
Selective, light-driven enzymatic dehalogenations of organic compounds†
Abstract
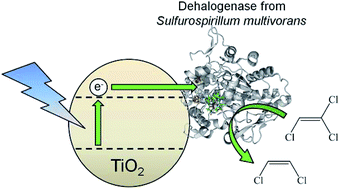
Tetrachloroethene reductive dehalogenase (PceA), a corrinoid-containing enzyme from Sulfurospirillum multivorans, is highly active for the sequential reduction of the organohalide tetrachloroethene (PCE) to trichloroethene (TCE), then regiospecifically to cis-1,2-dichloroethene (cDCE). We demonstrate direct electron transfer from graphite and semiconductor electrodes to PceA adsorbed onto the electrode surface. Colloidal TiO2 nanoparticles modified with PceA efficiently carry out the sequence of dehalogenation reactions under UV light irradiation.
- This article is part of the themed collection: Celebrating the 150th anniversary of the German Chemical Society
 Please wait while we load your content...
Please wait while we load your content...

