
Are noble gas molecules able to exhibit a superhalogen nature?†
Celina Sikorska*
Laboratory of Molecular Modeling, Department of Theoretical Chemistry, Faculty of Chemistry, University of Gdansk, Wita Stwosza 63, 80-308 Gdansk, Poland. E-mail: celina.sikorska@ug.edu.pl; Tel: +48 58 523 5351
First published on 25th October 2016
Abstract
Superhalogens are a class of highly electronegative molecules whose electron affinities even exceed those of the halogen elements. Since such species can serve as new oxidizing agents, biocatalysts, and building blocks of unusual salts, they are important to the chemical industry. The ability of noble gas (Ng) atoms to form stable mononuclear (NgF7−) and dinuclear (Ng2F13−) superhalogen anions has been reported. Theoretical considerations supported by ab initio calculations revealed that Ng atoms (i.e. Kr, Xe, Rn) should form strongly bound anionic systems when combined with fluorine atoms (acting as ligands). Particularly, those anionic molecules exhibit larger vertical electron detachment energies (6.97–9.56 eV) than that of the chlorine atom (3.62 eV), confirming their superhalogen nature. We believe that the results provided in this contribution will not only provide evidence of a new type of superhalogen molecule but also stimulate more research interest and efforts in the amazing superatom realm.
1. Introduction
Electron affinity is one of the major factors that govern molecular reactivity in chemistry. In particular, molecules with high EA usually act as strong oxidizing agents in chemical reactions and tend to form very stable negative ions. It is well known that halogen atoms possess the highest electron affinities (EA) among the elements (fluorine 3.40 eV, chlorine 3.62 eV).1 However, the EA of a polyatomic system may exceed the 3.62 eV limit due to collective effects. Although such species have been attracting chemists' attention since the early 60s, it was only in 1981 that Gutsev and Boldyrev2 proposed to term them superhalogens and introduced a simple formula for one class of these compounds, MXk+1, where M is a main group or transition metal atom, X is a halogen atom, and k is the maximal formal valence of the central atom (M). Once the extra electron is attached (which results in forming a superhalogen anion), it can delocalize over k + 1 halogen atoms, as opposed to one halogen atom. Consequently, the EA of the superhalogen is higher than that of the constituent halogen. Theoretical as well as experimental studies,3–10 which have been made to estimate the vertical electron detachment energies (VDEs) of various anions having superhalogens as their neutral parents, have shown that the presence of metals or halogens is not mandatory for a cluster to exhibit superhalogen properties. Examples include B(OF)4− (VDE = 7.08 eV)4 anion which follows the MXn+1− scheme but has no metal atom and halogen atoms have been replaced with fluoroxyl groups. The discovery of such species conduces to the development of oxidizing agents of new type.11,12The main purpose of exploring various novel superhalogen species is to provide reliable data and predictions considering the possible use of such compounds as electron acceptors (oxidizing agents). Superhalogens with strong oxidizing ability can be used to access the high oxidation states of metal atoms otherwise inaccessible in conventional chemistry. Explicitly, superhalogen can even oxidize counterparts with relatively high ionization potential, such as C60 nanoparticles12 and noble gas atoms.13 Moreover, the superhalogens exhibit great application potential in the synthesis of new chemical compounds, and in serving as promising building blocks of novel functional materials, such as nonlinear optical materials and magnetic materials.
Since the presence of metals or halogens is not mandatory for a cluster to exhibit superhalogen properties, the present study examines whether noble gas atoms are capable of forming stable superhalogen anions. Thus far, it has been proven that noble gas fluorides are able to act as fluoride acceptors themselves, and will add one or two fluorine anions. Examples includes the addition of F- to XeF4 to give the pentagonal planar XeF5− anion, or XeF6 to give either XeF7− or XeF82−, depending on the amount of fluoride added.14,15 In 2010, Vasdev et al. reported the spectroscopic evidence for the formation of the XeF3− anion in gas-phase suggesting its Y-shaped (C2v-symmetry) structure.16 The X-ray structural analyses performed by Ellern et al. revealed the existence of dinuclear Xe2F13− anion which structure resembles a XeF6 molecule bridged by two long Xe–F bonds to a XeF7− anion.17 More recently, the theoretical studies on the issue of the possible existence and stability of XeF7− anions were performed by Grant et al. (employing the coupled cluster methods at the CCSD(T) level) and resulted in the predicting the D5h-symmetry (pentagonal bipiramidal) structure of XeF7− anion.18 Even though the present knowledge considering noble gas atom compounds and their daughter anions seems substantial, the available description of some of their relatively structurally simple representatives is not satisfactory. In particular, most of the research in the chemistry of noble-gases has been limited to the compounds of the lighter element xenon. The chemistry of the heavier radon has not attached much attention probably due to its radioactivity. The ability to form stable anionic species, however, are expected to be very pronounced in the heavy element radon (with Z = 86). This lack od data has motivated us to take a closer look at the various anionic fluorides containing Kr, Xe or Rn as a central atom and to calculate their vertical detachment energies.
In this contribution, to examine the behavior of Ng in superhalogen design, we theoretically constructed and studied a series of NgF7− (Ng = Kr, Xe, Rn), NgF5− (Ng = Xe, Rn), NgF3− (Ng = Xe, Rn), and dinuclear Ng2F13− (Ng = Xe, Rn) anions. Also, we provide the Gibbs free energies of the most probable fragmentation reactions that the NgF7− and Ng2F13− anions might be susceptible to. The results show that these studied anions exhibit much larger VDE values than EAs of halogen elements, confirming their superhalogen identities. This finding provides a new way to design and synthesize superhalogens by decorating noble gases cores with fluorine ligands. The purpose of these efforts is to provide reliable predictions of their physical–chemical properties, considering the possible use of such compounds as electron acceptors (oxidizing agents) in various chemical processes as well as the role they can play in synthesis (e.g. in oxidation of counterpart systems with high ionization potentials (IP)).11,12
2. Computational methods
Molecular structures of anionic systems NgnF6n+1− (Ng = Kr, Xe, Rn; n = 1, 2) were combinatorial generated with the SuperhalogenCreator software.19 Since our main goal was to estimate the electronic stability (i.e. the vertical electron detachment energies, VDEs) for the KrF7−, XeF7−, RnF7−, Xe2F13−, and Rn2F13− anions, we limited our geometry optimization calculations to the closed-shell anionic species for which we also obtained harmonic vibrational frequencies at their minimum energy structures. For this purpose, we applied second-order Møller–Plesset (MP2) perturbational method with the 6-311++G(3df,3pd) basis sets.20,21 We applied the MP2/6-311+G(3df,3pd) approach (instead of more accurate but significantly more expensive methods (i.e. MP4 and CCSD(T))22–24) since the earlier report on the performance of selected ab initio methods and basis sets in estimating the VDEs of superhalogen anions confirms the ability of this method to reproduce the electronic properties of such anions with high accuracy.25 For the noble gas atoms, the effective core potential (ECP) Stuttgart/Dresden ECP-X-MWB basis set (incorporated in GAUSSIAN09 software)26 was employed (where X is the number of core electrons). For calculations involving krypton, xenon, and radon respectively ECP-28-MWB, ECP-46-MWB, and ECP78MWB basis sets were used. The final values of the vertical electron binding energies were obtained by employing the outer valence Green function OVGF method (B approximation)27–35 with the 6-311++G(3df,3pd) basis sets.20,21 The Green function methods are related to and essentially the same, when carried to the same order of analysis, as the propagator35–37 and equations of motion (EOM)32,38,39 methods. Since the OVGF approximation remains valid only for outer valence ionizations for which the pole strengths (PSs) are greater than 0.80–0.85,40 we verified that the PS values were sufficiently large to justify the use of the OVGF method for all states studied here (the smallest PS found for the states studied in this work was 0.84). The partial atomic charges (required for estimating polarity of Ng–F bonds) were fitted to the electrostatic potential according to the Merz–Singh–Kollman scheme.41To approximate the effect of hydration on the binding energies, we employed the polarized continuum (PCM) solvation model42–44 within a self-consistent reaction field treatment, as implemented in the Gaussian 09 program. The PCM calculations with a dielectric constant of water (ε = 78.39) were carried out with the same basis set as that for the isolated species.
All calculations were performed with the GAUSSIAN09 software package.26 In order to avoid erroneous results from the default direct SCF calculations, the keyword SCF = NoVarAcc was used and the two-electron integrals were evaluated (without prescreening) to a tolerance of 10−20 au. The optimizations of the geometries were performed using relatively tight convergence thresholds (i.e. 10−5 hartree/bohr (or radian) for the root mean square first derivative).
3. Results and discussion
To test the hypotheses whether noble gas atoms are capable of forming stable superhalogen anions, a systematic study of NgF7− (where Ng = Kr, Xe, and Rn) molecules was performed. The fluorine atoms were chosen as ligands since they are small in size (which allows one to perform high-level ab initio calculations), and their corresponding EA (3.40 eV)1 is high. It should be mentioned here that NgF7− systems' electron detachment energy have not been reported thus far.3.1. Equilibrium structures and thermodynamic stability of NgF7− (Ng = Kr, Xe, Rn) anions
The exploration of the potential energy surface (PES) of the anionic NgF7− systems (M = Kr, Xe, and Rn) leads to the minimum energy structures depicted in Fig. 1. The corresponding geometrical parameters along with harmonic vibrational frequencies for these structures are collected in Table 1. According to our findings KrCl7− and XeCl7− anions are geometrically unstable. The minimum energy structures of KrF7− and XeF7− anions possess D5h-symmetry. In addition, we checked that lower symmetry structures are not geometrically stable (i.e. we confirmed that the C3v- and C5v-symmetry NgF7− (Ng = Kr, Xe) systems do not correspond to the minimum energy structures and the deformation along their imaginary modes leads to pentagonal bipyramidal species). | ||
| Fig. 1 The equilibrium structures (on the left) and corresponding highest occupied molecular orbitals (on the right) of the NgF7− anions (Ng = Kr, Xe, Rn) obtained at the MP2/6-311++G(3df,3pd)+ECPs level. | ||
| Species (symmetry) | VDE (PS) | Geometrical parameters and atomic charges | Vibrational frequencies [cm−1] | ||
|---|---|---|---|---|---|
| a VDE in eV (pole strengths (PS) in parenthesis), bond lengths (r) in Å, atomic charges (q) in a.u., valence angles (α) and dihedral angles (ω) in degrees. | |||||
| KrF7− (D5h) | 7.508 (0.835) | r(Kr–Fax) = 1.880 | υ1,2 = 57 (e′′2) | υ8 = 283 (a′1) | υ15 = 393 (a′1) |
| r(Kr–Feq) = 2.007 | υ3,4 = 198 (e′′1) | υ9,10 = 305 (e′2) | υ16,17 = 546 (e′1) | ||
| qFax = −0.23 | υ5,6 = 201 (e′1) | υ11,12 = 317 (e′1) | υ18 = 629 (a′′2) | ||
| qFeq = −0.35 | υ7 = 257 (a′′2) | υ13,14 = 383 (e′2) | |||
| qKr = 1.22 | |||||
| XeF7− (D5h) | 6.967 (0.893) | r(Xe–Fax) = 1.987 | υ1,2 = 39 (e′′2) | υ8,9 = 287 (e′1) | υ15 = 521 (a′1) |
| r(Xe–Feq) = 2.026 | υ3,4 = 158 (e′1) | υ10,11 = 381 (e′2) | υ16,17 = 532 (e′1) | ||
| qFax = −0.36 | υ5 = 193 (a′′2) | υ12,13 = 430 (e′2) | υ18 = 565 (a′′2) | ||
| qFeq = −0.43 | υ6,7 = 199 (e′′1) | υ14 = 448 (a′1) | |||
| qXe = 1.89 | |||||
| RnF7− (Cs) | 8.136 (0.908) | r(Rn–F1) = 2.067 | α(F1RnF2) = 89.21 | υ1 = 4 (a′′) | υ10 = 323 (a′′) |
| r(Rn–F2) = 2.078 | α(F1RnF3) = 81.52 | υ2 = 27 (a′) | υ11 = 323 (a′) | ||
| r(Rn–F3) = 2.087 | α(F1RnF4) = 108.67 | υ3 = 160 (a′′) | υ12 = 471 (a′) | ||
| r(Rn–F4) = 2.092 | α(F1RnF5) = 171.37 | υ4 = 167 (a′) | υ13 = 476 (a′′) | ||
| r(Rn–F5) = 2.069 | ω(F1RnF2F3) = 81.41 | υ5 = 186 (a′) | υ14 = 489 (a′) | ||
| qF1,F5 = −0.42 | ω(F1RnF3F4) = 112.55 | υ6 = 225 (a′′) | υ15 = 523 (a′) | ||
| qF2,F3 = −0.46 | ω(F1RnF2F5) = 174.76 | υ7 = 231 (a′) | υ16 = 523 (a′′) | ||
| qF4 = −0.42 | υ8 = 241 (a′′) | υ17 = 532 (a′) | |||
| qRn = 2.17 | υ9 = 243 (a′) | υ18 = 535 (a′) | |||
According to our findings, in the pentagonal bipyramidal D5h-symmetry structure of KrF7− anion, the axial (ax) and equatorial (eq) Kr–F bond lengths read 1.880 and 2.007 Å, respectively. The lengths of the Xe–F bonds in the XeF7− anion were calculated as equal to 1.987 Å (ax) and 2.026 Å (eq); see Fig. 1, which is in good agreement with those predicted earlier by Grant et al. (ax 1.978 Å; eq, 2.016 Å)18 and the Xe–F separations observed in crystals (1.932–2.100 Å).17 The minimum energy structure of RnF7− anion was found to exhibit Cs-symmetry with the Rn–F bond lengths within the 2.067–2.092 Å range, as depicted in Fig. 1.
As far as the thermodynamic stability of the NgX7− anions is concerned, we calculated the free enthalpies (ΔH298r), entropies (ΔS298r), and Gibbs free energies (ΔG298r) for the reactions in gas phase (for T = 298.15 K and p = 1 atm) corresponding to the most likely fragmentation channels (see Table 2, where the ΔH298r, ΔS298r, and ΔG298r are collected). The KrF7− anion is stable with respect to fragmentations leading to the KrF6−, KrF4−, and Kr−, whereas it was found to be thermodynamically unstable when Kr or KrF2− loss is considered (the smallest ΔG298r value was predicted for the KrF7− → Kr + 3F2 + F− reaction and reads −19.96 kcal mol−1). From the other hand, the XeF7− and RnF7− species were found to be stable and not susceptible to any fragmentation. In particular, in XeF7− and RnF7− case, the smallest ΔG298r value was predicted for the F2 detachment (i.e. XeF7− → XeF5− + F2 and RnF7− → RnF5− + F2); however, both these Gibbs free energies were found to be positive (16.08 and 48.64 kcal mol−1, for XeF7− and RnF7− respectively), which confirms the thermodynamic stability of the XeF7− and RnF7− anions. According to the results gathered in Table 2, the fragmentations leading to the NgF6, NgF6−, NgF4, NgF4−, NgF3, NgF3−, NgF2, NgF2−, Ng, or Ng− (where Ng = Xe, Rn) are even less likely as the calculated ΔG298r values span the 66.20–556.99 kcal mol−1 range. Therefore, we conclude that the XeF7− and RnF7− anions are not susceptible to any fragmentations.
| Fragmentation path | ΔH298r | ΔS298r | ΔG298r |
|---|---|---|---|
| KrF7− → KrF6− + F | 34.42 | 31.90 | 24.91 |
| KrF7− → KrF4− + F2 + F | 27.85 | 71.18 | 6.63 |
| KrF7− → KrF2− + 2F2 + F | 30.55 | 102.87 | −0.12 |
| KrF7− → Kr + 3F2 + F− | 16.31 | 121.65 | −19.96 |
| KrF7− → Kr− + 3F2 + F | 134.24 | 124.41 | 97.15 |
| XeF7− → XeF6 + F− | 73.43 | 24.24 | 66.20 |
| XeF7− → XeF6− + F | 98.34 | 28.00 | 90.00 |
| XeF7− → XeF5− + F2 | 40.30 | 81.23 | 16.08 |
| XeF7− → XeF4 + F2 + F− | 98.78 | 61.75 | 80.37 |
| XeF7− → XeF4− + F2 + F | 114.70 | 65.77 | 95.09 |
| XeF7− → XeF3 +F− + F2 + F | 228.28 | 137.25 | 187.36 |
| XeF7− → XeF3− + 2F2 | 80.26 | 76.72 | 57.39 |
| XeF7− → XeF2 + 2F2 + F− | 124.08 | 94.09 | 96.03 |
| XeF7− → XeF2− + 2F2 + F | 149.58 | 102.05 | 119.15 |
| XeF7− → Xe + 3F2 + F | 150.60 | 120.94 | 114.54 |
| XeF7− → Xe− + 3F2 + F− | 308.86 | 123.70 | 271.98 |
| RnF7− → RnF6 + F− | 84.38 | 17.55 | 79.15 |
| RnF7− → RnF6− + F | 74.66 | 22.70 | 67.89 |
| RnF7− → RnF5− + F2 | 55.86 | 24.20 | 48.64 |
| RnF7− → RnF4 + F2 + F− | 123.38 | 54.32 | 107.19 |
| RnF7− → RnF4− + F2 + F | 133.18 | 63.03 | 114.38 |
| RnF7− → RnF3 +F− + F2 + F | 279.98 | 132.71 | 240.41 |
| RnF7− → RnF3− + 2F2 | 136.12 | 66.81 | 116.20 |
| RnF7− → RnF2 + 2F2 + F− | 161.04 | 85.80 | 135.46 |
| RnF7− → RnF2− + 2F2 + F | 190.99 | 94.20 | 162.91 |
| RnF7− → Rn + 3F2 + F− | 202.14 | 112.05 | 168.73 |
| RnF7− → Rn− + 3F2 + F | 591.22 | 114.80 | 556.99 |
Analysis of the ΔG298r values calculated for the most likely fragmentation reactions leads to following conclusions: (i) krypton atom forms thermodynamically unstable KrF7− anion; (ii) xenon and radon atoms are capable of forming thermodynamically stable respectively XeF7− and RnF7− anions. Albeit special attention should be paid to the species, which are not susceptible to spontaneous fragmentations (because their future experimental applications are much more likely), to provide the completeness of the results we decided to discuss the structure and electronic stability of all the anions (including the thermodynamically unstable anions). Furthermore, one should also keep in mind that even the thermodynamically unstable species discussed are both geometrically and electronically stable and any fragmentation process would likely require overcoming certain kinetic barriers (corresponding to bond breaking and structure reorganization). For instance, the kinetic barrier leading from the KrF7− to the fragmentation products was calculated to be 2.5 kcal mol−1 (Fig. 2) which means that this anion might exist at very low temperatures in the gas phase (assuming lack of any perturbing factors).
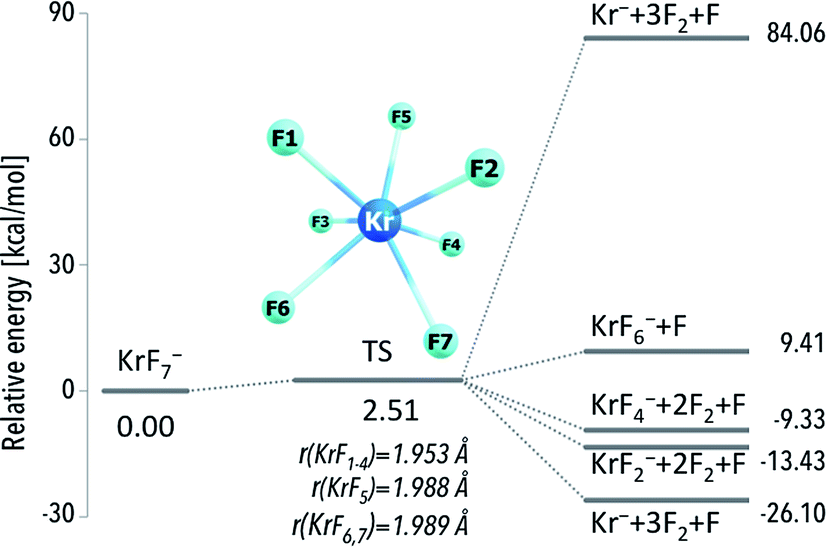 | ||
| Fig. 2 The CCST(T)/6-311++G(3df,3pd)+ECPs//MP2/6-311+G(3df)+ECPs energy diagram for various fragmentation paths of the KrF7− anion. Transition state (TS) is depicted as structure connecting the KrF7− anion and the fragmentation products. The corresponding geometrical parameters along with harmonic vibrational frequencies for the TS structure are provided in the ESI (Table ESI 1†). | ||
3.2. The formation of the XeF7− superhalogen anions
The formation of the NgF7− superhalogen anion can be explained by choosing one representative system and determining the eventual kinetic (i.e., activation) barriers that have to be overcome to create the resulting compound. We decided to discuss this issue by performing the additional calculations (followed by the results analysis) for arbitrarily chosen anionic XeF7− system.While analyzing the energy profile for the XeF6 + F− → XeF7− process, the excess electron has to be assigned to the F rather than XeF6 (for the separated F and XeF6 species) because of the significantly larger electron affinity of the fluorine atom. Indeed, as indicated by the localization of the highest energy molecular orbital (HOMO), the extra electron is located in the vicinity of the F species (as depicted in see Fig. 3). However, the part of the excess electron density is being transferred to the remaining F atoms as the originally distant F approaches the XeF6 to form the XeF7− (see Fig. 3 where also the HOMO for the equilibrium XeF7− structure is plotted). As a consequence, the excess electron density is equally distributed (due to symmetry reasons) among the seven F ligands in the XeF7− anion. The energy smoothly decreases as the “additional” F approaches the neutral XeF6 molecule and there is no kinetic barrier that has to be surmounted. Since the energy of the separated XeF6 and F− is much larger (by ca. 73 kcal mol−1, see the asymptote in Fig. 3) than the energy of XeF7− at its equilibrium structure, and taking into account that the XeF6 + F− → XeF7− process is predicted to be barrier-free, one may expect the XeF7− anions to be formed spontaneously in the gas phase (whenever the negatively charged fluorine atoms find themselves in the vicinity of XeF6 molecules).
3.3. Excess electron binding energies of NgF7− (Ng = Kr, Xe, Rn) anions
The vertical electron detachment energies of NgF7− (Ng = Kr, Xe, Rn) anions calculated at the OVGF/6-311++G(3df,3pd)+ECPs level are collected in Table 1 (in each case, the lowest VDE arises from OVGF improvements to the energy of HOMO). All of the calculated VDEs greatly exceed the electron affinity of the chlorine atom (3.62 eV), and thus the studied NgF7− species should be classified as the superhalogen anions. The largest VDE among systems considered was found for RnF7− (8.136 eV). The vertical electron detachment energies of the remaining anions are always exceeding 7 eV (see Table 1 and Fig. 1), which suggests that each of these species can potentially act as strong electron acceptors.In order to support our discussion, we present the three-dimensional pictures of the highest energy molecular orbitals (HOMO) for NgF7− anions (see Fig. 1). It is shown that for each anion, the HOMO reveals the antibonding character with respect to the ligand-central atom interaction. From the other hand the corresponding highly negative HOMO eigenvalues of −8.5, −7.8, and −9.1 eV for KrF7−, XeF7− and RnF7−, respectively, reflect the large electronic stabilities of these anions. The large electronic stability of NgF7− systems has to be related to the high polarity of Ng–F bonds with Ngδ+Fδ− charge distribution. Indeed, the population analysis performed (based on the partial atomic charges (qESP) fitted according to the Merz–Singh–Kollman scheme41 to reproduce the electrostatic potential) indicates the substantial charge separation (polarity) between the noble gas atom and each of F atoms. Namely, the ESP charges on Ng span the 1.22–2.17 a.u. range for NgF7− anions with the greatest qESP value for the RnF7− anion (qRn = 2.17 a.u.). Due to Ng–F bonds' high polarity, though the HOMOs are antibonding with respect to the central atom–ligand interaction the real contribution of the central atom AOs to the HOMO is small and the HOMOs of the NgF7− anions are practically nonbonding ligand MOs. Such situation was observed for the superhalogen anions studied in the past9 and enables the large stability of those systems.
As it is well established,45 the resulting VDE of a superhalogen anion depends mainly on the electronegativity of the ligands this anion is decorated with. In addition, the higher vertical detachment energy of an anion is expected for a large number of such ligands due to so called ‘collective effects’. Thus the unexpectedly large VDE value we obtained for the NgF7− anions is related to the fact that it contains seven strongly electronegative fluorine atoms serving as ‘ligands’. Explicitly, the excess electron in the NgX7− species occupies a molecular orbital that extends over all ligands and such an effective delocalization increases the electronic stability of this anion. The dependence of a number of ligands on an electronic stability of resulting compounds is yet to be discussed in the following section.
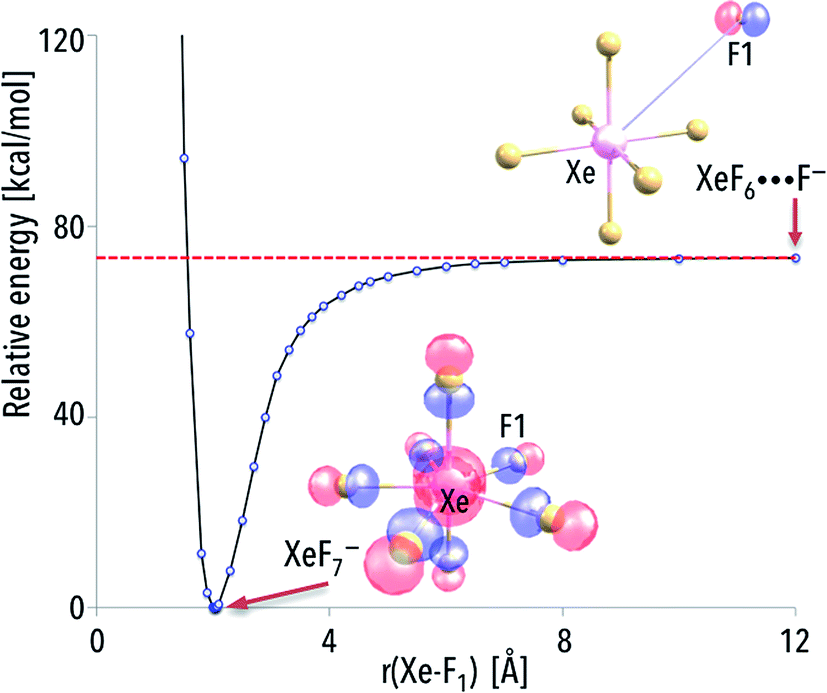 | ||
| Fig. 3 The MP2/6–311++G(3df,3pd)+ECPs energy profile for the formation of the XeF7− anion according to the XeF6 + F− → XeF7− reaction. The asymptote corresponds to the sum of the energies of isolated fragments [XeF6; F−] and reads 73.4 kcal mol−1. The highest occupied molecular orbitals (HOMO) holding the excess electron are depicted for the structures corresponding to r = 2.026 Å (equilibrium geometry) and r = 12 Å. | ||
3.4. Electronic stability of the anions matching the NgF3− and NgF5− formulas (Ng = Xe, Rn)
Due to the large electronic stabilities of anions designed to match the NgX7− formula we decided to verify if other negatively charged noble gas fluorides are also characterized by large VDEs (at the same level of theory, i.e. OVGF/6-311++G(3df,3pd)+ECPs). To investigate the possible electronic stability of the Ng's anions, we focus on the closed-shell anionic system utilizing Xe and Rn elements as central atoms and F atoms as ligands (as depicted in Fig. 4). As it found out, the vertical detachment energies calculated for NgF3− and NgF5− (Ng = Xe, Rn) anions span the ca. 5.2–7.8 eV range with the smallest (5.237 eV) values for the RnF3− and the largest (7.769 eV) for the XeF5− anion. Similar to NgX7− systems, despite of the fact that HOMOs reveal the antibonding character with respect to the ligand-central atom interaction, the corresponding highly negative HOMO eigenvalues of −6.2, −8.7, −5.9, and 9.0 eV for XeF3−, XeF5−, RnF3−, and RnF5−, respectively, reveal the large electronic stabilities of these anions (Fig. 4). Moreover, partial atomic charge on central atom reads 0.92, 1.46, 1.01, and 1.69 a.u. for respectively XeF3−, XeF5−, RnF3−, and RnF5−, which results in high polarity of Ng-F bonds and enables large electron binding energies of NgF3− and NgF5− (Ng = Xe, Rn) anions.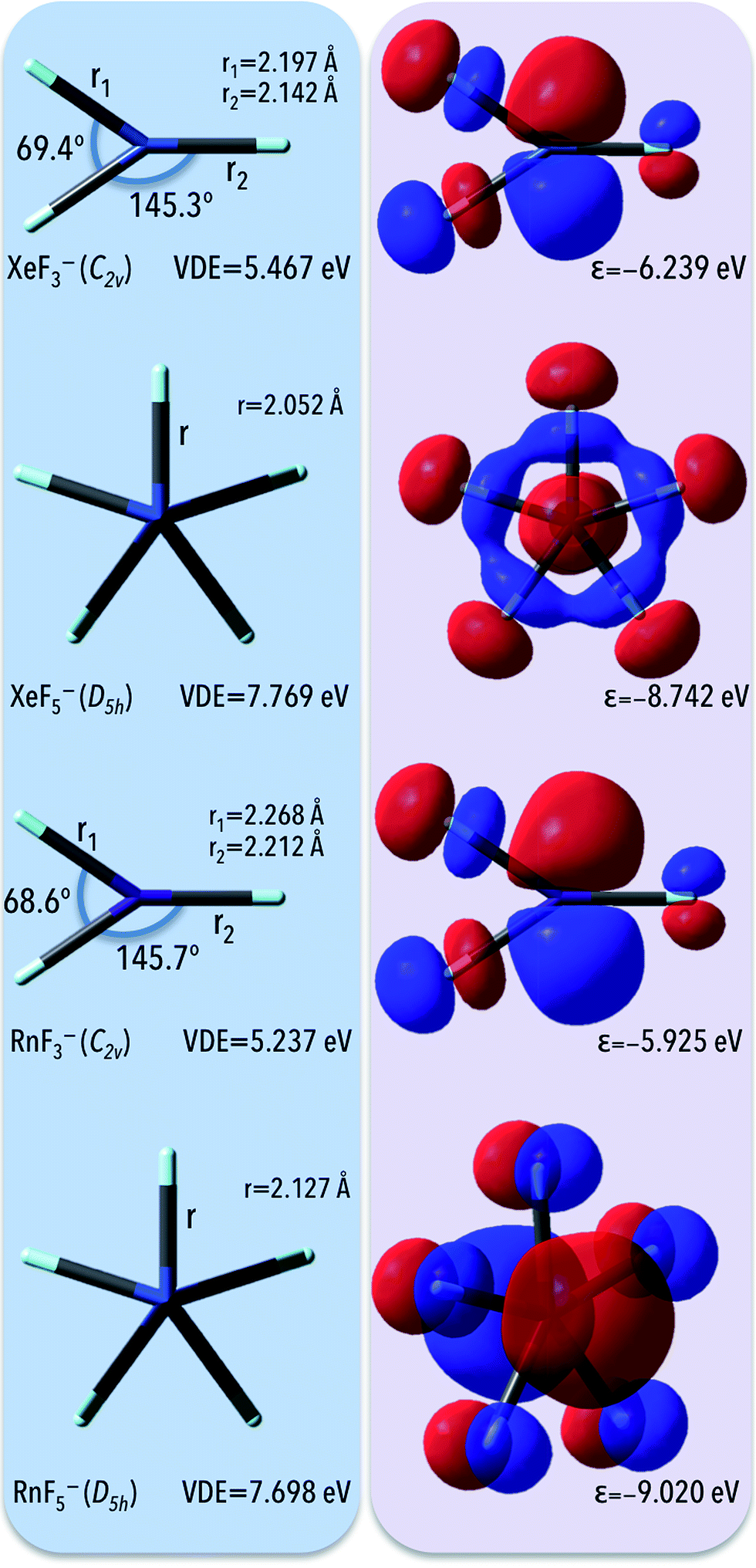 | ||
| Fig. 4 The equilibrium structures (on the left) and corresponding highest occupied molecular orbitals (on the right) of the NgF3− and NgF5− anions (Ng = Kr, Xe) obtained at the MP2/6-311++G(3df,3pd)+ECPs level. | ||
As expected,9,45 higher VDEs are predicted for larger (i.e., containing a larger number of fluorine ligands) species matching the RnFn− (n = 3, 5, 7) formula. Namely, in the series of the mononuclear fluorides RnF3−–RnF5−–RnF7− the VDE grows sharply when going from RnF3− (5.237 eV) to RnF5− (7.698 eV) and continuously increases when going to RnF7− (8.136 eV). In contrary, in the series of the XeF3−–XeF5−–XeF7− anions the VDE significantly increases when going from XeF3− (5.467 eV) to XeF5− (7.769 eV) and decreases when going to XeF7− (6.967 eV). This behavior indicates that an optimal combination of stabilization and destabilization factors for attaining the largest VDE of mononuclear XeFn− (n = 3, 5, 7) anions occurs in the pentacoordinate fluorides. The further increase of the number of ligands does not lead to increasing the VDE. In this case (i.e. XeF7− whose VDE is smaller than the vertical electron detachment energy of XeF5− anion by 0.802 eV), destabilization due to interligand repulsion prevails over the stabilization due to delocalization of the additional electron over a large number of the ligand atoms.
3.5. Effect of solvent on stability of NgF7− anions (Ng = Kr, Xe, Rn)
Although the gas phase calculations give a good measure of the raw stability of species, the geometric and energetic properties of compounds may be somewhat different in a dielectric environment where ε ≠ 1. To estimate these effects, the polarizable continuum model (PCM) calculations (at the MP2/6-311++G(3df,3pd)+ECPs level) were carried out in simulated solvent environments with fully optimized structures (Fig. 5). The free energies (ΔH298PCM), entropies (ΔS298PCM), and Gibbs free energies (ΔG298PCM) for the reactions in the liquid phase corresponding to the most likely fragmentation channels are collected in Table 3. For KrF7−, the effects of the polarizable surroundings decrease significantly Gibbs free energies of considered fragmentation channels (the smallest ΔG298PCM value was predicted for the KrF7− → Kr + 3F2 + F− reaction and reads −54.7 kcal mol−1). Thus, thermodynamic instability of KrF7− is observed both in the gas phase (Table 2) as well as in liquid solution. In contrary, the XeF7− and RnF7− species were found to be stable and not susceptible to any fragmentation. Similarly to gas phase, in liquid phase for XeF7− and RnF7− anions, the smallest Gibbs free energy value was predicted for the F2 detachment (i.e. XeF7− → XeF5− + F2 and RnF7− → RnF5− + F2); but both these Gibbs free energies were found to be positive (24.75 and 39.14 kcal mol−1, for XeF7− and RnF7− respectively), which confirms the thermodynamic stability of the XeF7− and RnF7− anions in water solution. Since the fragmentations leading to the NgF6, NgF6−, NgF4, NgF4−, NgF3, NgF3−, NgF2, NgF2−, Ng, or Ng− (where Ng = Xe, Rn) are even less likely (calculated ΔG298PCM values span the 29.5–542.3 kcal mol−1 range), we conclude that the XeF7− and RnF7− anions are not susceptible to any fragmentations in water solution. Consequently, analysis of the ΔG298PCM values calculated for the most likely fragmentation reactions confirms both (i) thermodynamic instability of KrF7− anion and (ii) xenon and radon atoms capability of forming thermodynamically stable NgF7− (Ng = Xe, Rn) anions.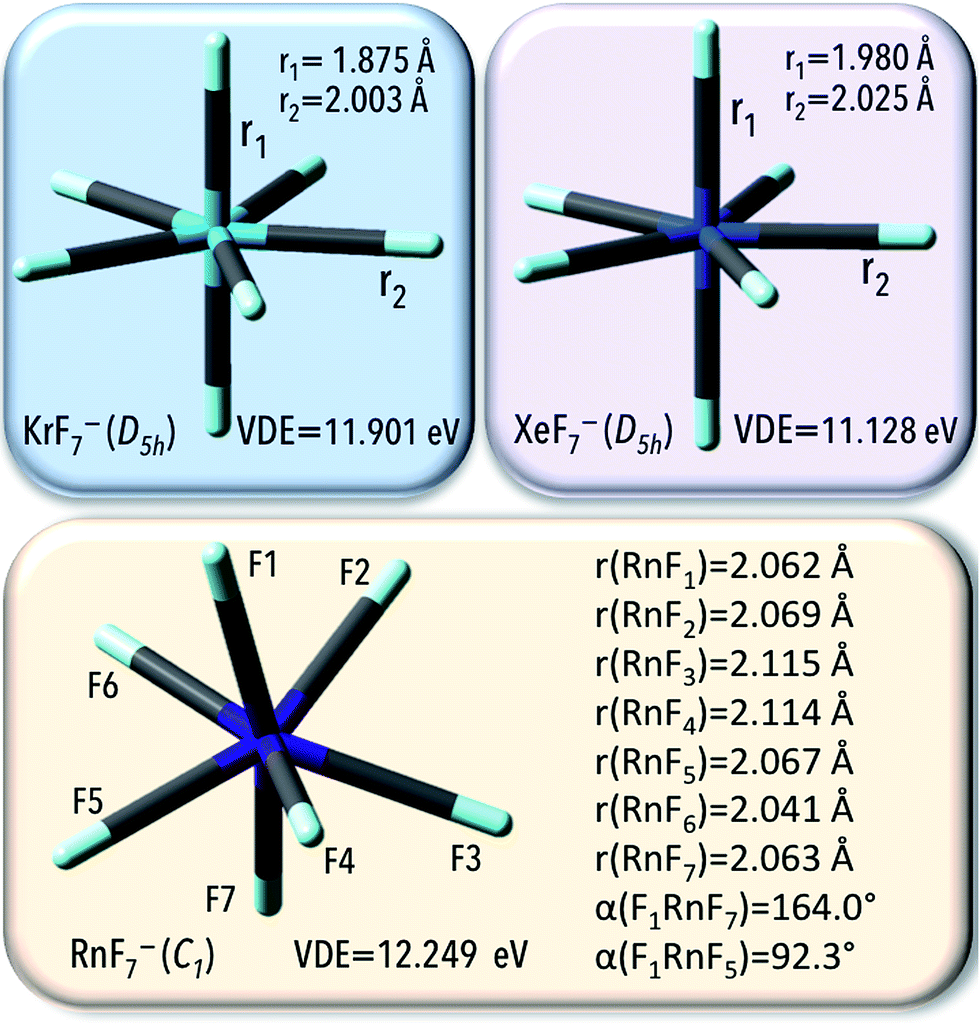 | ||
| Fig. 5 The equilibrium structures of the KrF7− (on the left), XeF7− (on the right), and RnF7− (on the bottom) anions obtained within polarizable continuum model (PCM) at the at the MP2/6-311++G(3df,3pd)+ECPs level. | ||
| Fragmentation path | ΔH298PCM | ΔS298PCM | ΔG298PCM |
|---|---|---|---|
| KrF7− → KrF6− + F | 32.25 | 27.91 | 23.93 |
| KrF7− → KrF4− + F2 + F | 5.79 | 96.13 | −22.87 |
| KrF7− → KrF2− + 2F2 + F | 19.44 | 98.01 | −9.78 |
| KrF7− → Kr + 3F2 + F− | −19.80 | 117.01 | −54.69 |
| KrF7− → Kr− + 3F2 + F | 19.44 | 98.01 | −9.78 |
| XeF7− → XeF6 + F− | 35.19 | 19.24 | 29.45 |
| XeF7− → XeF6− + F | 169.03 | 24.39 | 161.76 |
| XeF7− → XeF5− + F2 | 33.88 | 30.64 | 24.75 |
| XeF7− → XeF4 + F2 + F− | 56.67 | 56.51 | 39.83 |
| XeF7− → XeF4− + F2 + F | 102.77 | 68.77 | 82.27 |
| XeF7− → XeF3 +F− + F2 + F | 189.83 | 123.42 | 153.03 |
| XeF7− → XeF3− + 2F2 | 81.71 | 73.94 | 59.67 |
| XeF7− → XeF2 + 2F2 + F− | 82.12 | 88.54 | 55.72 |
| XeF7− → XeF2− + 2F2 + F | 136.86 | 96.79 | 108.00 |
| XeF7− → Xe + 3F2 + F | 113.34 | 115.16 | 79.00 |
| XeF7− → Xe− + 3F2 + F− | 300.56 | 117.92 | 256.41 |
| RnF7− → RnF6 + F− | 45.24 | 19.70 | 39.37 |
| RnF7− → RnF6− + F | 73.54 | 29.75 | 64.67 |
| RnF7− → RnF5− + F2 | 47.77 | 28.95 | 39.14 |
| RnF7− → RnF4 + F2 + F− | 79.69 | 55.76 | 63.06 |
| RnF7− → RnF4− + F2 + F | 126.77 | 56.73 | 109.86 |
| RnF7− → RnF3 +F− + F2 + F | 155.11 | 99.48 | 125.45 |
| RnF7− → RnF3− + 2F2 | 116.60 | 71.74 | 95.21 |
| RnF7− → RnF2 + 2F2 + F− | 117.49 | 86.84 | 91.60 |
| RnF7− → RnF2− + 2F2 + F | 176.68 | 96.01 | 148.05 |
| RnF7− → Rn + 3F2 + F− | 164.43 | 112.82 | 130.79 |
| RnF7− → Rn− + 3F2 + F | 576.72 | 115.58 | 542.26 |
An interaction with polarizable surroundings (represented by a water as a solvent) remarkably improves the electron bind ability of NgF7− (VDE increases up to 11.90, 11.13, and 12.25 eV for respectively KrF7−, XeF7−, and RnF7−), which is accompanied by Ng–F bonds decreased (by about 0.001–0.008 Å, Fig. 5). Those large VDE values imply that in the presence of the polarizable surroundings, NgF7− systems (Ng = Kr, Xe, Rn) are less sustainable to electron detachment.
In summary, the PCM results confirm that XeF7− and RnF7−anions are thermodynamically and geometrically stable compounds in water and their electronic stability increases (by ca. 4 eV) when the gas phase is replaced with the liquid phase. The KrF7− anion was found to be geometrically and electronically stable system, however, due to its thermodynamic instability, the KrF7− ends up as only a locally stable system.
3.6. Dinuclear anions Ng2F13− (Ng = Xe, Rn)
Gutsev and Boldyrev have concluded that designing polynuclear superhalogen anions give the possibility of increasing the number of ligands and avoiding the increase of interligand repulsion.9 Hence, construction dinuclear structures with more ligands will lead to achieving species with larger VDEs. In this contribution, dinuclear Ng2F13− (Ng = Xe, Rn) anions, were designed and investigated to verify whether such species have larger VDEs that the above-discussed mononuclear NgF7− anions. The exploration of the potential energy surface (PES) of the anionic Ng2F13− systems (M = Xe and Rn) leads to the minimum energy structures depicted in Fig. 6. The corresponding geometrical parameters along with harmonic vibrational frequencies for these structures are provided in the ESI (Table ESI 2†). The Rn2F13− anion was found to possess C2-symmetry and its structure resembles two RnF7 fragments oriented in an anti manner and sharing three fluorine atoms. The Rn–F bond lengths are in the 2.016–2.202 Å range except those in the Rn–F–Rn bridging fragments whose lengths are longer and read 2.305–2.460 Å. Recalling that the Rn–F separations in the Cs-symmetry RnF7− anion span the 2.067–2.092 Å range, it can be concluded that the Rn–F bonds in the dinuclear Rn2F13− anion are of similar lengths whereas the elongated bonds (by ca. 0.2–0.9 Å) are observed in the Rn–F–Rn bridging fragments. In addition, we checked that single- and double-bridged structures are not geometrically stable (i.e. we confirmed that the single- and double-bridged Rn2F13− systems do not correspond to the minimum energy structures and the deformation along their imaginary modes leads to triple-bridged species).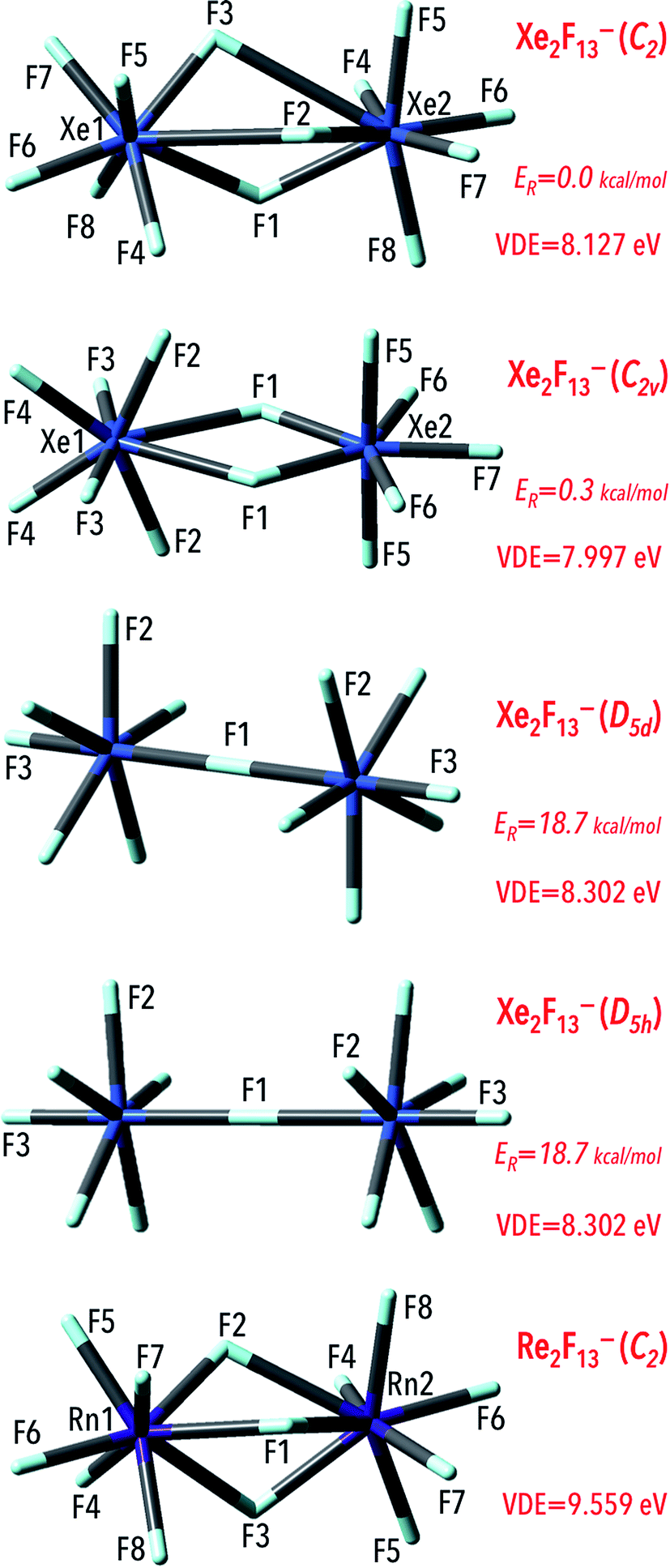 | ||
| Fig. 6 The equilibrium structures of the Ng2F13− anions (Ng = Xe, Rn) obtained at the MP2/6-311++G(3df,3pd)+ECPs level. The VDE values (in eV) and the relative energies (ER, in kcal mol−1) estimated for Xe2F13− isomers with respect to its corresponding global minimum are also provided. For all cases, the lowest VDE corresponds (from OVGF improvements) to the energy of HOMO. | ||
The structure of Xe2F13− anion is more complicated as four different isomers can be formed (see Fig. 6 where zero-point corrected energies of isomers are denoted as ER). Similarly to the Rn2F13−, the lowest energy structure (of C2-symmetry) resembles two XeF7 subunits (each of which contains the xenon atom surrounded by seven fluorine atoms) oriented in an anti manner and sharing three fluorine atoms. The values of the F–Xe–F bridging bond lengths were calculated to read 2.025–3.010 Å whereas the remaining Xe–F separations span relatively narrow 1.947–2.025 Å range. The resulting structure, albeit deficient in symmetry elements, corresponds to the global minimum on the ground state Xe2F13− anionic potential energy surface. The isomer having higher but similar energy (within 0.3 kcal mol−1) possesses C2v-symmetry and corresponds to the double-bridged (DB) form with the xenon atoms linked to each other through two fluorine (F1) atoms (Fig. 6). As expected,46–48 the Xe–F separation in DB connecting fragments are larger (by ca. 0.1–0.5 Å) than the Xe–F distances in the Xe–F terminal bonds (Table SI2†). The two other Xe2F13− isomers found are much higher in energy than the C2-symmetry lowest energy structure. Namely, the D5d- and D5h-symmetry single-bridged (SB) isomers were estimated to be energetically less stable by 18.7 kcal mol−1 (with respect to the C2-symmetry Xe2F13− structure) and as conformational isomers may likely rapidly interconverting at room temperature. In the SB isomers, the quasi-pentagonal bipiramidal XeF7 subunits can be distinguished (with the ω(F1KrF2F2) = 93.9° dihedral angles) oriented in the anti (D5d-symmetry isomer) or syn (which results in the D5h-symmetry configuration of the anion) manner (as depicted in Fig. 6). In conformational isomers the Xe central atoms are linked with fluorine atoms and the corresponding Xe–F bond lengths in bridging fragment were found to be equal to 2.308 Å. The remaining Xe–F bond lengths were estimated to be shorter (1.865–1.981 Å) in the both Xe2F13− local minima. Finally, the small energy gap (within 1 kcal mol−1) between double-bridged and triple-bridged isomers of Xe2F13− indicates that this anion may likely exist in both isomeric forms (the remaining single-bridges structures possess much higher energies and differences exceed 18 kcal mol−1), although adopting C2-symmetry structure is energetically more favorable.
Since the Ng2F13− (Ng = Xe, Rn) anionic systems thermodynamic stability needs to be confirmed, we decided to verify whether those species are susceptible to various fragmentations. In order to do this, we considered various most likely fragmentation channels in the case of each anionic system (see Table 4 for all the fragmentation reactions investigated). In particular, we tested the susceptibility of each Ng2F13− (Ng = Xe, Rn) anion to the closed-shell anionic NgFn− (n = 3, 5, 7) molecule loss. The analysis performed used the standard expressions for an ideal gas in the canonical ensemble and the results are gathered in Table 4. In general, the Gibbs free energies (ΔG298r) values for the reactions involving the systems considered are positive which indicates the thermodynamic stability thereof. The only negative ΔG298r value (−12 kcal mol−1) was found for the higher energy isomers (D5d- and D5h-symmetry) of the Xe2F13− anion, which indicates that the Xe2F13− → XeF7− + XeF6 process should be thermodynamically favorable. However, as discussed in the preceding paragraph, the D5d- and D5h-symmetry Xe2F13− isomers are much higher in energy (by 18.7 kcal mol−1) than the corresponding C2-symmetry isomer, hence its thermodynamic instability seems unimportant (as the formation of those particular isomers is not likely). Thus, we are confident that none of the anions studied (at its lowest-energy structure) is susceptible to fragmentation, as indicated by the positive ΔG298r values for the fragmentation channels considered.
| Species (symmetry) | Fragmentation path | ΔH298r | ΔS298r | ΔG298r |
|---|---|---|---|---|
| Xe2F13− (C2) | Xe2F13− → 2XeF6 + F− | 87.27 | 60.03 | 69.37 |
| Xe2F13− → XeF7− + XeF6 | 13.84 | 35.78 | 3.17 | |
| Xe2F13− → XeF5− + XeF6 + F2 | 54.15 | 68.69 | 33.67 | |
| Xe2F13− → XeF3− + XeF6 + 2F2 | 94.10 | 112.50 | 60.56 | |
| Xe2F13− (C2v) | Xe2F13− → 2XeF6 + F− | 86.88 | 57.36 | 69.78 |
| Xe2F13− → XeF7− + XeF6 | 13.45 | 33.11 | 3.58 | |
| Xe2F13− → XeF5− + XeF6 + F2 | 53.75 | 66.02 | 34.07 | |
| Xe2F13− → XeF3− + XeF6 + 2F2 | 93.71 | 109.83 | 60.97 | |
| Xe2F13− (D5d) | Xe2F13− → 2XeF6 + F− | 68.12 | 46.05 | 54.39 |
| Xe2F13− → XeF7− + XeF6 | −5.31 | 21.81 | −11.81 | |
| Xe2F13− → XeF5− + XeF6 + F2 | 35.00 | 54.71 | 18.68 | |
| Xe2F13− → XeF3− + XeF6 + 2F2 | 74.95 | 98.53 | 45.58 | |
| Xe2F13− (D5d) | Xe2F13− → 2XeF6 + F− | 68.13 | 46.90 | 54.15 |
| Xe2F13− → XeF7− + XeF6 | −5.30 | 22.66 | −12.05 | |
| Xe2F13− → XeF5− + XeF6 + F2 | 35.01 | 55.56 | 18.44 | |
| Xe2F13− → XeF3− + XeF6 + 2F2 | 74.96 | 99.38 | 45.33 | |
| Rn2F13− (C2) | Rn2F13− → 2RnF6 + F− | 106.93 | 65.86 | 87.30 |
| Rn2F13− → RnF7− + RnF6 | 22.55 | 48.31 | 8.14 | |
| Rn2F13− → RnF5− + RnF6 + F2 | 78.40 | 73.30 | 56.55 | |
| Rn2F13− → RnF3− + RnF6 + 2F2 | 158.67 | 115.12 | 124.35 |
The calculated values of the excess electron binding energies of the NgnF6n+1− (Ng = Xe, Rn) anions reveal a strong dependence on the number of central noble gas atoms (n). Namely, the vertical excess electron binding energy predicted for the Rn2F13− anion (9.559 eV) is larger by 1.423 eV than that of the RnF7− system (Table 1). Correspondingly, the most stable isomer (C2-symmetry) of the Xe2F13− exhibits the VDE of 8.127 eV, which is larger by 1.160 eV than the electronic stability of the XeF7− species. In addition, our results indicate that the VDE of the dinuclear superhalogen Xe2F13− system depends on its geometrical structure. Namely, the VDEs estimated for four Ng2F13− isomeric structures span the 7.997–8.302 eV range (Fig. 6). This observation is in agreement with previous theoretical studies indicating that VDE strongly depends on superhalogen's geometrical structure (if various isomeric structures are possible and stable).49
4. Summary
To conclude, this paper confirms the existence of hypervalent species utilizing noble gas elements as central atoms. The calculations performed at the OVGF/6-311++G(3df,3pd)+ECPs level of theory show that the vertical electron detachment energies of NgF7− anions (Ng = Kr, Xe, Rn) always exceed 7 eV. The electronic stability of the Ng's anions with decreased number of electronegative F ligands is also large as the VDEs calculated for NgF3− and NgF5− (Ng = Xe, Rn) anions span the 5.2–7.8 eV range. The VDEs of dinuclear Ng2F13− (Ng = Xe, Rn) anions are in the 8.1–9.6 eV range, which is significantly larger than this of corresponding mononuclear anions. Three main factors determining high VDEs of studied anions are: (i) the electronegative character of the ligands; (ii) the relatively large number of the ligands leading to extent of the delocalization of an extra electron; (ii) positive charge on the central atom, which provides an additional stabilization of an extra electron which is delocalized over ligands.Although this contribution focuses on Kr, Xe, and Rn elements as potential central atoms for superhalogen systems, the concept of using noble gas atoms to design strong electron acceptor is general and could be extended to other candidates. An important implication of these results is that if the right ligands could be identified, chemical compounds of smaller Ng's such as He and Ne could also be formed.
Acknowledgements
This work was supported by the Polish Ministry of Science and Higher Education (MNiSW) Grant No. 0560/IP3/2013/72. Calculations have been carried out using resources provided by Wroclaw Centre for Networking and Supercomputing (http://wcss.pl), grant No. 378.References
- H. Hotop and W. C. Lineberger, J. Phys. Chem. Ref. Data, 1985, 14, 731–750 CrossRef CAS.
- G. L. Gutsev and A. I. Boldyrev, Chem. Phys., 1981, 56, 277–283 CrossRef CAS.
- C. Sikorska and P. Skurski, Mol. Phys., 2012, 110, 1447–1452 CrossRef CAS.
- C. Sikorska, Chem. Phys. Lett., 2015, 638, 179–186 CrossRef CAS.
- A. K. Srivastava and N. Misra, RSC Adv., 2014, 4, 41260–41265 RSC.
- K. Pradhan, G. L. Gutsev, C. A. Weatherford and P. Jena, J. Chem. Phys., 2011, 134, 144305 CrossRef PubMed.
- W. M. Sun, D. Hou, D. Wu, X. H. Li, Y. Li, J. H. Chen, C. Y. Li and Z. R. Li, Dalton Trans., 2015, 44, 19901–19908 RSC.
- L. P. Ding, X. Y. Kuang, P. Shao, M. M. Zhong and Y. R. Zhao, RSC Adv., 2013, 3, 15449–15456 RSC.
- G. L. Gutsev and A. I. Boldyrev, Adv. Chem. Phys., 1985, 61, 169–221 CrossRef CAS.
- G. N. Reddy and S. Giri, RSC Adv., 2016, 6, 47145–47150 RSC.
- C. Sikorska and P. Skurski, Chem. Phys. Lett., 2010, 500, 211–216 CrossRef CAS.
- C. Sikorska, Phys. Chem. Chem. Phys., 2016, 18, 18739–18749 RSC.
- N. Bartlett, Proc. Chem. Soc., London, 1962, 218 CAS.
- R. D. Peacock, I. Sheft and H. Selig, Proc. Chem. Soc., London, 1964, 285 CAS.
- R. D. Peacock, H. Selig and I. Sheft, J. Inorg. Nucl. Chem., 1966, 28, 2561–2567 CrossRef CAS.
- N. Vasdev, M. D. Moran, H. M. Tuononen, R. Chirakal, R. J. Suontamo, A. D. Bain and G. J. Schrobilgen, Inorg. Chem., 2010, 49, 8997–9004 CrossRef CAS PubMed.
- A. Ellern, A. R. Mahjoub and K. Seppelt, Angew. Chem., Int. Ed., 1996, 35, 1123–1125 CrossRef CAS.
- D. J. Grant, T. H. Wang, D. A. Dixon and K. O. Christe, Inorg. Chem., 2010, 49, 261–270 CrossRef CAS PubMed.
- K. Sikorska and C. Sikorska, SuperhalogenCreator, Gdansk, 2015, http://celinasikorska.pl/superhalogencreator/.
- A. D. Mclean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- K. Raghavachari and J. A. Pople, Int. J. Quantum Chem., 1978, 14, 91–100 CrossRef.
- G. E. Scuseria, C. L. Janssen and H. F. Schaefer, J. Chem. Phys., 1988, 89, 7382–7387 CrossRef CAS.
- G. E. Scuseria and H. F. Schaefer, J. Chem. Phys., 1989, 90, 3700–3703 CrossRef CAS.
- C. Sikorska, D. Ignatowska, S. Freza and P. Skurski, J. Theor. Comput. Chem., 2011, 10, 93–109 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2009 Search PubMed.
- D. J. Rowe, Rev. Mod. Phys., 1968, 40, 153–166 CrossRef.
- J. Simons, J. Chem. Phys., 1972, 57, 3787 CrossRef CAS.
- L. S. Cederbaum, J. Phys. B: At., Mol. Opt. Phys., 1975, 8, 290–303 CrossRef CAS.
- J. V. Ortiz, J. Chem. Phys., 1988, 89, 6353–6356 CrossRef CAS.
- J. Simons, J. Chem. Phys., 1971, 55, 1218-& Search PubMed.
- J. Simons and W. D. Smith, J. Chem. Phys., 1973, 58, 4899–4907 CrossRef CAS.
- V. G. Zakrzewski and J. V. Ortiz, Int. J. Quantum Chem., 1994, 23–27 CrossRef CAS.
- V. G. Zakrzewski and J. V. Ortiz, Int. J. Quantum Chem., 1995, 53, 583–590 CrossRef CAS.
- V. G. Zakrzewski, J. V. Ortiz, J. A. Nichols, D. Heryadi, D. L. Yeager and J. T. Golab, Int. J. Quantum Chem., 1996, 60, 29–36 CrossRef.
- J. Linderberg and Y. Ohrn, Int. J. Quantum Chem., 1977, 12, 161–191 CrossRef.
- J. V. Ortiz, J. Chem. Phys., 1988, 89, 6348–6352 CrossRef CAS.
- N. W. Winter, A. Laferrie and V. Mckoy, Phys. Rev. A, 1970, 2, 49–60 CrossRef.
- V. Mckoy, Phys. Scr., 1980, 21, 238–241 CrossRef CAS.
- V. G. Zakrzewski, O. Dolgounitcheva and J. V. Ortiz, J. Chem. Phys., 1996, 105, 5872–5877 CrossRef CAS.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239–245 CrossRef CAS.
- C. Sikorska, Chem. Phys. Lett., 2015, 625, 157–163 CrossRef CAS.
- B. Yin, J. L. Li, H. C. Bai, Z. Y. Wen, Z. Y. Jiang and Y. H. Huang, Phys. Chem. Chem. Phys., 2012, 14, 1121–1130 RSC.
- B. Yin, T. Li, J. F. Li, Y. Yu, J. L. Li, Z. Y. Wen and Z. Y. Jiang, J. Chem. Phys., 2014, 140, 094301 CrossRef PubMed.
- J. F. Li, Y. Y. Sun, H. C. Bai, M. M. Li, J. L. Li and B. Yin, AIP Adv., 2015, 5, 067143 CrossRef.
- C. Sikorska and P. Skurski, Chem. Phys. Lett., 2012, 536, 34–38 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra21933k |
| This journal is © The Royal Society of Chemistry 2016 |