
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aqueous biphasic iron-catalyzed asymmetric transfer hydrogenation of aromatic ketones†
K. Z.
Demmans
,
O. W. K.
Ko
and
R. H.
Morris
*
Department of Chemistry, University of Toronto, 80 Saint George Street, Toronto, Ontario M5S3H6, Canada. E-mail: rmorris@chem.utoronto.ca
First published on 9th September 2016
Abstract
For the first time, an iron(II) catalyst is used in the biphasic asymmetric transfer hydrogenation (ATH) of ketones to enantioenriched alcohols employing water and potassium formate as the proton and hydride source, respectively. The precatalyst [FeCl(CO)(P–NH–N–P)]BF4 (P–NH–N–P = (S,S)-PPh2CH2CH2NHCHPhCHPhNCHCH2PPh2) in the organic phase with the substrate is activated by base to produce a system that rivals the best ruthenium biphasic ATH catalysts in activity but not enantioselectivity. Biorenewable 2-methyltetrahydrofuran as a cosolvent and biodegradable TWEEN80 as a surfactant were added to the reaction mixture to greatly decrease the mass-transfer limitations caused by the biphasic reaction mixture. The enantioselectivity of the reduction was as high as 76% depending on the substitution pattern of the arylketone employed. NMR studies verified the formation of an iron hydride [FeH(CO)(PPh2CH2CH2NHCHPhCHPhNCHCHPPh2)] intermediate as was observed in our 2-propanol-based ATH studies.
Introduction
The asymmetric hydrogenation of ketones is a widely studied area of chemistry that has seen a recent surge in iron-catalyzed reactions employing various tridentate and tetradentate ligands.1 Our group has focused on the asymmetric transfer hydrogenation (ATH) of ketones, a strategy that removes the dangers of using pressurized hydrogen by utilizing 2-propanol as the proton and hydride source.2Over the past two decades, the library of proton and hydride sources has been expanded to include azeotropic mixtures of formic acid and triethylamine for catalysts active in acidic conditions,3 sodium formate and water for catalysts active in basic conditions,3c,4 and even using glycerol as the proton and hydride source.5 All of these studies involve the use of expensive ruthenium, rhodium, or iridium catalysts to obtain enantiomerically pure alcohols. Alcohols produced in this way can be valuable intermediates for the synthesis of pharmaceuticals.4c,6 Interestingly, a recent study by Hu et al. employs an iron(II) PONOP pincer catalyst with sodium formate in methanol to reduce various aldehydes quantitatively.7 Concurrently, we sought to replace our 2-propanol-based ATH with water and sodium formate to shift the equilibrium to favour full conversion to the product alcohol as shown schematically in Fig. 1.
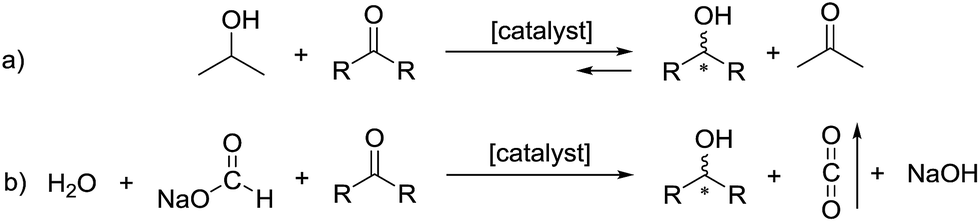 | ||
| Fig. 1 (a) The reversible ATH of ketones when using 2-propanol and (b) the irreversible ATH when using water and sodium formate as the proton and hydride sources. | ||
In the ATH with 2-propanol, acetone is formed which can be reduced back into 2-propanol. However, when employing sodium formate, the CO2 that is produced can be purged from the system thus favouring the forward reaction and reducing the ketone substrate completely. In principle, the CO2 could be hydrogenated in a separate process to regenerate the formate. It is important to note that the reaction becomes more basic as sodium hydroxide is formed as a byproduct of the deprotonation of water. This can affect the enantioselectivity of the process as discussed in this study.
Another disadvantage was the formation of a biphasic reaction mixture; the substrate and catalyst form an organic top layer while the bottom was a polar basic aqueous layer. Fortunately, extensive mechanistic studies about the ATH in water with ruthenium have been reported by several groups.4a,8
With these studies in mind, the iron catalyst 1 shown in Fig. 2 was successfully used in conjunction with water and sodium formate for the enantioselective reduction of ketones for the first time. Due to its large abundance and lower toxicity, iron serves as a greener alternative to ruthenium, rhodium, and iridium, metals that are typically used in such a process.4c,6,8,9
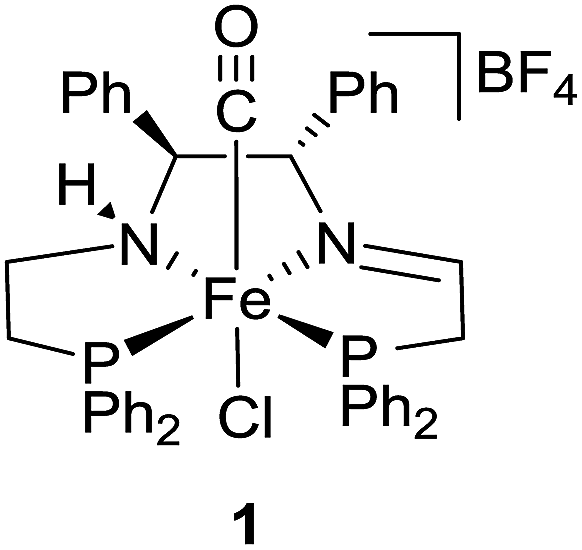 | ||
| Fig. 2 Iron(II) catalyst precursor trans-(S,S)-[FeCl(CO)(P–NH–N–P)][BF4].2d | ||
Results and discussion
Starting reaction conditions and hypothesis of reaction mechanism
Our previously reported conditions for the ATH with 2-propanol were adapted to a mixture of water, sodium formate, and a strong base such as potassium hydroxide (to ensure activation of 1). This solution was placed in a flask and sealed under argon gas against a mercury bubbler set at about 1.5 atm pressure. In contrast to the homogeneous 2-propanol system, the mass-transfer limitations caused by the formation of the biphasic system require the addition of a phase transfer catalyst such as tetrabutylammonium bromide (TBA+Br−) to ease the activation of the non-polar iron catalyst 1 by the basic water.Fig. 3 presents a possible mechanism of biphasic catalysis. It is based on the homogeneous ATH mechanism using 2-propanol as the solvent and reductant as elucidated earlier.2d,10 In the two phase system, the iron-containing species are thought to remain in the organic phase on the basis of the colour of the phases while salts such as KOH, KCl, and KHCOO remain almost exclusively in the aqueous phase as indicated by the blue boxes.
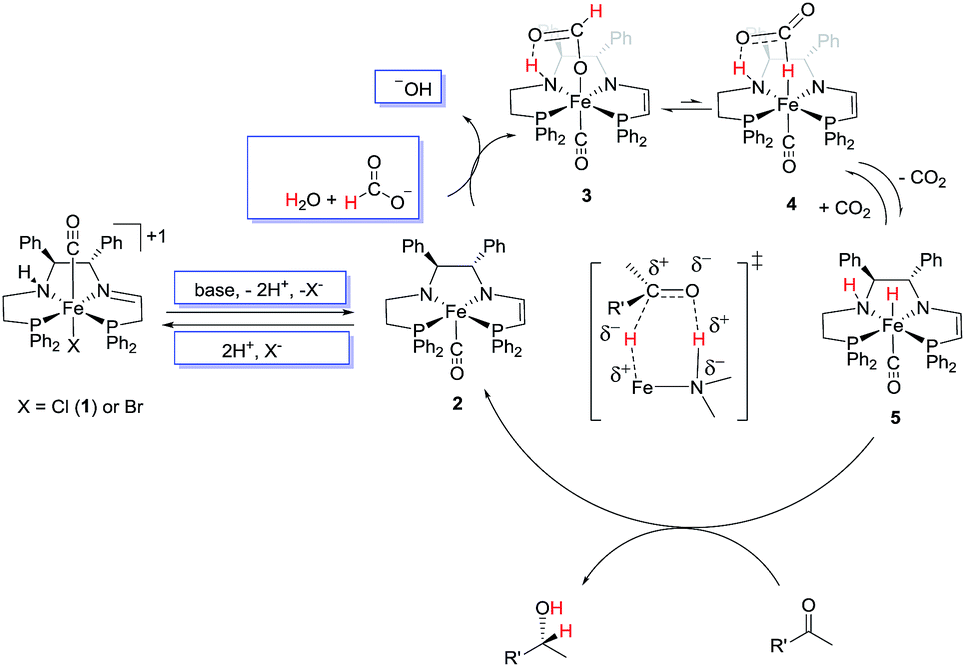 | ||
| Fig. 3 Proposed catalytic cycle. | ||
In the mechanism, the catalyst precursor iron chloride complex 1 reacts with base, either hydroxide or formate from the aqueous phase, and loses one equivalent of HCl to form the amido–eneamido complex 2; in the original 2-propanol mechanism, potassium 2-propoxide acts as the base.
The results below are discussed in the light of this proposed mechanism.
Optimization of the reaction conditions
To begin, the sodium formate concentration was adjusted to 1000, 4000, and 8000 equivalents relative to 1 (Table 1, entries 1–3).|
|
||||
|---|---|---|---|---|
| Entry | TBA+X− | Equiv. | Conversion (%) | e.e. (%) |
a All values are in equivalents relative to the iron catalyst: 0.006 mmol 1, 1000 eq. NaHCOO, 8 eq. KOH, 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetophenone:catalyst ratio, 4 mL water, 600 RPM stirring rate. The (R) enantiomer is always the main product.
b 4000 eq. of NaHCOO.
c 8000 eq. of NaHCOO.
d No KOH was added.
e 32 eq. of KOH.
f 100:1 acetophenone:catalyst ratio. :1 acetophenone:catalyst ratio, 4 mL water, 600 RPM stirring rate. The (R) enantiomer is always the main product.
b 4000 eq. of NaHCOO.
c 8000 eq. of NaHCOO.
d No KOH was added.
e 32 eq. of KOH.
f 100:1 acetophenone:catalyst ratio.
|
||||
| 1 | X = Br | 1 | 9 | 76 |
| 2b | 1 | 20 | 79 | |
| 3c | 1 | 25 | 79 | |
| 4 | 2 | 7 | 77 | |
| 5 | 5 | 5 | 74 | |
| 6 | 0 | 10 | 77 | |
| 7d | 1 | 9 | 77 | |
| 8e | 1 | 9 | 67 | |
| 9 | X = BF4 | 1 | 18 | 80 |
| 10 | 5 | 15 | 78 | |
| 11f | 1 | 23 | 79 | |
As expected, the conversion greatly increases with the concentration of formate anions in the water. Increasing the equivalents of TBA+Br− (1, 2, and 5) decreases the conversion in accordance with what has been previously observed (Table 1, entries 1, 4, 5).4a,8 Interestingly, when TBA+Br− was omitted from the reaction a slight increase in catalytic activity was observed (Table 1, entry 6). We concluded that the introduction of more halide anions could deactivate the catalyst by coordinating to the open site as shown in the proposed catalytic reaction mechanism (Fig. 3). Therefore the phase transfer catalyst's counteranion was switched to tetrafluoroborate to match that of 1, resulting in a doubling of activity (Table 1, entry 9). Once again as the concentration of TBA+BF4− is increased beyond the optimal one equivalent, the activity decreases. Lastly, removing the potassium hydroxide had no impact on the activity which demonstrated that the water layer was basic enough to activate 1 (Table 1, entry 7 compared to entry 1). Furthermore, adding a large excess of potassium hydroxide causes a sharp decrease in enantiomeric excess (e.e.) (Table 1, entry 8), presumably by some modification of the catalyst structure. Therefore potassium hydroxide was not used in subsequent studies.
With all of the aforementioned catalytic reactions, regardless of how extreme the reaction conditions were pushed, an asymptote of approximately 25% conversion was observed. Even lowering the acetophenone:catalyst ratio to 100:1 did not lead to a high conversion (Table 1, entry 11). The experimental set-up was not allowing for the carbon dioxide being produced in the reaction flask to be purged from the system efficiently. Therefore, an argon flow through a condenser to the external bubbler (filled with mineral oil) was added to the experimental set-up, as well as decreasing the reaction volume to 1 mL and using 2000 eq. of sodium formate to match the concentration used in Table 1, entry 3. These changes increased the conversion to 74% after 4 h (Table 2, entry 1).
| Entry | Time (h) | Conversion (%) | e.e. (%) |
|---|---|---|---|
|
a Constant argon flow was used to purge the reaction flask. All values are in equivalents relative to the iron catalyst: 0.006 mmol 1, 2000 eq. NaHCOO, 1 eq. TBA+BF4−, 100:1 acetophenone:catalyst ratio, 1 mL water, 600 RPM stirring rate, 65 °C.
b MeTHF was added (1 mL).
c 2000 eq. of KHCOO was used.
|
|||
| 1 | 4 | 74 | 53 |
| 2b | 1 | 83 | 57 |
| 3b,c | 1 | >99 | 57 |
While the conversion greatly increased, a large increase in the amount of hydroxide anions produced lead to a decrease in e.e. from 78% to 53%. This was proposed to be due to the modification of 2 into alternate iron complexes with a lower enantioselectivity under very basic conditions.2a,d,9e Most of the active ruthenium biphasic ATH catalysts described in the literature are found in the aqueous layer along with a formate salt while the substrate is found in an organic layer.3c,4 This allows the ruthenium catalyst to be in constant contact with the proton and hydride source. In contrast, the biphasic system studied herein has the substrate and 1 in the non-polar layer, with sodium formate and water in the polar layer. This causes the transfer of the proton and hydride from water and sodium formate to 1 to be slow while subsequent transfer of this dihydrogen equivalent from the iron catalyst to the ketone substrate in the organic layer should be very rapid if the mechanism is the same as the one established for ATH with 2-propanol. For this reason a cosolvent for the organic layer was added to increase the size of the interphase region and promote the interaction of 2 with the proton and hydride sources. 2-Methyltetrahydrofuran (MeTHF) was chosen over tetrahydrofuran as the cosolvent due to its higher boiling point, allowing for higher reaction temperatures, and for its bio-renewability as it is derived from C6 sugars.11 With MeTHF, 83% conversion was obtained in only an hour (Table 2, entry 2). Switching the hydride source to potassium formate allowed for the quantitative conversion in an hour (Table 2, entry 3) and therefore the potassium salt was used hereafter. The difference in reactivity of the two salts cannot be attributed to solubility differences in the organic phase. We measured the concentration of a saturated solution of KHCOO or NaHCOO in a dry MeTHF/acetone mixture (v/v = 4/0.2) at 65 °C to be approximately 0.01 M. However the sodium salt would be expected to be more strongly ion paired in the organic phase and therefore less reactive. It has been shown in the hydrogenation of CO2 that the densely positive charge from a lithium cation eases the release of the κ-O-formato from the iron complex to form the product lithium formate.12 Therefore the catalytic cycle and the reaction rate found herein should also be cation sensitive.
Substrate scope
With the optimized reaction conditions in hand, a substrate scope was conducted using bio-renewable MeTHF as the cosolvent (Fig. 4).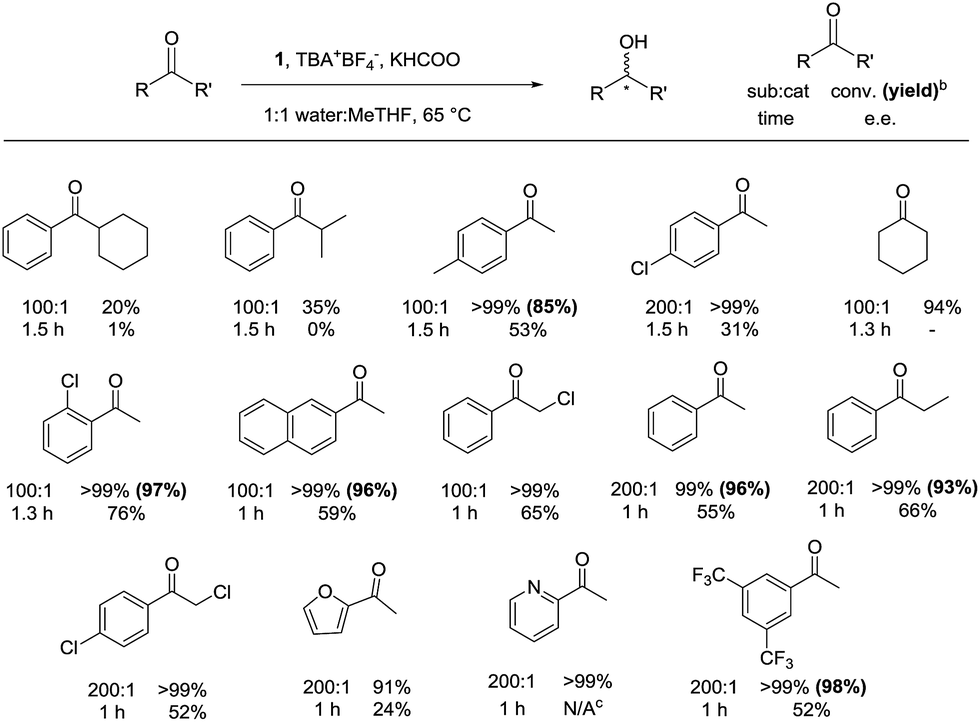 | ||
| Fig. 4 Substrate scope under optimized conditionsa. a Constant argon flow was used to purge the reaction flask. All values are in equivalents relative to the iron catalyst: 0.006 mmol 1, 2000 eq. KHCOO, 1 eq. TBA+BF4−, 2 mL reaction volume (1:1 water:MeTHF), 600 RPM stirring rate. b Isolated yield using the specifications found in the ESI.†c Gas chromatography could not separate the (R) and (S) alcohols accurately. | ||
The ketones with bulky substrates on the right side as drawn in Fig. 4, such as cyclohexylphenylketone and isobutyrophenone, were the only substrates that were not reduced to quantitative conversion. In all other cases, the ketones were quantitatively converted to their respective alcohols with moderate enantioselectively. An exceptional case displaying high enantioselectivity was 2′-chloroacetophenone with 76%. Iron precatalyst 1 displayed some functional group tolerance, converting 2-acetylfuran and 2-acetylpyridine to the alcohols to near quantitative yields within one hour. A major benefit of the protocol presented here was the reduction of ketones that are soluble in MeTHF but not soluble in 2-propanol, such as 2,4′-dichloroacetophenone; the use of 2-propanol gave no conversion (see ESI, S15†). 3′,5′-Bis(trifluoromethyl)acetophenone was converted at 200:1 substrate:catalyst ratio in one hour, albeit with a moderate enantioselectivity. The product alcohol is a precursor to aprepitant, a drug to relieve nausea caused by chemotherapy treatment.13
The scale of the reaction can be increased tenfold to successfully convert 1.4 mL of acetophenone in an hour while maintaining the e.e. at 55%. An increased RPM and larger stirring bar must be used to efficiently mix the two immiscible layers.
Moderate reaction conditions and study on additives
All aforementioned studies involved in the optimization of this system involved a constant flow of argon through the reaction mixture to purge the CO2 produced and shift the equilibrium towards the alcohol product. This wastes a large volume of argon over the course of an hour. By only purging the system at the beginning of catalysis for 10 seconds then leaving the reaction flask open to an oil bubbler with no subsequent purging, the conversion decreased from >99% to 43% (Table 3, entry 3).| Entry | Flow | Additive | Conversion (%) | e.e. (%) |
|---|---|---|---|---|
|
a All values are in equivalents relative to the iron catalyst: 0.006 mmol 1, 2000 eq. KHCOO, 1 eq. TBA+BF4− or 5 eq. TWEEN80, 200:1 acetophenone:catalyst ratio, 2 mL reaction volume (1:1 water:MeTHF), 600 RPM stirring rate, 65 °C, 1 h.
|
||||
| 1 | Constant argon | TBA+BF4− | >99 | 55 |
| 2 | Constant nitrogen | TBA+BF4− | 53 | 40 |
| 3 | Static | TBA+BF4− | 43 | 50 |
| 4 | Constant argon | TWEEN80 | >99 | 57 |
| 5 | Constant nitrogen | TWEEN80 | 69 | 59 |
| 6 | Static | TWEEN80 | 97 | 59 |
Purging with nitrogen does not yield a high conversion as nitrogen may coordinate to the open site of the active catalyst 2. Purging with a non-coordinating, heavier gas such as argon was necessary to achieve greater reaction rates. Fortunately when 5 eq. of the biodegradable THF-based surfactant, TWEEN80, was used the activity increased readily allowing for near quantitative conversion without constant argon purging. Upon the addition of TWEEN80, micelles may form which greatly increase the interphase area and decreases mass-transfer limitations. A study comparing the hydrophilic–lipophilic balance of many surfactants determined that TWEEN80 is highly soluble in water due to the long polyethylene glycol chains and also helps solubilize organics containing alkenes or aromatic groups due to the long chain alkene found in the structure of TWEEN80.14 This surfactant has been used in a catalytic hydroxylation reaction.15
To confirm that TWEEN80 was not providing protons or hydrides for the ketone reduction process, a few blank tests were run to determine its role in catalysis (Table 4).
| Entry | Condition | TWEEN80 (eq.) | Conversion (%) |
|---|---|---|---|
|
a All values are in equivalents relative to the iron catalyst: 0.006 mmol 1, 200:1 acetophenone:catalyst ratio, 1 mL MeTHF, 600 RPM stirring rate, 65 °C, 1 h.
|
|||
| 1 | 1 mL water, 2000 eq. KHCOO | 5 | >99 |
| 2 | No water, no KHCOO | 5 | 1 |
| 3 | No water, 2000 eq. KHCOO | 5 | 6 |
| 4 | No water, 2000 eq. KHCOO | 100 | 33 |
| 5 | No water, 2000 eq. KHCOO | 200 | 49 |
Comparing to the standard catalytic run (Table 4, entry 1), there was little to no conversion when both the proton and hydride source are removed from the mixture, while only 6% conversion was obtained by solely removing the proton source. As the amount of TWEEN80 was increased, the conversion also increases, demonstrating that TWEEN80 may act as a proton source; although it mainly contributes as a surfactant to increase conversion employing water as the proton source.
In order to determine whether the lower activity of the iron catalyst compared to the 2-propanol system was due to the formation of a biphasic reaction mixture or due to using formate as the hydride source, HDBU formate was synthesized according to literature procedures.16 HDBU formate dissolves in MeTHF at 65 °C, forming a single phase containing the required proton and hydride source. Since only 10% conversion was obtained in an hour, it does appear that there is an inherent barrier to the transfer of the hydride from formate to the iron.
Lastly, an attempt to decrease the temperature to 40 °C led to a large decrease in activity.
Cosolvent screen
Acetophenone was used as a representative substrate for the catalysis using water and potassium formate with various cosolvents by studying the activity with an increased substrate:catalyst ratio of 500:1. The conversion and e.e. were studied (Table 5).
| Entry | Cosolvent | Conversion (%) | e.e. (%) |
|---|---|---|---|
|
a Constant argon flow was used to purge the reaction flask. All values are in equivalents relative to the iron catalyst: 0.006 mmol 1, 2000 eq. KHCOO, 1 eq. TBA+BF4−, 500:1 acetophenone:catalyst ratio, 2 mL reaction volume (1:1 water:cosolvent), 600 RPM stirring rate, 65 °C, 1 h.
b Uses 5 eq. of TWEEN80 instead of TBA+BF4−.
c Uses both 1 eq. of TBA+BF4− and 5 eq. of TWEEN80.
|
|||
| 1 | DMF | 2 | 73 |
| 2 | MeOH | 11 | 64 |
| 3 | Toluene | 14 | 19 |
| 4 | MeTHF | 23 | 40 |
| 5 | MeCN | 30 | 59 |
| 6b | MeTHF | 35 | 55 |
| 7c | MeTHF | 67 | 57 |
| 8 | DCM | 41 | 59 |
| 9 | Heptanes | 44 | 41 |
| 10 | Et2O | 45 | 46 |
| 11 | THF | 49 | 61 |
While no immediate trend was observed based on the polarity of the cosolvent, there were a few important observations. DMF had the lowest conversion because of its ability to coordinate to the open site of 2 and deactivate the catalyst. There is a weak trend in increasing conversion with decreasing cosolvent boiling point. This is most likely due to an increase in the catalyst concentration over time as the cosolvent evaporates. While MeTHF was chosen as the cosolvent for most of the studies here, THF provides the best activity and the highest enantioselectivity. This can be explained by the formation of the active hydrido–eneamido iron complex (5 in Fig. 3). When THF was used as the cosolvent, the 1H NMR revealed the presence of a similar iron hydride species to the active species observed in our 2-propanol studies,2d while three iron hydride species were formed using MeTHF (see ESI, S16†). These extra hydride species may also be active in transfer hydrogenation, but produce alcohols with a lower e.e. (Table 5, entries 4 and 11).
Switching to TWEEN80 lead to increased conversions of 35%, while using the phase-transfer catalyst and the surfactant in combination lead to a drastic increase in activity with 67% conversion in an hour (Table 5, entries 4, 6, and 7).
Literature comparison of catalytic activity
Various biphasic systems using sodium formate and formic acid with water were collected in Table 6 to compare the turnover frequency (TOF). The ligands are shown in Fig. 5.| Entry | Precatalyst | Temp. (°C) | TOFa (h−1) | e.e. (%) | Ref. |
|---|---|---|---|---|---|
a
 , where the time was taken at near quantitative conversion. , where the time was taken at near quantitative conversion.
|
|||||
| 1 | [IrCl2Cp*]2 + L1 | 24 | 7 | 73 (R) | 17 |
| 2 | [RhCl2Cp*]2 + L2 | 25 | 94 | 91 (S) | 18 |
| 3 | [RhCl2Cp*]2 + L3 | 28 | 96 | 94 (R) | 19 |
| 4 | [RhClL4] | 28 | 67 | 96 (R) | 3b |
| 5 | [RuCl(p-cymene)L5] | 40 | 4 | 56 (S) | 20 |
| 6 | [RhCl2Cp*]2 + L6 | 40 | 70 | 99 (R) | 21 |
| 7 | [RuCl(p-cymene)L7] | 40 | 110 | 97 (R) | 3a |
| 8 | [RhCl2Cp*]2 + L8 | 40 | 199 | 93 (R) | 22 |
| 9 | [Ir(H2O)Cp*L9]2+ | 40 | 248 | — | 23 |
| 10 | [RhClCp*L10] | 40 | 310 | 97 (R) | 24 |
| 11 | [IrClCp*L11] | 40 | 388 | 99 (R) | 25 |
| 12 | [RuCl2(p-cymene)]2 + L12 | 60 | 9 | 82 (S) | 26 |
| 13 | 1 | 65 | 199 | 55 (R) | This work |
| 14 | [RuCl2(p-cymene)]2 + L13 | 80 | 20 | 28 (R) | 9f |
| 15 | [IrClCp*L14] | 80 | 1780 | — | 9d |
| 16 | [RuCl(p-cymene)L15]+ | 90 | 16 | — | 27 |
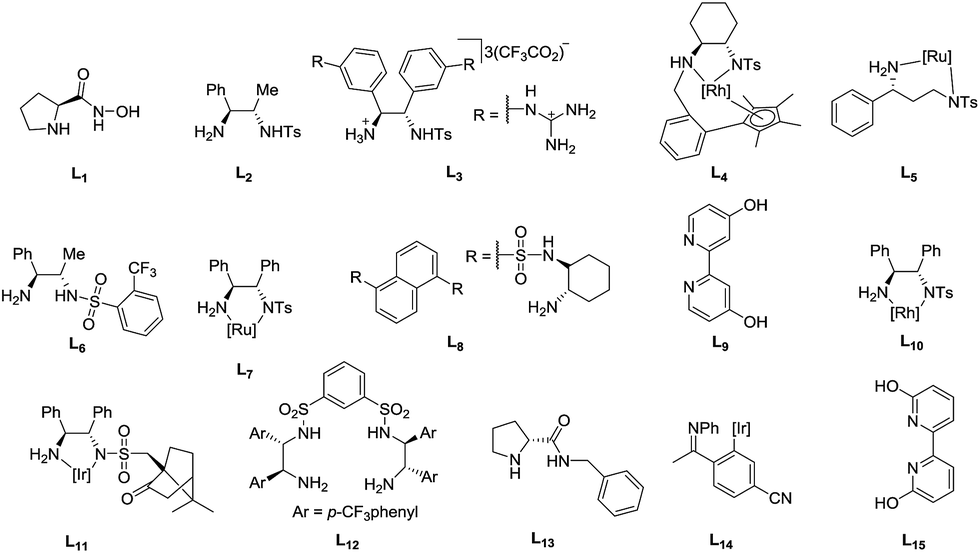 | ||
| Fig. 5 Ligand reference library for Table 6. | ||
With each temperature increment as shown in Table 6, ruthenium complexes tend to be the least active. In general, the activity increases from ruthenium to rhodium to iridium as demonstrated by Xiao et al. with a series of metal complexes employing L11.25 While it is hard to directly compare the activity of these complexes at different temperatures, iron catalyst 1 was shown to be more active than the ruthenium complexes from entries 5, 12, 14, 16. Several rhodium complexes were active at low temperatures and quite active at 40 °C, while the iridium complexes (entries 11 and 15) had the largest TOF of 388 h−1 at 40 °C and 1780 h−1 at 80 °C. Xiao et al. offer a more comprehensive review of transfer hydrogenation which includes use of supported catalysts, biomimetic catalysts working in conjunction with NADH, as well as Brønsted acid organocatalysts.13
The proposed catalytic cycle
The cycle is entered by the dehydrohalogenation of the chloro or bromo precursor complex 1 (Fig. 3) in the same fashion as the activation of the catalyst for use in ATH using 2-propanol as the reductant.2d,10 The addition of excess halide apparently reverses this process and causes a reduction in catalytic activity (Table 1). The major difference between this biphasic mechanism (Fig. 3) and mechanisms that utilize 2-propanol is the conversion of 2 to the iron hydride 5. In the current mechanism an equivalent of formic acid is postulated to add to 2 to form 3 with a formate bound via oxygen to the iron and via a hydrogen bond to the N–H group that is formed by protonation of the amido nitrogen of 2 (see ESI S15†). These steps may not be concerted, as suggested by kinetic isotope effect studies of a similar reaction by Xiao et al.4a A similar reaction and bonding mode was observed in the structure of RuH(O2CH)(NH2CMe2CMe2NH2)(PPh3)2.28 The κ-O-formate complex 3 would be very stable due to the hydrogen bond between the unbound oxygen of the formato and the amino group of the ligand. As such, the transformation from 3 to the required to κ-H-formate complex 4 would not be thermodynamically favoured, and indeed literature detailing the mechanisms of the reverse reaction (i.e. the hydrogenation of CO2 to formate) suggests this.9k,29 This barrier is estimated to be larger than the proton and hydride donation in our 2-propanol system (17 kcal mol−1), and this would contribute to the lower rates which are observed in the biphasic system and in the system where DBU-formate is used in one phase.9j Subsequently, complex 4 loses CO2 to give the hydride complex 5. The reaction between 3 and 5 is thought to be reversible because we observed that a pressure of CO2 (for example from conducting the reaction in a closed flask) slows, and even stops, catalysis. CO2 might also form an adduct with the basic amido nitrogen of 2, an adduct with the eneamido carbon next to phosphorus of 2 or 5 as observed by Milstein and co-workers30 or even reversibly functionalize a C–H bond of the eneamido of 2 or 5 as observed by Song and co-workers.31 We have no evidence for these ideas as yet. If present, these processes would have to be reversible because catalysis is restored when the pressure of CO2 is released by purging with an inert gas.From the active iron hydride species 5, the ketone is reduced in an outer-sphere mechanism via a six-membered ring intermediate with subsequent release of the alcohol product and formation of 2 thus completing the cycle. This enantioselective ketone hydrogenation step is the same as that detected in the 2-propanol ATH mechanism.2d,32 When the reaction is conducted in the presence of DMF, the catalysis is thought to be slower due to the coordination of DMF to 2 or to 1 with displacement of a halide.
Conclusions
A highly active, air-sensitive iron P–NH–N–P catalyst for ATH in basic 2-propanol was tested for its activity in a biphasic reaction employing water and potassium formate as the proton and hydride sources, respectively. The catalyst was found to be less active for the reduction of ketones in a biphasic system, but quite active using as little as 0.5 mol% catalyst loading. The observed reactivity rivals that of most ruthenium-based catalysts employing a biphasic water and formate-based system, albeit with a lower enantioselectivity. Due to the nature of the biphasic mixture, the product alcohols can be easily separated by removing the MeTHF layer from the reaction vessel and extracting with as little as 2 mL of MeTHF, but purification of the product alcohols still required a silica plug with hexanes and ethylacetate to remove the iron catalyst. While argon purging was not necessary at lower substrate:catalyst loadings when employing TWEEN80 as a surfactant, the use of argon purging does lead to increased TOF. Future improvement to this system includes using a cosolvent which does not produce multiple active hydrides or removing the need for a cosolvent entirely. The catalyst stability could also be improved by maintaining the pH of the reaction at 7.5. Hydroxide anions are produced as the reaction proceeds and the iron catalyst is known to be sensitive to harsher basic conditions. These changes would lead to an even more sustainable and environmentally benign catalytic process that might use formate produced in a solar process33 and iron complexes with low synthetic E factors.
Experimental
General remarks
All manipulations were performed under an inert atmosphere of argon using Schlenk or standard glovebox techniques, unless otherwise stated. Solvents and liquid substrates were dried and degassed under standard procedures prior to use. All solid substrates were heated to 80 °C under vacuum to remove any traces of water before being stored in the glovebox. HDBU formate was synthesized according to literature.16 NMR spectra were recorded at ambient temperature and pressure using an Agilent DD2 600 MHz spectrometer [1H (600 MHz)]. The conversions and enantiomeric excess for each reaction were obtained on a Perkin Elmer Clarus 400 Chromatograph equipped with a chiral column (CP chirasil-Dex CB 25 m × 2.5 mm) and using hydrogen gas as the mobile phase. All experiments were repeated three times for accuracy using 1,4-di-tert-butylbenzene (DTBB) as an external standard. All extraction solvents were distilled for reuse.General procedure for asymmetric transfer hydrogenation of acetophenone (Table 1)
In a glovebox, sodium formate (808 mg, 11.90 mmol) and potassium hydroxide (6 mg, 0.10 mmol) were placed in a 25 mL Schlenk flask which was then removed from the glovebox. Distilled water (8 mL) was added to form the basic water stock solution containing a concentration of 101 mg mL−1 and 0.75 mg mL−1 of sodium formate and potassium hydroxide, respectively. This solution was then freeze–pump–thawed three times to remove any oxygen. Concurrently in a glovebox, a stock solution of 1 (10 mg, 0.012 mmol) and TBA+Br− or TBA+BF4− (4 mg, 0.006 mmol, in both cases) in dichloromethane (1 mL) was prepared. This was evenly split into two 10 mL Schlenk flasks charged with a stir bar, then the solvent was removed in vacuo. Two syringes with acetophenone (143 mg, 1.189 mmol each) were speared into a rubber stopper. Everything was removed from the glovebox. The basic water stock solution was heated to 65 °C, while the Schlenk flasks were placed under argon. Once the temperature was reached, the acetophenone was injected into the Schlenk flask, quickly followed by the hot basic water solution (4 mL). The stirring plate was set to 600 RPM to begin the reaction. After 16 h, the reaction was removed from heat and exposed to oxygen. The product was extracted with MeTHF (3 × 6 mL).Note: each Schlenk flask contained sodium formate (404 mg, 1000 eq.), potassium hydroxide (3 mg, 8 eq.), TBA+Br− or TBA+BF4− (2 mg, 1 eq.), and acetophenone (143 mg, 200 eq.). Various stock solutions were made to change the equivalents for the studies found herein.
General procedure for the argon purged asymmetric transfer hydrogenation of ketones (Tables 2–5 and Fig. 4)
Procedure follows above with the following changes. The basic stock solution was either 808 mg mL−1 of sodium formate in water or 1000 mg mL−1 of potassium formate in water. The substrates were dissolved in the cosolvent (1 mL), then speared into a rubber stopper. All Schlenk flasks and syringes were removed from the glovebox. The basic water solution was heated to 65 °C, while the Schlenk flasks were placed under argon. Once the temperature was reached, the substrate and cosolvent were injected into the Schlenk flask, quickly followed by the hot basic water solution (1 mL). A condenser (outsourced to an external bubbler) was attached to each Schlenk flask, which was then submerged into the oil bath at 65 °C. The stirring plate was set to 600 RPM to begin the reaction. After the allotted time, the reaction was removed from heat and exposed to oxygen. All ketones were extracted with MeTHF (3 × 6 mL), then diluted to 20 mL. For a reaction using 200:1 substrate:catalyst ratio 0.1 mL of the product alcohol solution was added to a GC vial containing 0.9 mL of a 0.011 M solution of DTBB. For a reaction using 100:1 substrate:catalyst ratio 0.2 mL of the product alcohol solution was added to a GC vial containing 0.8 mL of a 0.0125 M solution of DTBB. In this way, the final concentration of DTBB was 0.01 M in the GC vial.
Alterations include: addition of TWEEN80 (39 mg, 5 eq., 0.029 mmol) or HDBU formate (2.36 g, 11.90 mmol), purging with nitrogen, or purging with argon for 10 seconds then shutting off the argon.
Hydride 1H NMR study
In a glovebox, 1 (10 mg, 0.012 mmol), TBA+BF4− (2 mg, 0.003 mmol, 1 eq.), and potassium formate (10 mg, 0.120 mmol, 10 eq.) were dissolved in either THF (1 mL) or MeTHF (1 mL) in a Schlenk flask. This was brought out onto the Schlenk line and stirred under argon at 65 °C for 5 minutes. Degassed distilled water (1 mL) was added and the reaction was stirred for another 5 minutes. The solvent was removed in vacuo and the Schlenk flask was brought into the glovebox. The residue was dissolved in benzene-d6 and filtered through a 13 mm syringe filter with a 0.45 μm PTFE membrane. 1H NMR was taken immediately afterwards.Acknowledgements
R. H. Morris thanks NSERC for a Discovery Grant and the Canada Council for the Arts for a Killam Research Fellowship. NSERC is thanked for a PGS D awarded to K. Z. Demmans. The authors wish to acknowledge the Canadian Foundation for Innovation, project number 19119, and the Ontario Research Fund for funding of the Centre for Spectroscopic Investigation of Complex Organic Molecules and Polymers. S. A. M. Smith is thanked for her help with these studies, as well as W. Zuo for his help preparing the iron catalyst.Notes and references
- (a) A. Buchard, H. Heuclin, A. Auffrant, X. F. Le Goff and P. Le Floch, Dalton Trans., 2009, 1659–1667 RSC; (b) G. Wienhoefer, F. A. Westerhaus, K. Junge and M. Beller, J. Organomet. Chem., 2013, 744, 156–159 CrossRef CAS; (c) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS; (d) K. Junge, K. Schroeder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC.
- (a) S. A. M. Smith and R. H. Morris, Synthesis, 2015, 1775–1779 CAS; (b) W. Zuo and R. H. Morris, Nat. Protoc., 2015, 10, 241–257 CrossRef CAS PubMed; (c) P. E. Sues, K. Z. Demmans and R. H. Morris, Dalton Trans., 2014, 43, 7650–7667 RSC; (d) W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080–1083 CrossRef CAS.
- (a) X. Wu, X. Li, F. King and J. Xiao, Angew. Chem., Int. Ed., 2005, 44, 3407–3411 CrossRef CAS; (b) D. S. Matharu, D. J. Morris, G. J. Clarkson and M. Wills, Chem. Commun., 2006, 30, 3232–3234 RSC; (c) X. Wu, C. Wang and J. Xiao, Platinum Met. Rev., 2010, 54, 3–19 CrossRef CAS.
- (a) X. Wu, J. Liu, D. Di Tommaso, J. A. Iggo, C. R. A. Catlow, J. Bacsa and J. Xiao, Chem.–Eur. J., 2008, 14, 7699–7715 CrossRef CAS; (b) Y. Ma, H. Liu, L. Chen, X. Cui, J. Zhu and J. Deng, Org. Lett., 2003, 5, 2103–2106 CrossRef CAS; (c) Z. Luo, F. Qin, S. Yan and X. Li, Tetrahedron: Asymmetry, 2012, 23, 333–338 CrossRef CAS; (d) R. Qin, J. Wang, W. Xiong, J. Feng, D. Liu and H. Chen, Cuihua Xuebao, 2012, 33, 1146–1153 CAS.
- O. Prakash, H. Joshi, K. N. Sharma, P. L. Gupta and A. K. Singh, Organometallics, 2014, 33, 3804–3812 CrossRef CAS.
- N. A. Cortez, G. Aguirre, M. Parra-Hake and R. Somanathan, Tetrahedron: Asymmetry, 2013, 24, 1297–1302 CrossRef CAS.
- S. Mazza, R. Scopelliti and X. Hu, Organometallics, 2015, 34, 1538–1545 CrossRef CAS.
- M. Zhu, Appl. Catal., A, 2014, 479, 45–48 CrossRef CAS.
- (a) A. Zoabi, S. Omar and R. Abu-Reziq, Eur. J. Inorg. Chem., 2015, 2101–2109 CrossRef CAS; (b) Y. Wei, X. Wu, C. Wang and J. Xiao, Catal. Today, 2015, 247, 104–116 CrossRef CAS; (c) I. Szatmari, G. Papp, F. Joo and A. Katho, Catal. Today, 2015, 247, 14–19 CrossRef CAS; (d) Y. Wei, D. Xue, Q. Lei, C. Wang and J. Xiao, Green Chem., 2013, 15, 629–634 RSC; (e) X. Xu, R. Wang, J. Wan, X. Ma and J. Peng, RSC Adv., 2013, 3, 6747–6751 RSC; (f) S. Denizalti, D. Mercan, B. Sen, A. G. Gokce and B. Cetinkaya, J. Organomet. Chem., 2015, 779, 62–66 CrossRef CAS; (g) N. Madern, B. Talbi and M. Salmain, Appl. Organomet. Chem., 2013, 27, 6–12 CrossRef CAS; (h) H. Tuerkmen, I. Kani and B. Cetinkaya, Eur. J. Inorg. Chem., 2012, 4494–4499 CrossRef CAS; (i) B. Seashore-Ludlow, F. Saint-Dizier and P. Somfai, Org. Lett., 2012, 14, 6334–6337 CrossRef CAS; (j) W. Zuo, D. E. Prokopchuk, A. J. Lough and R. H. Morris, ACS Catal., 2016, 6, 301–314 CrossRef CAS; (k) S. Ramakrishnan, K. M. Waldie, I. Warnke, A. G. De Crisci, V. S. Batista, R. M. Waymouth and C. E. D. Chidsey, Inorg. Chem., 2016, 55, 1623–1632 CrossRef CAS.
- (a) D. E. Prokopchuk and R. H. Morris, Organometallics, 2012, 31, 7375–7385 CrossRef CAS; (b) A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266–12280 CrossRef CAS.
- (a) H. Heeres, R. Handana, D. Chunai, C. Borromeus Rasrendra, B. Girisuta and H. Jan Heeres, Green Chem., 2009, 11, 1247–1255 RSC; (b) J. A. Melero, J. Iglesias and A. Garcia, Energy Environ. Sci., 2012, 5, 7393–7420 RSC.
- Y. Zhang, A. D. MacIntosh, J. L. Wong, E. A. Bielinski, P. G. Williard, B. Q. Mercado, N. Hazari and W. H. Bernskoetter, Chem. Sci., 2015, 6, 4291–4299 RSC.
- C. Wang, X. Wu and J. Xiao, Chem.–Asian J., 2008, 3, 1750–1770 CrossRef CAS.
- Croda Europe Ltd., Span and Tween, www.croda.com/europe (accessed May 18, 2016).
- S. C. Maurer, K. Kuehnel, L. A. Kaysser, S. Eiben, R. D. Schmid and V. B. Urlacher, Adv. Synth. Catal., 2005, 347, 1090–1098 CrossRef CAS.
- J. Sun, X. Yao, W. Cheng and S. Zhang, Green Chem., 2014, 16, 3297–3304 RSC.
- K. Ahlford and H. Adolfsson, Catal. Commun., 2011, 12, 1118–1121 CrossRef CAS.
- S. H. Deshpande, V. S. Shende, S. K. Shingote, D. Chakravarty, V. G. Puranik, R. V. Chaudhari and A. A. Kelkar, RSC Adv., 2015, 5, 51722–51729 RSC.
- Y. Tang, X. Li, C. Lian, J. Zhu and J. Deng, Tetrahedron: Asymmetry, 2011, 22, 1530–1535 CrossRef CAS.
- G. Facchetti, R. Gandolfi, M. Fuse, D. Zerla, E. Cesarotti, M. Pellizzoni and I. Rimoldi, New J. Chem., 2015, 39, 3792–3800 RSC.
- N. A. Cortez, G. Aguirre, M. Parra-Hake and R. Somanathan, Tetrahedron: Asymmetry, 2008, 19, 1304–1309 CrossRef CAS.
- R. Montalvo-Gonzalez, D. Chavez, G. Aguirre, M. Parra-Hake and R. Somanathan, Synth. Commun., 2009, 39, 2737–2746 CrossRef CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, S. Miyazawa, H. Sugihara, T. Hirose and K. Kasuga, Chem.–Eur. J., 2008, 14, 11076–11081 CrossRef CAS.
- X. Wu, X. Li, A. Zanotti-Gerosa, A. Pettman, J. Liu, A. J. Mills and J. Xiao, Chem.–Eur. J., 2008, 14, 2209–2222 CrossRef CAS.
- X. Li, J. Blacker, I. Houson, X. Wu and J. Xiao, Synlett, 2006, 1155–1160 CAS.
- X. Liu, T. Zhang, Y. Hu and L. Shen, Catal. Lett., 2014, 144, 1289–1295 CrossRef CAS.
- I. Nieto, M. S. Livings, J. B. Sacci, L. E. Reuther, M. Zeller and E. T. Papish, Organometallics, 2011, 30, 6339–6342 CrossRef CAS.
- K. Abdur-Rashid, S. E. Clapham, A. Hadzovic, J. N. Harvey, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2002, 124, 15104–15118 CrossRef CAS.
- (a) J. Kothandaraman, M. Czaun, A. Goeppert, R. Haiges, J.-P. Jones, R. B. May, G. K. S. Prakash and G. A. Olah, ChemSusChem, 2015, 8, 1442–1451 CrossRef CAS; (b) Y.-Y. Ohnishi, Y. Nakao, H. Sato and S. Sakaki, Organometallics, 2006, 25, 3352–3363 CrossRef CAS; (c) R. Marcos, L. Xue, R. Sánchez-De-Armas and M. S. G. Ahlquist, ACS Catal., 2016, 6, 2923–2929 CrossRef CAS; (d) F. Bertini, N. Gorgas, B. Stoger, M. Peruzzini, L. F. Veiros, K. Kirchner and L. Gonsalvi, ACS Catal., 2016, 6, 2889–2893 CrossRef CAS; (e) H. Gao, L. Chen, J. Chen, Y. Guo and D. Ye, Catal. Sci. Technol., 2015, 5, 1006–1013 RSC; (f) H. W. Suh, T. J. Schmeier, N. Hazari, R. A. Kemp and M. K. Takase, Organometallics, 2012, 31, 8225–8236 CrossRef CAS; (g) N. Kumar, D. M. Camaioni, M. Dupuis, S. Raugei and A. M. Appel, Dalton Trans., 2014, 43, 11803–11806 RSC; (h) S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. v. Stein, U. Englert, M. Holscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC; (i) C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS; (j) G. A. Filonenko, D. Smykowski, B. M. Szyja, G. Li, J. Szczygiel, E. J. M. Hensen and E. A. Pidko, ACS Catal., 2015, 5, 1145–1154 CrossRef CAS; (k) S. Savourey, G. Lefevre, J. C. Berthet, P. Thuery, C. Genre and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 10466–10470 CrossRef CAS; (l) A. Urakawa, M. Iannuzzi, J. Hutter and A. Baiker, Chem.–Eur. J., 2007, 13, 6828–6840 CrossRef CAS; (m) J. Li, J. Li, D. Zhang and C. Liu, ACS Catal., 2016, 6, 4746–4754 CrossRef CAS; (n) I. Osadchuk, T. Tamm and M. S. G. Ahlquist, ACS Catal., 2016, 6, 3834–3839 CrossRef CAS; (o) M. Z. Ertem, Y. Himeda, E. Fujita and J. T. Muckerman, ACS Catal., 2016, 6, 600–609 CrossRef CAS.
- O. Rivada-Wheelaghan, A. Dauth, G. Leitus, Y. Diskin-Posner and D. Milstein, Inorg. Chem., 2015, 54, 4526–4538 CrossRef CAS PubMed.
- V. T. Annibale, D. A. Dalessandro and D. Song, J. Am. Chem. Soc., 2013, 135, 16175–16183 CrossRef CAS PubMed.
- W. Zuo, S. Tauer, D. E. Prokopchuk and R. H. Morris, Organometallics, 2014, 33, 5791–5801 CrossRef CAS.
- S. Sato, T. Arai and T. Morikawa, Inorg. Chem., 2015, 54, 5105–5113 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GC data, IR spectrum of 3, 1H NMR spectra of hydride complexes. See DOI: 10.1039/c6ra22538a |
| This journal is © The Royal Society of Chemistry 2016 |