phenanthroline ligands, two different crystalline phases were obtained and fully characterized. A general behavior was observed with the formation of the different phases, where the “LREE” metals (La–Eu) gave rise to the formation of phase 1, while phase 2 is obtained with the “HREE” lanthanides (Tb–Yb). As was demonstrated, Eu3+ can be described as the inflection point which divides the two phases. Structural differences are caused by the loss of one coordinated water molecule, triggering the rearrangement of the packing and symmetry changes. The 2D supramolecular network was topologically simplified as sql and 3,5L52 for phases 1 and 2, respectively. Moreover, nanoindentations on single crystals showed anisotropic elastic modulus and hardness. This is the first example of such a type of characterization in sulfonate-coordination polymers. The degree of anisotropy along the crystallographic faces could be directly correlated with the crystalline structure. This was evidence for the presence of strong covalent bonding interactions (Ln–O–Ln) accompanied by weak interactions within the same framework which could lead to highly directional mechanical responses.A comprehensive characterization of the PL properties of the reported phases was performed. The optical performance is mainly dominated by LCL accompanied by 4f-transitions. The quenching of the 4f emissions could be explained by two non-radiative mechanisms: multiphonon relaxation, and concentration quenching of luminescence. At this point, it is important to mention that an almost white-light Dy emission was obtained. Also, the activity of the Tb compound as a chemical sensor was evaluated. The intensity ratio parameter was used as a tool for distinguishing the presence of different aromatic compounds. Moreover, a hypsochromic shift transition of a Tb-CP has been observed.
With all these results in mind, it was possible to establish structural–mechanical–optical relationships. The combination of sensor activity along with elastic and hardness anisotropy makes these compounds multifunctional materials with emerging applications.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Acknowledgements
R. D. acknowledges Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior for the CAPES/PNPD scholarship from the Brazilian Ministry of Education, to M. Soto-Monsalve and R. de Almeida Santos for measurements and FAPESP (2009/54011-8) for providing Apex-II equipment. J. E. is grateful to CNPq for the research fellowships. G. E. G. acknowledges CONICET (Consejo Nacional de Investigaciones Científicas y Técnicas) for the postdoctoral fellowship and INTEQUI-UNSL for the TGA analysis. D. F. L. acknowledges CONICET doctoral fellowship. G. J. A. A. S. I. and M. C. F. are members of CIC-CONICET.References
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- C. B. Aakeroy, N. R. Champness and C. Janiak, CrystEngComm, 2010, 12, 22–43 RSC.
- R. F. D'Vries, V. A. de la Peña-O’Shea, Á. Benito Hernández, N. Snejko, E. Gutiérrez-Puebla and M. A. Monge, Cryst. Growth Des., 2014, 14, 5227–5233 Search PubMed.
- R. F. D'Vries, G. E. Gomez, J. H. Hodak, G. J. A. A. Soler-Illia and J. Ellena, Dalton Trans., 2016, 45, 646–656 RSC.
- A. E. Platero-Prats, M. Iglesias, N. Snejko, A. n. Monge and E. Gutiérrez-Puebla, Cryst. Growth Des., 2011, 11, 1750–1758 CAS.
- F. Gándara, E. G. r. Puebla, M. Iglesias, D. M. Proserpio, N. Snejko and M. A. n. Monge, Chem. Mater., 2009, 21, 655–661 CrossRef.
- R. F. D'Vries, M. Iglesias, N. Snejko, E. Gutiérrez-Puebla and M. A. Monge, Inorg. Chem., 2012, 51, 11349–11355 CrossRef PubMed.
- F. Gándara, E. Gutiérrez-Puebla, M. Iglesias, N. Snejko and M. A. n. Monge, Cryst. Growth Des., 2009, 10, 128–134 Search PubMed.
- J. Perles, M. Iglesias, M.-Á. Martín-Luengo, M. Á. Monge, C. Ruiz-Valero and N. Snejko, Chem. Mater., 2005, 17, 5837–5842 CrossRef CAS.
- K. Müller-Buschbaum, F. Beuerle and C. Feldmann, Microporous Mesoporous Mater., 2015, 216, 171–199 CrossRef.
- R. F. D'Vries, S. Alvarez-Garcia, N. Snejko, L. E. Bausa, E. Gutierrez-Puebla, A. de Andres and M. A. Monge, J. Mater. Chem. C, 2013, 1, 6316–6324 RSC.
- F. Gandara, N. Snejko, A. d. Andres, J. R. Fernandez, J. C. Gomez-Sal, E. Gutierrez-Puebla and A. Monge, RSC Adv., 2012, 2, 949–955 RSC.
- S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS PubMed.
- R. F. D'Vries, I. Camps and J. Ellena, Cryst. Growth Des., 2015, 15, 3015–3023 Search PubMed.
- R. F. D'Vries, V. A. de la Peña-O’Shea, N. Snejko, M. Iglesias, E. Gutiérrez-Puebla and M. A. Monge, J. Am. Chem. Soc., 2013, 135, 5782–5792 CrossRef PubMed.
- J. C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- A. Gouldstone, N. Chollacoop, M. Dao, J. Li, A. M. Minor and Y.-L. Shen, Acta Mater., 2007, 55, 4015–4039 CrossRef CAS.
- J. C. Tan, J. D. Furman and A. K. Cheetham, J. Am. Chem. Soc., 2009, 131, 14252–14254 CrossRef CAS PubMed.
- I. Bruker-AXS, APEX2 Software Suite, 2, Madison, WI, 2006 Search PubMed.
- I. Bruker-Siemens, SAINT, V 6.28A, Madison, WI, 1997 Search PubMed.
- Z. Otwinowski and W. Minor, Methods in Enzymology, Academic Press, New York, 1997 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- H. P. K. Brandenburg, DIAMOND-Crystal and Molecular Structure Visualization, Crystal Impact: Kreuzherrenstr, Bonn, Germany, 2006, vol. 102, p. 53227 Search PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van De Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- J. C. Tan and A. K. Cheetham, Chem. Soc. Rev., 2011, 40, 1059–1080 RSC.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- R. F. D'Vries, V. A. de la Peña-O’Shea, N. Snejko, M. Iglesias, E. Gutiérrez-Puebla and M. Á. Monge, Cryst. Growth Des., 2012, 12, 5535–5545 Search PubMed.
- R. F. D'Vries, N. Snejko, M. Iglesias, E. Gutiérrez-Puebla and M. A. Monge, Cryst. Growth Des., 2014, 14, 2516–2521 Search PubMed.
- P. Henderson, in Developments in Geochemistry, ed. P. Henderson, Elsevier, 1984, vol. 2, pp. 1–32 Search PubMed.
- Z. L.-G. XIAN Chun-Ying and Y. U. Qing-Sen, Chem. J. Chin. Univ., 1999, 20, 1504–1508 Search PubMed.
- N. G. Connelly, T. Damhus, R. M. Hartshorn and A. T. Hutton, Nomenclature of Inorganic Chemistry - IUPAC Recommendations 2005, RSC Publishing, Cambridge, UK, 2005 Search PubMed.
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC.
- J. C. Tan, C. A. Merrill, J. B. Orton and A. K. Cheetham, Acta Mater., 2009, 57, 3481–3496 CrossRef CAS.
- J.-C. Tan, P. J. Saines, E. G. Bithell and A. K. Cheetham, ACS Nano, 2012, 6, 615–621 CrossRef CAS PubMed.
- J. C. Tan, T. D. Bennett and A. K. Cheetham, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9938–9943 CrossRef CAS PubMed.
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356–1359 CrossRef CAS PubMed.
- G. E. Gomez, M. C. Bernini, E. V. Brusau, G. E. Narda, D. Vega, A. M. Kaczmarek, R. Van Deun and M. Nazzarro, Dalton Trans., 2015, 3417–3429 RSC.
- M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2011, 112, 1126–1162 CrossRef PubMed.
- E. C. Spencer, J. Zhao, N. L. Ross, M. B. Andrews, R. G. Surbella and C. L. Cahill, J. Solid State Chem., 2013, 202, 99–104 CrossRef CAS.
- Y. Cui, F. Zhu, B. Chen and G. Qian, Chem. Commun., 2015, 51, 7420–7431 RSC.
- G. Accorsi, A. Listorti, K. Yoosaf and N. Armaroli, Chem. Soc. Rev., 2009, 38, 1690–1700 RSC.
- G. E. Gomez, A. M. Kaczmarek, R. Van Deun, E. V. Brusau, G. E. Narda, D. Vega, M. Iglesias, E. Gutierrez-Puebla and M. Á. Monge, Eur. J. Inorg. Chem., 2016, 2016, 1577–1588 CrossRef CAS.
- S. R. Russell, C. Gosset, X. Agache, C. Volkringer, N. Henry, R. Decadt, R. Van Deun, M. Visseaux and T. Loiseau, CrystEngComm, 2016, 18, 3594–3605 RSC.
- K. L. Wong, G. L. Law, Y. Y. Yang and W. T. Wong, Adv. Mater., 2006, 18, 1051–1054 CrossRef CAS.
- B. Chen, L. Wang, F. Zapata, G. Qian and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718–6719 CrossRef CAS PubMed.
- C. Daiguebonne, N. Kerbellec, O. Guillou, J.-C. Bünzli, F. Gumy, L. Catala, T. Mallah, N. Audebrand, Y. Gérault, K. Bernot and G. Calvez, Inorg. Chem., 2008, 47, 3700–3708 CrossRef CAS PubMed.
- Z.-J. Lin, B. Xu, T.-F. Liu, M.-N. Cao, J. Lü and R. Cao, Eur. J. Inorg. Chem., 2010, 2010, 3842–3849 CrossRef.
- P. J. Saines, M. Steinmann, J.-C. Tan, H. H. M. Yeung and A. K. Cheetham, CrystEngComm, 2013, 15, 100–110 RSC.
- G. E. Gomez, M. C. Bernini, E. V. Brusau, G. E. Narda, W. A. Massad and A. Labrador, Cryst. Growth Des., 2013, 13, 5249–5260 CAS.
- G. Kickelbick, in Hybrid Materials, Wiley-VCH Verlag GmbH & Co. KGaA, 2007, pp. 1–48 Search PubMed.
- C.-Y. Sun, X.-L. Wang, X. Zhang, C. Qin, P. Li, Z.-M. Su, D.-X. Zhu, G.-G. Shan, K.-Z. Shao, H. Wu and J. Li, Nat. Commun., 2013, 4, 2717 Search PubMed.
- P. Falcaro and S. Furukawa, Angew. Chem., Int. Ed., 2012, 51, 8431–8433 CrossRef CAS PubMed.
- M. R. N. Soares, M. J. Soares, A. J. S. Fernandes, L. Rino, F. M. Costa and T. Monteiro, J. Mater. Chem., 2011, 21, 15262–15265 RSC.
- A. M. Kaczmarek, K. Van Hecke and R. Van Deun, Inorg. Chem., 2014, 53, 9498–9508 CrossRef CAS PubMed.
- B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585–1595 CrossRef.
- Y. H. Niu, M. S. Liu, J. W. Ka, J. Bardeker, M. T. Zin, R. Schofield, Y. Chi and A. K. Y. Jen, Adv. Mater., 2007, 19, 300–304 CrossRef CAS.
- H.-C. Su, H.-F. Chen, F.-C. Fang, C.-C. Liu, C.-C. Wu, K.-T. Wong, Y.-H. Liu and S.-M. Peng, J. Am. Chem. Soc., 2008, 130, 3413–3419 CrossRef CAS PubMed.
- B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693–1696 CrossRef CAS.
- H. Xu, X. Rao, J. Gao, J. Yu, Z. Wang, Z. Dou, Y. Cui, Y. Yang, B. Chen and G. Qian, Chem. Commun., 2012, 48, 7377–7379 RSC.
- Y. Yu, J.-P. Ma and Y.-B. Dong, CrystEngComm, 2012, 14, 7157–7160 RSC.
- S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed.
- Y. Takashima, V. M. Martínez, S. Furukawa, M. Kondo, S. Shimomura, H. Uehara, M. Nakahama, K. Sugimoto and S. Kitagawa, Nat. Commun., 2011, 2, 168 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental X-ray powder patterns, TG and DSC analysis, FTIR spectra and assignment of the excitation and emission transitions. CCDC 1477857–1477863. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra23516f |
| This journal is © The Royal Society of Chemistry 2016 |