
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient dual acidic carbo-catalyst for one-pot conversion of carbohydrates to levulinic acid†
Dinesh
Gupta
ab,
Sabuj
Kundu
b and
Basudeb
Saha
*ac
aDepartment of Chemistry, Laboratory of Catalysis, University of Delhi, Delhi 11007, India. E-mail: bsaha@udel.edu
bDepartment of Chemistry, Indian Institute of Technology Kanpur, India
cCatalysis Center for Energy Innovation, University of Delaware, Newark, DE 19716, USA
First published on 21st October 2016
Abstract
Levulinic acid (LA) is an important platform intermediate for several high value renewable bioproducts. One-pot synthesis of LA from carbohydrates is disadvantaged by the lack of proximity of the active sites required for sequential isomerization, dehydration and rehydration reactions. Herein, we report an efficient one-pot conversion of C6 sugars to LA using a carbo-catalyst (ZrHyC), containing Lewis acidic zirconium and Brønsted acidic carboxyl and hydroxyl sites. The proximity of the two acid sites on a hydrothermally prepared carbon support makes the catalyst highly effective enabling a maximum 73% LA yield from glucose conversion. Fructose conversion achieved a higher LA yield (89%) because of a facile reaction without involving an isomerization step. The effects of solvents, the glucose to catalyst ratio, and the reaction time on glucose conversion and LA yield are systemically examined. Detailed characterization of the catalyst using FTIR, Raman spectroscopy, XPS, XRD, FESEM and NH3-TPD techniques are presented to elucidate the presence of the two acid sites and zirconium in the +4 oxidation state.
1. Introduction
Growing demand for energy in emerging economies in recent years has created opportunities for researchers to shift their focus to the development of alternate energy resources based on abundant non-food biomass and solar energy.1 Selective and efficient conversions of biomass feedstock to sustainable chemicals and liquid fuels are considered to be the most promising pathways to solve the energy crisis for future generations, owing to their abundance and sustainability.2,3 This approach of producing clean energy and bio-products will also mitigate the environment concerns related to the emissions of greenhouse gases. However, development of cost-effective processes for producing such products is still a challenge, although significant advancements have been achieved in the past decade.4 In this context, development of solid acid catalysts has been one of the thrust areas in materials science for biomass transformation due to distinct advantages of such materials for easy separation from the product stream, recyclability, and regeneration.5–75-Hydroxymethylfurfural (HMF) and furfural are top ten biomass derived platform chemicals and have applications as a feedstock for petroleum replacement advanced fuels, and commodity and specialty chemicals including monomers for the next generation polymeric products.8 Levulinic acid (LA) is one such product that is formed from HMF and its analogous furfural and furfural alcohol via acid catalyzed rehydration reaction.9 LA is an important precursor for biorenewable fuel additives, performance products, polyacrylate, polycarbonates, biodegradable herbicides, photosynthesis promoters and aliphatic carboxylic acid (Scheme 1).10,11 LA synthesis from lignocellulose involves several intermediate steps, namely (1) lignocellulose pretreatment and hydrolysis to monosaccharides, (2) acid catalyzed isomerization and dehydration of monosaccharides to furfurals and (3) subsequent rehydration of furfurals with acid catalysts.12,13 Depending on the reaction conditions for HMF and furfural production, some amount of LA is also formed as a by-product. Heeres et al. have reported two approaches for LA production from carbohydrates in which the first approach involves the use of high concentration of mineral acid catalyst at low temperature and the second approach is the use of dilute mineral acid catalyst at higher temperature.14 However, both approaches use homogeneous acid, which is corrosive, expensive to separate from the product stream and poses environmental concern.
 | ||
| Scheme 1 Applications of LA for other bio-renewable chemicals. | ||
The use of Brønsted and Lewis acidic materials separately (mineral acid, ionic liquids, metal halides) or a physical mixture of the two components have reported for carbohydrates conversion to LA.15–17 Hanna et al. used 0.3 wt% aqueous sulphuric acid for the conversion of extruded starch at 200 °C which yielded maximum 47% LA for 1 h.9 Sulfuric acid has also been used in an earlier report for kinetic study of HMF rehydration to LA, which demonstrated that maximum 94% molar yield of LA can be achieved in 3 h under HMF to H2SO4 molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (0.1 M HMF and 1 M H2SO4).18 Recently, Lewis acidic InCl3 (2.5 mol%) catalysis of fructose in water reported 45% LA yield at 180 °C for 1 h.19 The latter reaction progressed via dehydration of fructose to HMF in the first step followed by rehydration of HMF. Among the reported catalysts, the dual acidic materials, containing Lewis and Brønsted acidic sites, are beneficial for high yield of LA.20 For example, dual acidic CrCl3–HY-zeolite catalyst containing CrCl3 and HY-zeolite in 1:1 weight ratio yielded 62% LA from glucose in water at 160 °C for 3 h.21 In this reaction, Lewis acid sites facilitated glucose isomerization and Brønsted sites catalyzed dehydration and subsequent rehydration steps. A mixture of Lewis (ZnBr2) and Brønsted (HCl) acidic sites also facilitated glucose conversion under microwave irradiation, enabling 53 wt% LA in 6 min.22 Direct conversion of cellulose with Brønsted acidic –HSO3 functionalized ionic acid catalysts yielded 43% LA under microwave-irradiated heating at 170 °C for 30 min.23 Cellulose conversion has also been reported with sulfonated hyperbranched poly(arylene oxindole) catalyst which achieved 30% LA yield in water at 170 °C for 24 h.14
:10 (0.1 M HMF and 1 M H2SO4).18 Recently, Lewis acidic InCl3 (2.5 mol%) catalysis of fructose in water reported 45% LA yield at 180 °C for 1 h.19 The latter reaction progressed via dehydration of fructose to HMF in the first step followed by rehydration of HMF. Among the reported catalysts, the dual acidic materials, containing Lewis and Brønsted acidic sites, are beneficial for high yield of LA.20 For example, dual acidic CrCl3–HY-zeolite catalyst containing CrCl3 and HY-zeolite in 1:1 weight ratio yielded 62% LA from glucose in water at 160 °C for 3 h.21 In this reaction, Lewis acid sites facilitated glucose isomerization and Brønsted sites catalyzed dehydration and subsequent rehydration steps. A mixture of Lewis (ZnBr2) and Brønsted (HCl) acidic sites also facilitated glucose conversion under microwave irradiation, enabling 53 wt% LA in 6 min.22 Direct conversion of cellulose with Brønsted acidic –HSO3 functionalized ionic acid catalysts yielded 43% LA under microwave-irradiated heating at 170 °C for 30 min.23 Cellulose conversion has also been reported with sulfonated hyperbranched poly(arylene oxindole) catalyst which achieved 30% LA yield in water at 170 °C for 24 h.14
Among the other reported solid acid catalysts for LA production, notable examples are ZrP and Amberlyst-70 catalysts, yielding 17% and 28% LA, respectively, from glucose in aqueous medium at 160 °C for 4 h.24,25 The latter reactions formed about 63% humin at 160 °C. While the other reports did not quantify the amount of humins, it is evident from the yields of LA that the loss of carbon in the form of humins was significant. One reason for high humins could be the use of high temperatures (150–200 °C) for the reactions, which aggravates humins formation in the presence of acid catalysts.26 Herein, we report the synthesis and characterization of a carbonaceous catalyst, containing Lewis acidic zirconium sites and Brønsted acidic carboxyl and hydroxyl groups, under hydrothermal method. As-synthesized ZrHyC material, containing monoclinic crystalline planes and Zr in +4 oxidation state, shows high catalytic activity in the conversion of glucose to LA, affording 67 mol% LA with nearly 100% selectivity in biphasic solvent at 65 °C and short reaction time (20 min). Fructose conversion recorded higher yield of LA (89%) under comparable reaction conditions.
2. Experimental section
2.1 Materials
Zirconium(IV) oxychloride (ZrOCl2·8H2O) was purchased from Sigma-Aldrich. Glucose, fructose and sucrose were obtained from SRL, India. Acetonitrile, methyl isobutyl ketone (MIBK) and ethanol were purchased from Spectrochem, India and Changshu Yangyuan, China. Dimethylsulfoxide (DMSO), sodium chloride, tetrahydrofuran (THF) and ethyl acetate were purchased from Sigma-Aldrich. All chemicals were used as received.The catalyst, ZrHyC, was prepared by a two-step process. In the first step, glucose (2.5 g) was dissolved in 100 mL DI water in a 100 mL Teflon-lined stainless steel autoclave. The reactor was sealed and heated in a muffle furnace at 160 °C for 12 h. After hydrothermal treatment of aqueous solution of glucose, the autoclave was cooled down to room temperature and black carbonaceous solid was dried at 60 °C overnight. The dried material was powdered using a mortar and pestle, which is henceforth, referred as hydrothermal carbon (HyC) support. In the second step, ZrOCl2 was incorporated on the HyC support hydrothermally. In this step, 150 mg of ZrOCl2·8H2O was dissolved in 50 mL DI water and 1 g HyC support was slowly added into the ZrOCl2 solution with continuous stirring for 30 minute. The mixture was then transferred into a 100 mL Teflon-lined stainless steel reactor which was heated at 160 °C for 12 h in a muffle furnace. Upon heating for the set time, the reactor was cooled down to room temperature and the material was filtered to obtain solid powder. The filtered material was subsequently washed with DI water and ethyl alcohol, and oven dried at 80 °C overnight. As-synthesized material, refereed here to as ZrHyC.
2.2 Instrumentations
The material properties of as-synthesized ZrHyC were analyzed by X-ray photoelectron spectra (XPS, PHI5000 Versa Prob II, FEI Inc.) equipped with Auger Electron Spectroscopy (AES) module and C60 sputter gun. The sample charging during the measurement was compensated by an electron flood gun. The electron take off angle with respect to the sample surface was 45°. The XPS spectra were collected in a fixed analyser transmission mode at 89.45 and 35.75 eV for recording survey and high resolution spectra, respectively. Field Emission Scanning Electron Microscopy (FESEM, JSM-7100F; JEOL) equipped with energy-dispersive X-ray (EDX) mapping device, Transmission Electron Microscopy (TEM, FEI Technai G2 12 Twin TEM 120 KV) and powder X-ray diffractometer (PXRD, Panalytical, Cu, Ka radiation) were used for recording XRD pattern and morphological images. Fourier transform infrared (FT-IR) spectra of the sample were collected on a Perkin Elmer (Spectrum II) spectrometer using KBr disk. Elemental analysis of the sample was performed using a CHNS analyzer (Perkin Elmer series-II). N2 adsorption–desorption isotherms were obtained on an Autosorb iQ-BET surface area analyzer instrument. The sample was degassed at 150 °C for 16 h under vacuum prior to N2 sorption analysis. Pore size was determined by Barrett–Joyner–Halenda (BJH) method using NOVA 2000e pore size analyser at 77 K. NH3-TPD analysis was conducted on an Autosorb iQ acidity analyzer in the temperature range of 100–750 °C by using a thermal conductivity detector. For TPD measurement, the sample was activated at 100 °C inside the reactor of the TPD furnace under a He flow. After cooling to 100 °C, 20% NH3 balanced with He was injected, and the system was allowed to equilibrate. The temperature was then raised at a heating rate of 10 °C min−1 up to 750 °C.2.3 Catalysis
The catalytic effectiveness of ZrHyC for the conversion of carbohydrates (glucose, fructose and sucrose) to LA were carried out in a mixture of water and THF under microwave-irradiation (CEM Matthews WC Discover of model SP14170 equipped with programmable pressure and temperature controller) by adding desired amount of substrate, solvent and catalyst in a 50 mL glass reactor. In a typical experiment, the reactor was loaded with 20 mg of catalyst, 100 mg of substrate and 6 mL of solvent (5 mL THF and 1 mL water). The loaded reactor was then placed inside the microwave reactor pre-set at the desired temperature and reaction time. After completion of reaction for the set time, the reaction mass was cooled down to room temperature and the catalyst was filtered. To the filtrate, 100 mg of NaCl was added for facilitating the separation of water and THF layers. The THF layer was decanted into a pre-weighed empty glass vessel. The aqueous layer was washed with THF (three times, 2 mL each time) to extract residual organic product and the extractant was collected into the same THF vessel. THF was evaporated under vacuum by using a rotary evaporator and the vessel containing the crude product was weighed again to obtain isolated yield of LA. The purity and yield of LA was further measured by 1H-NMR, using known amount of mesitylene as an internal standard. In case of reaction in MIBK–H2O biphasic solvent, MIBK phase containing organic product was decanted into a separate vessel and the aqueous phase was washed with MIBK to extract residual product. The crude product was obtained by following the similar procedure mentioned above.2.4 Determination of LA yield and selectivity
The yields of LA were determined by 1H NMR spectroscopic method. In this method, known amount of mesitylene (internal standard) was added into the LA crude product solution in CDCl3. The 1H NMR spectrum of the crude product in CDCl3 shows signals for two –CH2 and –CH3 groups of LA (Fig. S1 and S2†). The yields of HMF and LA were calculated from integrated values of –CHO and –CH2 protons (δ = 2.6 ppm) of HMF and LA and three aromatic ring protons of mesitylene (δ = 6.79 ppm). The NMR yields were further validated by HPLC analysis of the product solutions for some reactions. An Agilent HPLC equipped with a C18 reverse phase and Hi-Plex H columns and a refractive index (RI) detector as well as a photodiode array (PDA) detector was used. PDA detector was used for analysis of HMF and LA using water-AN solvent. The characteristic peaks for organic products were identified from their retention times. Each peak was integrated, and the actual concentrations of organic products were calculated from their respective pre-calibrated plots of peak areas vs. concentrations. Selectivities were calculated based on total detected carbon yields (yield of an individual product × 100)/(total yield of carbon products).3. Results and discussion
3.1 Structural and morphology studies of ZrHyC
The wide angle XRD pattern of ZrHyC (Fig. 1) shows peaks at 2θ values of 32, 46 and 61, matching with monoclinic ZrO2 (d111,220,310) (JCPDS file card no. 17-923, 27-997).27 The strong and weak diffraction peaks at 26.5° and 43° are for (002) and (100) planes of the HyC support matches with graphite carbon (JCPDS file no. 08-0415),28,29 suggesting HyC has some graphitic characteristic. The interlayer spacing at 2θ = 26.5° is calculated as 0.36 nm.30 The FT-IR spectrum (Fig. 2) shows –O–H and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration bands at 3350–3500 and 1575 cm−1.31,32 Other peaks at 1707, 1374 and 1083 cm−1 correspond to stretching vibrations for –CO (carboxylic acid), –C–OH and –C–O–C functional groups of the HYC support.33,34 The spectrum shows two distinct stretching vibration bands for –Zr–O and Zr–O–Zr at 655 and 475 cm−1.35,36 This confirms that zirconium is present on the HyC support. Two other bands at 1568 and 1437 cm−1 can be assigned to the stretching vibration of quinonoid and benzenoid rings that are present in the support.29
C stretching vibration bands at 3350–3500 and 1575 cm−1.31,32 Other peaks at 1707, 1374 and 1083 cm−1 correspond to stretching vibrations for –CO (carboxylic acid), –C–OH and –C–O–C functional groups of the HYC support.33,34 The spectrum shows two distinct stretching vibration bands for –Zr–O and Zr–O–Zr at 655 and 475 cm−1.35,36 This confirms that zirconium is present on the HyC support. Two other bands at 1568 and 1437 cm−1 can be assigned to the stretching vibration of quinonoid and benzenoid rings that are present in the support.29
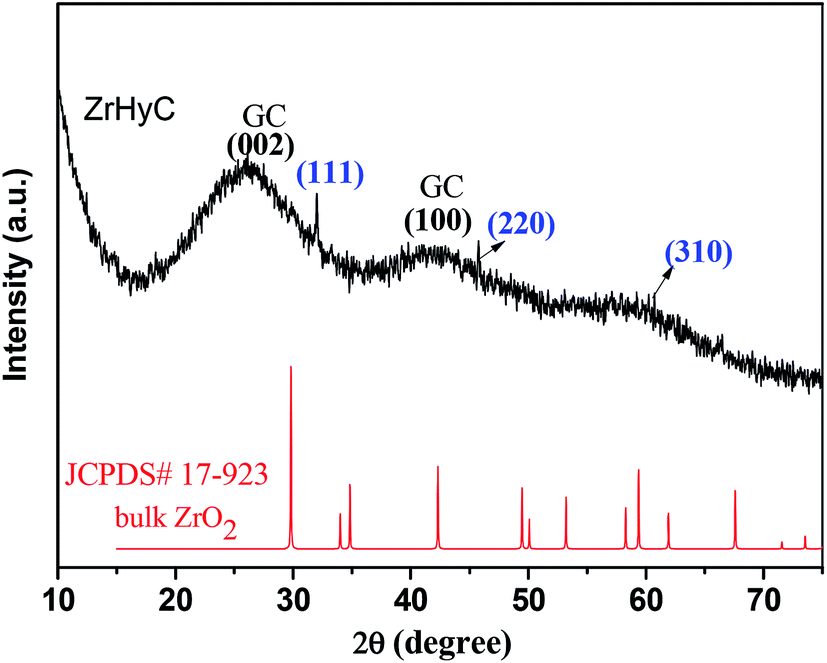 | ||
| Fig. 1 Powder XRD pattern of ZrHyC. | ||
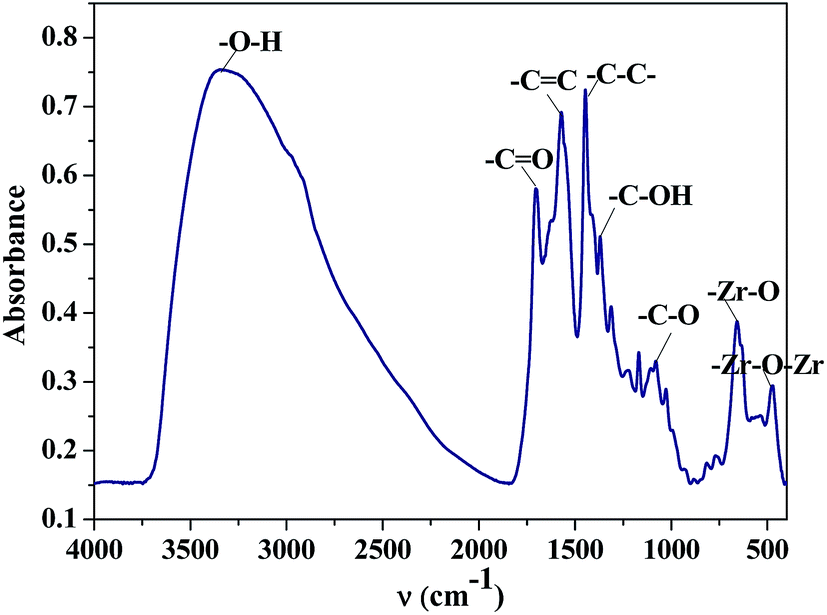 | ||
| Fig. 2 FT-IR spectrum of the as synthesized ZrHyC catalyst. | ||
In order to conclusively reveal the oxidation state of zirconium in the catalyst and its characteristic features, the X-ray photoelectron spectroscopy (XPS) of ZrHyC was recorded. The wide range spectra of ZrHyC show the presence of C 1s (59.8%), O 1s (35.1%), Zr 3d (4.5%), and Cl 2p (0.6%), respectively (Fig. 3). Two pairs of spin–orbit-split peaks correspond to two phases of zirconia, one phase denoted with Zr 3d5/2 peak at 181.24 eV and other with Zr 3d3/2 peak at 183.61 eV.37 In the wide range spectra, the binding energy of 181.24 eV and 183.61 eV (Fig. 3a) are for Zr–C and Zr–O–C (Zr 3d5/2,3/2), respectively, which manifests normal oxidation state of zirconium as Zr4+ in the as-synthesized ZrHyC.38 The binding energy peaks at 283.32 eV, 286.90 eV and 530.34 eV are consistent with –C–C, –C–OH and C–OO− functionalities of carbon skeleton as observed previously.29,39,40
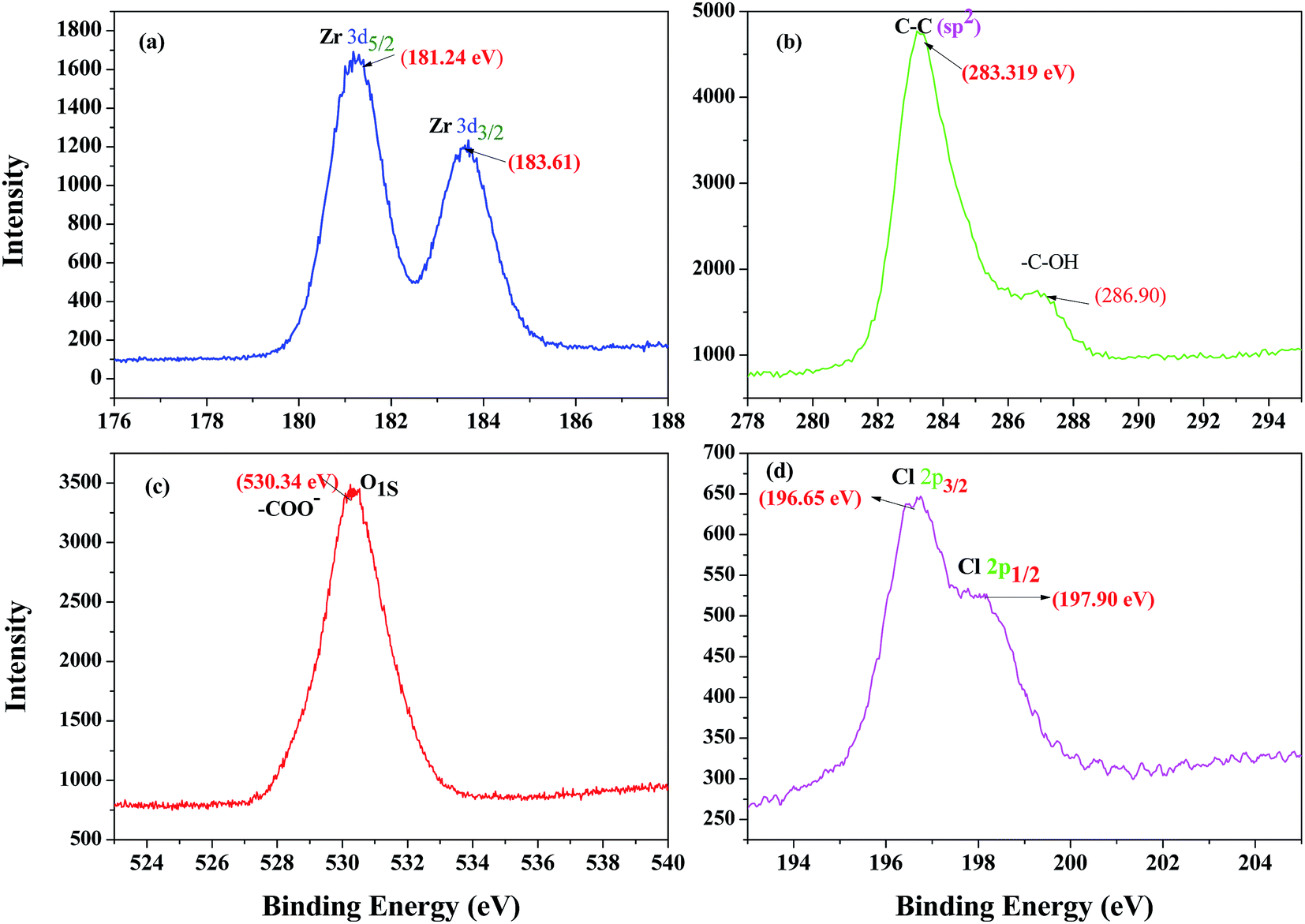 | ||
| Fig. 3 Broad scan XPS spectra of the as-synthesized ZrHyC, (a) zirconium (3d5/2,3/2) (b) carbon (C 1s, sp2), –C–O, (c) oxygen (O 1s) and (d) chlorine (Cl 2p3/2,1/2). | ||
Based on characterization of the –OH and –COOH functional groups on the support, and further confirmation of the presence of Zr4+ in the as-synthesized material, we illustrate the possible interactions of Zr ions with –OH and –COOH groups in Fig. 4. This illustration also suggests that ZrHyC contains Lewis and Brønsted acid sites. Previous reports also demonstrated Lewis acidity for Zr4+ on the surface of the heterogeneous catalysts.41,42
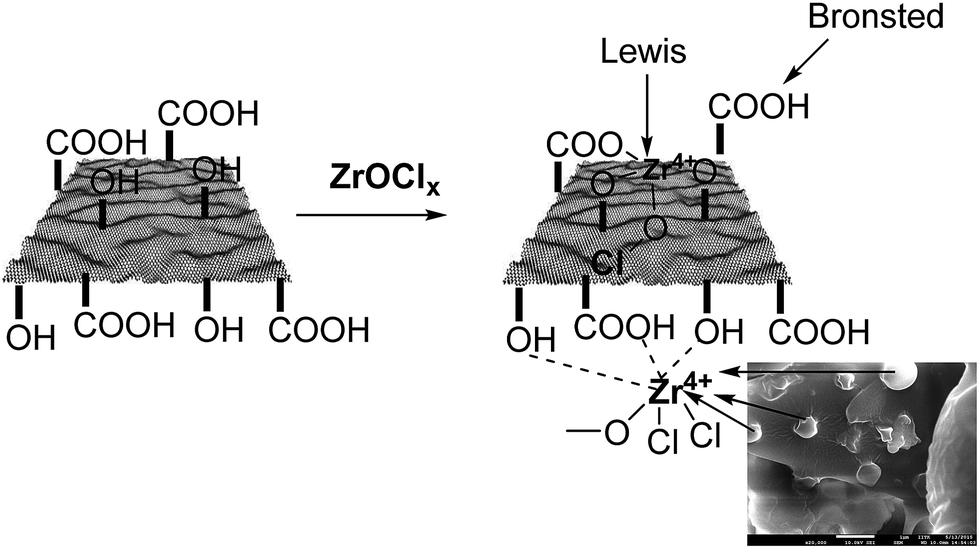 | ||
| Fig. 4 An illustration for Zr4+ incorporation on the HyC support. | ||
FESEM images show graphitic sheet like morphology of carbon and spherical shape of zirconium (Fig. 5). It shows that randomly aggregated, thin and crumpled sheets of the as-synthesized materials are closely associated with each other to form a disordered solid. It is further observed that the graphitic sheet is composed of randomly assembled thin nanosheets with a thickness of several nanometers (Fig. 5a, crumbled area). Fig. 5c shows spherical to plate shape morphology of zirconium oxide particles on the graphitic sheet (red circles in Fig. 5c). The low-magnification FESEM image (Fig. 5b) indicates that graphitic sheets are uniformly loaded with zirconium particles. The elemental composition by elemental mapping via FESEM-EDS confirms that ZrHyC contains Zr, C, O and Cl (Fig. 5d and S3 and S4†) and these elements are uniformly distributed throughout the material (Fig. S7†). CHNS elemental analysis and XPS data further supports the presence of H, C and Cl on the HyC support (Fig. S5†).
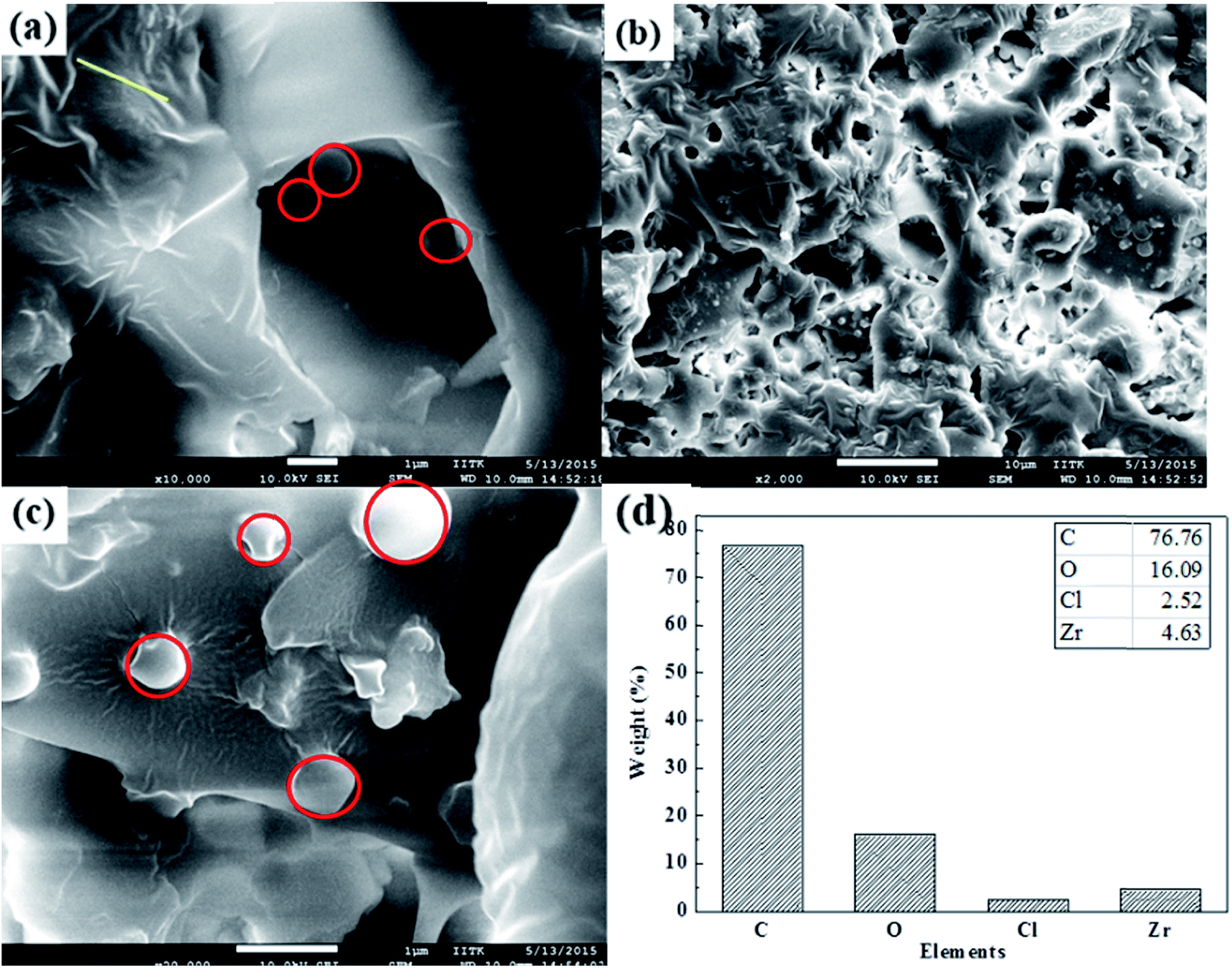 | ||
| Fig. 5 (a–c) FESEM images at different resolution and (d) FESEM-EDS of ZrHyC. | ||
The porosity and BET surface area of ZrHyC were determined from N2 adsorption–desorption study at 77 K (Fig. 6). This isotherm can be classified as a type IV isotherm with hysteresis loop of combination of H2 and H3, which correspond to micro- and mesoporous materials based on their adsorption isotherm at a low P/Po.43 The BET surface area, measured from the BET plot using 20 point BET equation, is 5.6 m2 g−1.13 Low surface area of ZrHyC may be due to agglomeration of graphitic sheets. The pore size, calculated from Barrett–Joyner–Halenda (BJH) model, varies from 2.2 to 3.3 nm.
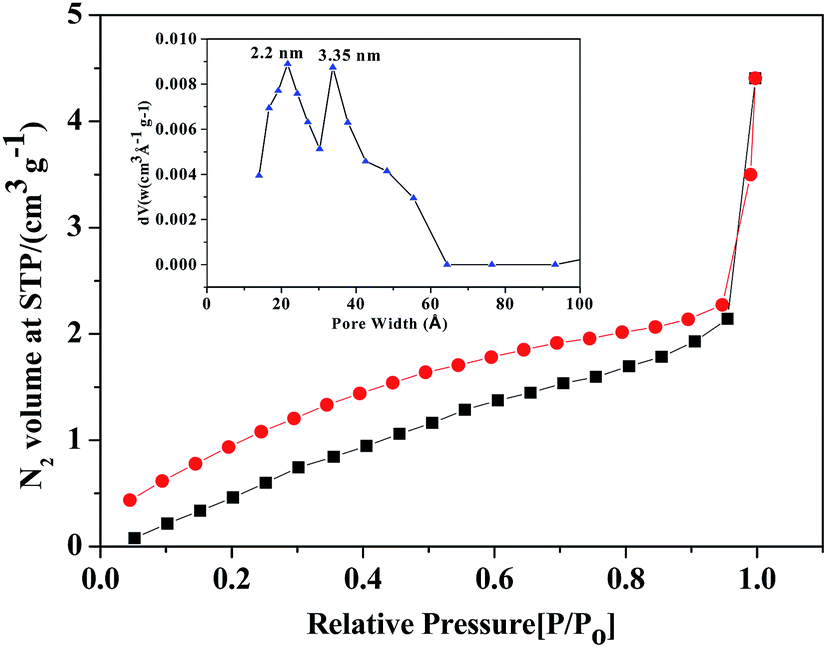 | ||
| Fig. 6 N2 adsorption–desorption isotherm of ZrHyC. Inset: BJH pore size distribution shows two types of pores. | ||
Acid density of ZrHyC was quantified by NH3-TPD measurement. In this experiment, the catalyst surface was first degassed and activated as discussed in the Experimental section, and NH3 was allowed to absorb on the catalyst surface and then desorbed as the temperature of the NH3-adsorbed material gradually increased. Usually desorption of NH3 at lower and higher temperatures occur from weaker and stronger acidic sites. The results in Fig. 7 show three peaks at 179, 389 and 474 °C. Based on previous literatures,44–46 we assign the peak at 179 °C for Lewis acidic Zr4+, and the other two peaks at higher temperatures for Brønsted acid sites (–COOH, –OH). Our interpretation of Lewis and Brønsted acid sites correlates with XPS and FTIR data. Calculated acid densities of the two major peaks are 1.35 and 1.17 mmol g−1, respectively; thus total acid density is 2.52 mmol g−1.
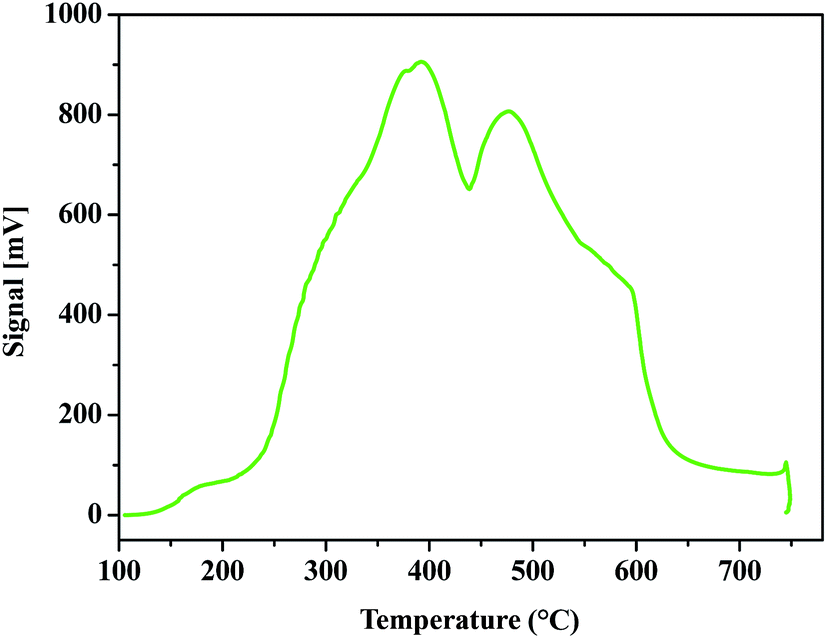 | ||
| Fig. 7 NH3-TPD profile of ZrHyC. | ||
4. Catalysis
4.1 Glucose conversion
The catalytic effectiveness of ZrHyC for glucose conversion was examined because of its high acid density (2.52 mmol g−1) and dual acid sites that can facilitate sequential glucose isomerization and dehydration.47 A preliminary reaction of 100 mg glucose with 20 mg catalyst in water/THF biphasic solvent (1:5 v/v) at 65 °C yields 22 mol% LA for 5 min under microwave irradiated heating. In terms of selectivity, this reaction gives more HMF (75%) than LA (25%). It is evident from the selectivity data that HMF is formed first via Zr4+ catalyzed isomerization of glucose to fructose, followed by Brønsted acid catalyzed dehydration of fructose, which is then rehydrated to LA via acid catalyzed ring-opening (RO) of furan ring. Light yellowish color of the product solution suggests that the reaction did not form humin by-products, in soluble and insoluble forms, although Brønsted acid density of the catalyst is quite high. It is possible that lower reaction temperature minimizes such by-products formation.
To understand the effectiveness of the catalyst further, we repeated this reaction under identical conditions for a longer time (10 min) which yields LA with higher selectivity (39%) at the expense of lower HMF selectivity (50%). Further increase of reaction time from 10 to 20 min results in more LA (67%) with absolute selectivity at quantitative conversion of glucose. However, there is a loss of about 33% carbon on molar basis which could be due to humins by-products as 1H NMR spectrum of the crude products did not show any signals for HMF or other lower ranged carbon products. It is worthy to mention that black color sticky material was observed when catalyst was separation by filtration, which is likely humins. Assuming that fructose was formed an as intermediate via isomerization of glucose (vide supra), we hypothesized that a reaction using fructose as a starting substrate would be more efficient. To examine this fact we start a reaction between 100 mg fructose and 20 mg catalyst under comparable reaction conditions at 65 °C which yields 89 mol% LA at quantitative conversion of fructose. The latter reaction with fructose explains a few important reaction insights. Firstly, the reaction gives 22% more LA when compared with LA yield from glucose conversion, which was 67%. This suggests the LA yield from glucose, which progressed via its isomerization of fructose followed by dehydration, is limited by the isomerization step, not by the dehydration step. Secondly, carbon mass loss in fructose conversion, presumably in the form of humins, is significantly less (11%) as compared to 33% carbon loss from glucose conversion. While humins formation pathways via self-condensation of HMF or its condensation of HMF with fructose and glucose have been reported in the literature48 and the involvement of an exact pathway is still a debate, our results show that carbon loss is more when we started the reaction from glucose, which may be an indication that condensation of HMF with glucose dominates than that with fructose. As shown in Fig. 8, the yield of LA remains at about 67% when the reaction time for glucose conversion with 20 mg catalyst at 65 °C was further increased up to 45 min.
 | ||
| Fig. 8 Dependence of LA and HMF selectivity on reaction time from ZrHyC catalyzed glucose conversion. Other reaction conditions: 100 mg glucose, H2O:THF (1:5 v/v), 65 °C, 20 mg catalyst and microwave power of 100 W. | ||
When compared with prior arts, the present reaction with ZrHyC, giving 67 mol% LA yield at lower temperature (65 °C) is significant. The prior reports using solid acid catalysts (sulfonated hyperbranched poly(arylene oxindole), ZrP and Amberlyst-70) yielded LA in the range of 20–30% at significantly higher temperatures (150–170 °C). A CrCl3/AlCl3 catalyzed conversion of glucose reported 46% yield of LA for 6 h at 140 °C.49 A zirconium phosphate catalyzed reaction demonstrated 40% glucose conversion with 17% LA yield at 160 °C for 3 h.25 Recently, Fe/HY zeolite catalyzed conversion of glucose yielded 62% LA for 3.3 h at 173 °C.50
4.2 The effect of catalyst loading
The catalyst variation experiments show that glucose conversion and LA selectivity increases with an increase in catalyst loading from 5 mg to 50 mg (Fig. 9). At low catalyst loading (5–10 mg), HMF selectivity is high as low concentration of acid sites of the catalyst was not able to further rehydrate HMF to LA. As the Brønsted acid sites in the reaction system increased at 20 mg catalyst loading, LA selectivity increased at the expense of HMF selectivity. A further increase of catalyst loading from 20 mg to 50 mg had no significant effect on LA selectivity. Therefore, additional experiments of glucose dehydration were carried out using 20 mg catalyst.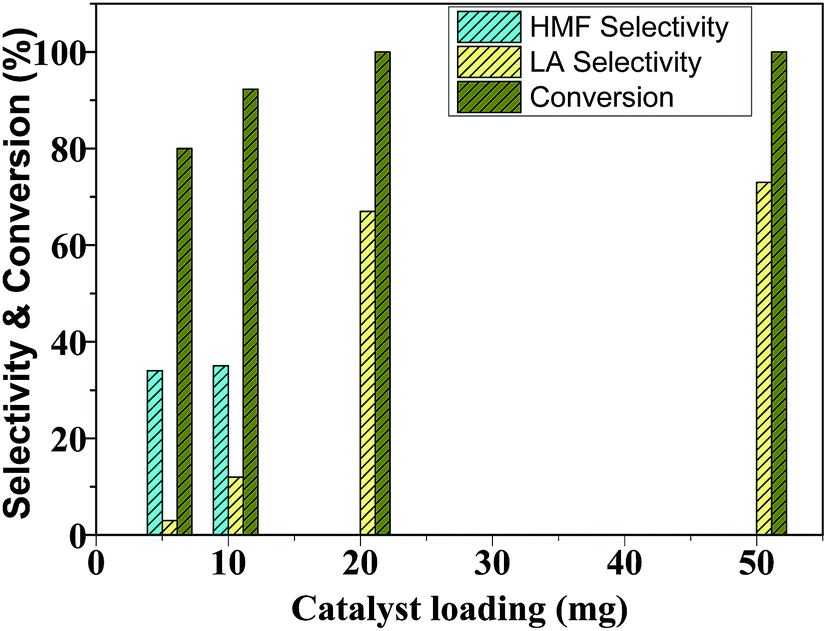 | ||
| Fig. 9 Glucose conversion and products selectivity as a function of catalyst loading. Reaction condition: 100 mg glucose, 20 min reaction time, H2O:THF (1:5 v/v) and 65 °C. | ||
4.3 The effect of glucose amount
To understand the effect of LA yield on glucose concentration, we varied the amount of glucose from 50 mg to 300 mg (0.27–1.6 mmol) for reaction with 20 mg catalyst. The results show a significant variation in LA yield with the substrate concentrations (Fig. 10). The reaction with 50 mg glucose gives the maximum amount of LA (77 mol%), which decreased with an increase in glucose amount. Almost three-fold decrease in the molar yield of LA is noted for decreasing initial glucose amount from 50 mg to 300 mg. Based on the acidity data, 20 mg catalyst contains 0.027 mmol Lewis acid sites, 0.022 mmol Brønsted acid sites and 0.049 mmol total acidity. Thus, it is apparent that the reaction becomes less efficient when the molar ratio of the catalyst's total acid density to glucose decreased from 0.18 (0.27 mmol glucose reaction) to 0.03 (1.6 mmol glucose reaction). As the ratio of Lewis to Brønsted acid sites also decreases in the same trend, this factor seems to have minimal effect on the reaction, involving sequential isomerization, dehydration and rehydration pathways. However, the reaction rate depends on the total acid concentration and the reaction at lower catalyst to glucose ratio is simply limited by lower acid concentration.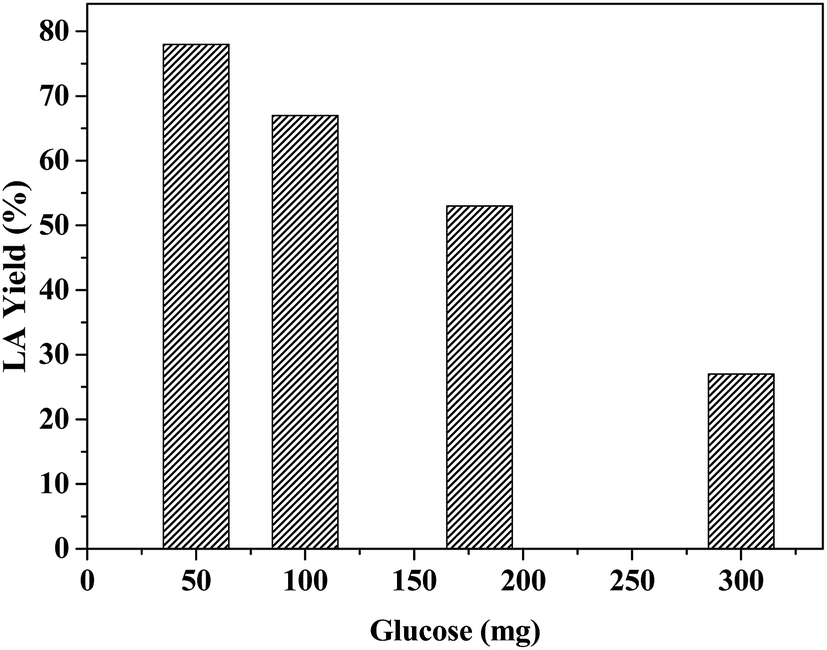 | ||
| Fig. 10 The effect of LA yields on the initial glucose amount from ZrHyC catalyzed conversion of glucose. Reaction condition: 20 mg ZrHyC, 65 °C and 20 min reaction time. | ||
4.4 The effect of solvents
We have also tested the effectiveness of ZrHyC for glucose conversion in other biphasic solvent system, for example water/MIBK, to elucidate the effect of partitioning of organic products (HMF, LA) in to the organic phase and hence the overall yields. Under comparable conditions, a reaction in water/MIBK yields 23 mol% HMF and 31 mol% LA at 89% glucose conversion at 65 °C for 20 min. The yield of LA in water/MIBK is lower than that observed in water/THF (67 mol% LA). While total yields of HMF and LA together (54%) in water/MIBK is closer to the LA yield in water/THF, the difference in observed results could be due to the effect of the organic solvent. In case of water/THF system, THF is miscible with water without NaCl in the aqueous phase. As NaCl was added after the reaction for partitioning the two phases, HMF formed as an intermediate from glucose, was in the same phase with the reactive medium during the reaction and hence HMF could effectively rehydrated to LA by the acid sites of the catalyst. However, MIBK is immiscible in water. Thus, HMF could efficiently extracted by MIBK during progression of reaction. This could results in less contact of HMF with the acid sites of the catalyst in the reactive phase for its rehydration to LA.4.5 Reaction pathway
Glucose conversion to LA generally progressed via acid catalyzed isomerization and dehydration of glucose to HMF followed by acid catalyzed rehydration of HMF to LA (Scheme 2). Several mechanisms for dehydration of glucose invoking acyclic and cyclic intermediates have been proposed.51 In the present reaction, Zr4+ acts as Lewis acid to isomerize glucose to fructose intermediate. Zr4+ catalyzed isomerization is reported in our earlier publication.52 In the subsequent step, Brønsted acidic –COOH and –OH sites facilitate dehydration step and subsequently rehydration of HMF to LA via ring opening. Pore size of the catalyst is reasonably high to perform these sequential reactions. To understand the reaction intermediates and to get a more insight into the reaction steps, we monitored glucose conversion to LA by 1H-NMR spectroscopic technique in which glucose (20 mg) was reacted with ZrHyC (5 mg) in DMSO-d6 (0.5 mL) in a J-Young NMR tube at 130 °C in oil bath. The first spectrum was recorded at time zero without heating the reaction mass. The subsequent spectra at 5, 20, 60, 180, 300, 480 and 600 min were recorded upon heating the reaction mass in the NMR tube in a pre-heated oil bath and immediately quenching the solution in an ice bath. The stack plot of the 1H NMR spectra (Fig. S6†) show signals for –OH protons of 1,6-anhydro-β-D-glucopyranose (AGP), 1,6-anhydro-β-D-glucofuranose (AGF) and 1,6-anhydro-α-D-fructofuranose (AFF) in the beginning of the reaction (5 min). A previous computational study proposed that AGP and AGF are formed irreversibly in glucose-to-fructose isomerization.53 As reaction progresses, the –OH proton signals gradually diminishes and HMF (δ 9.5), FA (δ 8.1) and LA (δ 2.2) are formed. Based on results from NMR studies, we propose plausible reaction pathways in Scheme 2.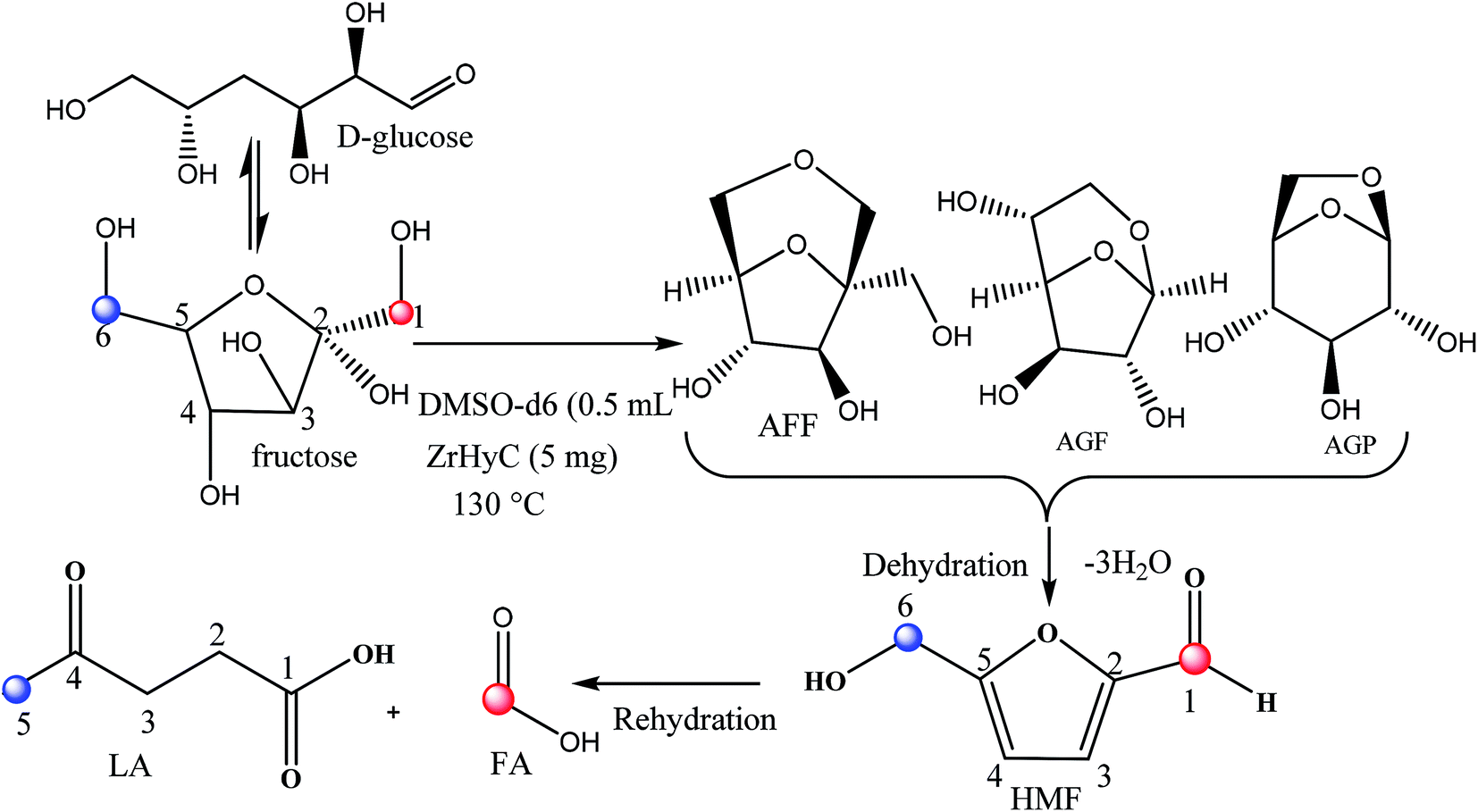 | ||
| Scheme 2 Reaction pathway for glucose conversion to LA. | ||
4.6 Catalyst recyclability study
The stability of ZrHyC was examined by conducting glucose conversion at 65 °C under MI heating for 20 min in water/THF. After completion of reaction in each cycle, solid catalyst was separated by filtration and reused for the next cycle. The sample in each cycle was analyzed using 1H NMR by following the same procedure described in the Experimental section. The results (Fig. S7†) show a decrease in LA selectivity from cycle 1 to 4, while total yield of HMF and LA remained similar because of an increase in HMF selectivity from cycle 1 to 4. This suggests that the catalyst exhibits comparable isomerization and dehydration activities, resulting in similar HMF yields, in all cycles, however the catalyst becomes less effective in rehydration of HMF to LA. Thus, the observed HMF yield from cycle 1 to 4 gradually increases at the expense of its less conversion to LA. A decrease of rehydration activity of the reused catalyst could be due to (i) a loss of surface Brønsted acid sites of the reused catalyst or (ii) block of Brønsted acid sites by polymeric humin residues.5. Conclusions
A hydrothermal carbon supported zirconium catalyst ZrHyC was synthesized for conversion of glucose to levulinic acid in biphasic solvent systems. The characterization of catalyst by FTIR, XPS, NH3-TPD, FESEM and XRD methods suggest that the material contains Lewis acidic Zr4+ ions and Brønsted acidic oxygen functional groups and the total acid density is 2.52 mmol g−1. The support material of interlayer thickness of 0.36 nm is uniformly loaded with Zr ions. Glucose conversion with the catalyst achieved a maximum 73% LA yield at mild reaction conditions (65 °C) under MI in 20 min. Fructose conversion under comparable conditions produced 89% LA. The effects of solvents, catalyst loading, reaction time, substrate concentration on the conversion of glucose and selectivity of HMF and LA are examined. The concentrations of Brønsted acid sites have a profound effect on LA yield and selectivity. The reactions at lower catalyst loading form HMF while at higher catalyst loading undergoes rehydration of HMF to LA with high selectivity. Mechanistic studies show that glucose conversion progresses via 1,6-anhydro-β-D-glucopyranose (AGP), 1,6-anhydro-β-D-glucofuranose (AGF) and 1,6-anhydro-α-D-fructofuranose (AFF) intermediates.Acknowledgements
DG acknowledges Department of Chemistry, Indian Institute of Technology, Kanpur, India, for a Postdoctoral fellowship (P.F. No. PDF-42) and the Advanced Centre for Materials Science (ACMS) for material characterization. BS acknowledges Catalysis Center for Energy Innovation, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award number DE-SC0001004 for partly supporting at later stages.References
- C. Ortiz-Cervantes, M. Flores-Alamo and J. J. GarcÃ-a, ACS Catal., 2015, 5, 1424–1431 CrossRef CAS.
- F. M. A. Geilen, B. Engendahl, A. Harwardt, W. Marquardt, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2010, 49, 5510–5514 CrossRef CAS PubMed.
- E. F. Iliopoulou, S. Stefanidis, K. Kalogiannis, A. C. Psarras, A. Delimitis, K. S. Triantafyllidis and A. A. Lappas, Green Chem., 2014, 16, 662–674 RSC.
- S. Dutta, ChemSusChem, 2012, 5, 2125–2127 CrossRef CAS PubMed.
- P. Gupta and S. Paul, Green Chem., 2011, 13, 2365–2372 RSC.
- R. Sahu and P. L. Dhepe, ChemSusChem, 2012, 5, 751–761 CrossRef CAS PubMed.
- R. Rinaldi and F. Schuth, Energy Environ. Sci., 2009, 2, 610–626 CAS.
- B. Saha, D. Gupta, M. M. Abu-Omar, A. Modak and A. Bhaumik, J. Catal., 2013, 299, 316–320 CrossRef CAS.
- J. Y. Cha and M. A. Hanna, Ind. Crops Prod., 2002, 16, 109–118 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- B. Saha and M. M. Abu-Omar, Green Chem., 2014, 16, 24–38 RSC.
- B. R. Caes, R. E. Teixeira, K. G. Knapp and R. T. Raines, ACS Sustainable Chem. Eng., 2015, 3, 2591–2605 CrossRef CAS.
- M. G. Mazzotta, D. Gupta, B. Saha, A. K. Patra, A. Bhaumik and M. M. Abu-Omar, ChemSusChem, 2014, 7, 2342–2350 CrossRef CAS PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- L. Qi, Y. F. Mui, S. W. Lo, M. Y. Lui, G. R. Akien and I. n. T. Horváth, ACS Catal., 2014, 4, 1470–1477 CrossRef CAS.
- G.-T. Jeong, Ind. Crops Prod., 2014, 62, 77–83 CrossRef CAS.
- J. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979–1985 CrossRef CAS PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Green Chem., 2006, 8, 701–709 RSC.
- Y. Shen, Y. Xu, J. Sun, B. Wang, F. Xu and R. Sun, Catal. Commun., 2014, 50, 17–20 CrossRef CAS.
- S. Van de Vyver, J. Thomas, J. Geboers, S. Keyzer, M. Smet, W. Dehaen, P. A. Jacobs and B. F. Sels, Energy Environ. Sci., 2011, 4, 3601–3610 CAS.
- N. Ya'aini, N. A. S. Amin and S. Endud, Microporous Mesoporous Mater., 2013, 171, 14–23 CrossRef.
- V. B. Kumar, I. N. Pulidindi and A. Gedanken, RSC Adv., 2015, 5, 11043–11048 RSC.
- H. Ren, Y. Zhou and L. Liu, Bioresour. Technol., 2013, 129, 616–619 CrossRef CAS PubMed.
- R. Weingarten, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2012, 5, 7559–7574 CAS.
- R. Weingarten, Y. T. Kim, G. A. Tompsett, A. Fernández, K. S. Han, E. W. Hagaman, W. C. Conner Jr, J. A. Dumesic and G. W. Huber, J. Catal., 2013, 304, 123–134 CrossRef CAS.
- X. Hu, C. Lievens, A. Larcher and C.-Z. Li, Bioresour. Technol., 2011, 102, 10104–10113 CrossRef CAS PubMed.
- F. Kazemi, A. Saberi, S. Malek-Ahmadi, S. Sohrabi, H. Rezaie and M. Tahriri, Ceram.–Silik., 2011, 55, 26–30 CAS.
- M. A. Mellmer, J. M. R. Gallo, D. Martin Alonso and J. A. Dumesic, ACS Catal., 2015, 5, 3354–3359 CrossRef CAS.
- L. Tang, Y. Wang, Y. Li, H. Feng, J. Lu and J. Li, Adv. Funct. Mater., 2009, 19, 2782–2789 CrossRef CAS.
- K. Krishnamoorthy, M. Veerapandian, K. Yun and S.-J. Kim, Carbon, 2013, 53, 38–49 CrossRef CAS.
- X. Tang, L. Hu, Y. Sun, G. Zhao, W. Hao and L. Lin, RSC Adv., 2013, 3, 10277–10284 RSC.
- G. D. Yadav and A. R. Yadav, Microporous Mesoporous Mater., 2014, 195, 180–190 CrossRef CAS.
- S. Giri, D. Ghosh and C. K. Das, Adv. Funct. Mater., 2014, 24, 1312–1324 CrossRef CAS.
- H. Xu, S. Yuan, Z. Wang, Y. Zhao, J. Fang and L. Shi, RSC Adv., 2014, 4, 8472–8480 RSC.
- H. Yao, D. Jia and H. Zhang, Ceram. Int., 2015, 41, 1531–1534 CrossRef CAS.
- S. A. Steiner III, T. F. Baumann, B. C. Bayer, R. Blume, M. A. Worsley, W. J. MoberlyChan, E. L. Shaw, R. Schlögl, A. J. Hart, S. Hofmann and B. L. Wardle, J. Am. Chem. Soc., 2009, 131, 12144–12154 CrossRef PubMed.
- H. Xu, S. Yuan, Z. Wang, Y. Zhao, J. Fang and L. Shi, RSC Adv., 2014, 4, 8472–8480 RSC.
- D. Guo, Y. Lu, Y. Zhao and X. Zhang, RSC Adv., 2015, 5, 11738–11744 RSC.
- C. Fu, G. Zhao, H. Zhang and S. Li, Int. J. Electrochem. Sci., 2013, 8, 6269–6280 CAS.
- J. Kang, A. Hirata, H. J. Qiu, L. Chen, X. Ge, T. Fujita and M. Chen, Adv. Mater., 2014, 26, 269–272 CrossRef CAS PubMed.
- A. Ramanathan, M. C. Castro Villalobos, C. Kwakernaak, S. Telalovic and U. Hanefeld, Chem.–Eur. J., 2008, 14, 961–972 CrossRef CAS PubMed.
- F. Su and Y. Guo, Green Chem., 2014, 16, 2934–2957 RSC.
- M. Sasidharan and A. Bhaumik, ACS Appl. Mater. Interfaces, 2013, 5, 2618–2625 CAS.
- M. G. Mazzotta, D. Gupta, B. Saha, A. K. Patra, A. Bhaumik and M. M. Abu-Omar, ChemSusChem, 2014, 7, 2342–2350 CrossRef CAS PubMed.
- M. I. Alam, S. De, B. Singh, B. Saha and M. M. Abu-Omar, Appl. Catal., A, 2014, 486, 42–48 CrossRef CAS.
- V. V. Ordomsky, J. van der Schaaf, J. C. Schouten and T. A. Nijhuis, ChemSusChem, 2012, 5, 1812–1819 CrossRef CAS PubMed.
- T. D. Swift, H. Nguyen, Z. Erdman, J. S. Kruger, V. Nikolakis and D. G. Vlachos, J. Catal., 2016, 333, 149–161 CrossRef CAS.
- X. Zhang, B. B. Hewetson and N. S. Mosier, Energy Fuels, 2015, 29, 2387–2393 CrossRef CAS.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed.
- N. A. S. Ramli and N. A. S. Amin, Energy Convers. Manage., 2015, 95, 10–19 CrossRef CAS.
- M. Wolfrom, R. Schuetz and L. F. Cavalieri, J. Am. Chem. Soc., 1948, 70, 514–517 CrossRef CAS PubMed.
- S. Dutta, S. De, M. I. Alam, M. M. Abu-Omar and B. Saha, J. Catal., 2012, 288, 8–15 CrossRef CAS.
- G. Marcotullio and W. De Jong, Carbohydr. Res., 2011, 346, 1291–1293 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra18513d |
| This journal is © The Royal Society of Chemistry 2016 |