
Microstructure investigation, optical properties and magnetic phase transition of Tm3+ substituted nanocrystalline ZnO (Zn0.95Tm0.05O)
Abstract
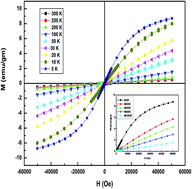
The magnetic phase transition, optical and dielectric properties of nanocrystalline Tm3+-doped zinc oxide (Zn0.95Tm0.05O), prepared by the co-precipitation method, were determined for the first time. The oxygen vacancy of the as prepared sample was enhanced by annealing at 200 °C for 6 h in an argon atmosphere. The formation of defects/oxygen vacancy is reflected in the Raman, FTIR and PL spectral analyses of the doped sample. Paramagnetic to ferromagnetic phase transition of Zn0.95Tm0.05O was observed at and below ∼30 K. The onset of ordering is successfully explained by the vacancy assisted bound-magnetic-polaron (BMP) model. The results of dielectric measurements suggest that the dielectric response of Zn0.95Tm0.05O is significantly enhanced, compared to that of pristine ZnO, and this is due to the presence of large amounts of oxygen vacancies, and the nanostructure of the sample.
 Please wait while we load your content...
Please wait while we load your content...

