
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of gas atmospheres and ceria on the stability of nanoporous gold studied by environmental electron microscopy and in situ ptychography†
Sina Baiera,
Arne Wittstockb,
Christian D. Damsgaardcd,
Ana Diaze,
Juliane Reinhardtf,
Federico Benzia,
Junjie Shib,
Torsten Scherergh,
Di Wanggh,
Christian Kübelgh,
Christian G. Schroerfi and
Jan-Dierk Grunwaldt*aj
aInstitute for Chemical Technology and Polymer Chemistry, Karlsruhe Institute of Technology, 76131 Karlsruhe, Germany. E-mail: grunwaldt@kit.edu; Fax: +49-721-608-44820; Tel: +49-721-608-42120
bInstitute of Applied and Physical Chemistry, University of Bremen, 28359 Bremen, Germany
cCenter for Electron Nanoscopy, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark
dCenter for Individual Nanoparticle Functionality, Department of Physics, Technical University of Denmark, 2800 Kgs. Lyngby, Denmark
ePaul Scherrer Institut, 5232 Villigen PSI, Switzerland
fDeutsches Elektronen-Synchrotron DESY, Notkestr. 85, 22607 Hamburg, Germany
gInstitute of Nanotechnology, Karlsruhe Institute of Technology, 76021 Karlsruhe, Germany
hKarlsruhe Nano Micro Facility (KNMF), Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, Germany
iDepartment Physik, Universität Hamburg, Luruper Chaussee 149, 22761 Hamburg, Germany
jInstitute of Catalysis Research and Technology, Karlsruhe Institute of Technology, 76344 Eggenstein-Leopoldshafen, Germany
First published on 22nd August 2016
Abstract
A novel complementary approach of electron microscopy/environmental TEM and in situ hard X-ray ptychography was used to study the thermally induced coarsening of nanoporous gold under different atmospheres, pressures and after ceria deposition. The temperature applied during ptychographic imaging was determined by IR thermography. While using elevated temperatures (room temperature – 400 °C) and realistic gas atmospheres (1 bar) we achieved for the first time a spatial resolution of about 20 nm during hard X-ray ptychography. The annealing of pure and ceria stabilized nanoporous gold in different atmospheres revealed that the conditions have a tremendous influence on the coarsening. The porous structure of the samples was stable up to approximately 800 °C in vacuum, whereas pronounced changes and coarsening were observed already at approximately 300 °C in oxygen containing atmospheres. A layer of ceria on the nanoporous gold led to an improvement of the stability, but did not alleviate the influence of the gas atmosphere. Different behaviors were observed, such as coarsening and even material loss or migration. The results suggest that additional mechanisms beyond surface diffusion need to be considered and that microscopic studies aimed at more realistic conditions are important to understand the behavior of such materials and catalysts.
1. Introduction
During the last few decades, nanostructures such as nanoporous gold (np-Au) have attracted a lot of attention in various applications, including catalysis,1–4 sensing4–6 or as a biomaterial interface.7 One of the most established methods for the fabrication of np-Au is the dealloying of a gold–silver alloy in nitric acid.8 During dealloying, a three-dimensional sponge-like structure is formed, which exhibits ligaments and pores sized in the range of a few tens of nanometers (mostly about 30 nm). The dimensions of this porous network are important for applications in catalysis, as the amount of surface defects such as kinks and steps influences the reactivity, and in turn depends on the size of the ligaments.3,4 The latter can be controlled either by varying parameters of the dealloying process directly,9 or via post-synthetic treatment methods like annealing.10–12 During annealing, usually a coarsening sets in, which leads to both an increased pore size and an increased ligament size.10,13–15 Notably the coarsening is known to be dependent on the annealing atmospheres,10,16 or on the heating power applied e.g. during laser induced heating.11,13Up to now, the coarsening effects of np-Au have mostly been studied by electron microscopy, following ex situ treatment in various atmospheres.10,11,14,17,18 According to these studies, the observed coarsening is almost exclusively explained by surface diffusion of gold,10,14,18,19 which is driven by minimization of surface energy. Besides these processes, Kolluri and Demkowicz20 discussed the effect of volume diffusion, which plays a role during the collapse of neighboring ligaments, especially in the early state of coarsening during corrosion. For nanoporous palladium, similar mechanisms have been discussed by Klein et al.21
Stabilization of the np-Au structures at temperatures of several hundred degrees was achieved by the deposition of metal oxides such as TiO2 or CeO2. These modified structures can be used as catalysts during catalytic processes like the water-gas shift reaction even up to 500 °C.2,22,23 However, also in this case a clear understanding of the underlying mechanisms is missing due to the lack of in situ studies. In addition, although gold is often considered as inert metal, a stabilization has been further attributed to surface adsorbates.16 Recently, Kuwano-Nakatani et al.24 found different coarsening behavior in vacuum or argon, compared to nitrogen or oxygen atmosphere. From different activation energies, it was concluded that different coarsening mechanisms dominate depending on the atmosphere. Lattice diffusion of gold was suggested as an alternative mechanism in this case. Furthermore, both Kuwano-Nakatani et al.24 and Sun et al.18 did not observe any strong coarsening in vacuum. Kuwano-Nakatani et al.24 did not detect any coarsening behavior at 400 °C in argon either, whereas Chen et al.10 reported a pronounced coarsening at 600 °C. Moreover, a stabilization of the structures could be reached by the latter in CO atmosphere and Biener et al.16 reported a stabilization by treatment with ozone compared to annealing in helium.
The discussion on the dependence on the atmosphere, which points to a strong influence of adsorbates on the coarsening mechanism, shows that further studies are required to understand the underlying mechanisms, as not all observations can be explained by the generally proposed surface diffusion mechanism. This is partly a consequence of the only recent availability of techniques which enable studies of the annealing behavior of np-Au in situ. These are required to understand the coarsening mechanisms, especially at higher temperature in the aforementioned atmospheres. Higher temperatures require special equipment such as high-temperature tolerant Transmission Electron Microscopy (TEM) grids or special TEM heating chips.25,26 Furthermore, to realize in situ studies with high spatial resolution under ambient pressure, (hard) X-ray microscopy can be advantageous.27 Scanning coherent X-ray diffraction imaging, also known as X-ray ptychography, is particularly suited for this purpose.28,29 Hard X-ray ptychography under ex situ conditions offers a spatial resolution below 10 nm (ref. 30 and 31) in 2D, while a resolution around 40 nm has been demonstrated under in situ conditions at atmospheric pressure and elevated temperature.32 Hard X-ray ptychography offers the possibility to track sample changes inside a reaction chamber32,33 under realistic pressures not only for thin samples (<100 nm), but also relatively thick samples.
In this study, the annealing behavior of 100 nm thick np-Au was studied at temperatures up to 800 °C by complementary in situ (E)TEM and hard X-ray ptychography. Samples were analyzed under different atmospheres, pressures and with/without the presence of ceria coating. In particular, an improved in situ setup to perform X-ray ptychography with a spatial resolution of about 20 nm and in combination with IR thermography for temperature determination is presented as an integral component of these studies. Before and after the annealing treatment, electron microscopy was performed at the same position on each sample, to gain a detailed and complementary insight into various effects occurring during the annealing process of 100 nm thick np-Au and CeO2/np-Au samples.
2. Experimental
Different samples were studied using a variety of microscopic methods. The different steps involved in sample preparation are summarized in Fig. 1. Detailed information about each step is given in the following paragraphs.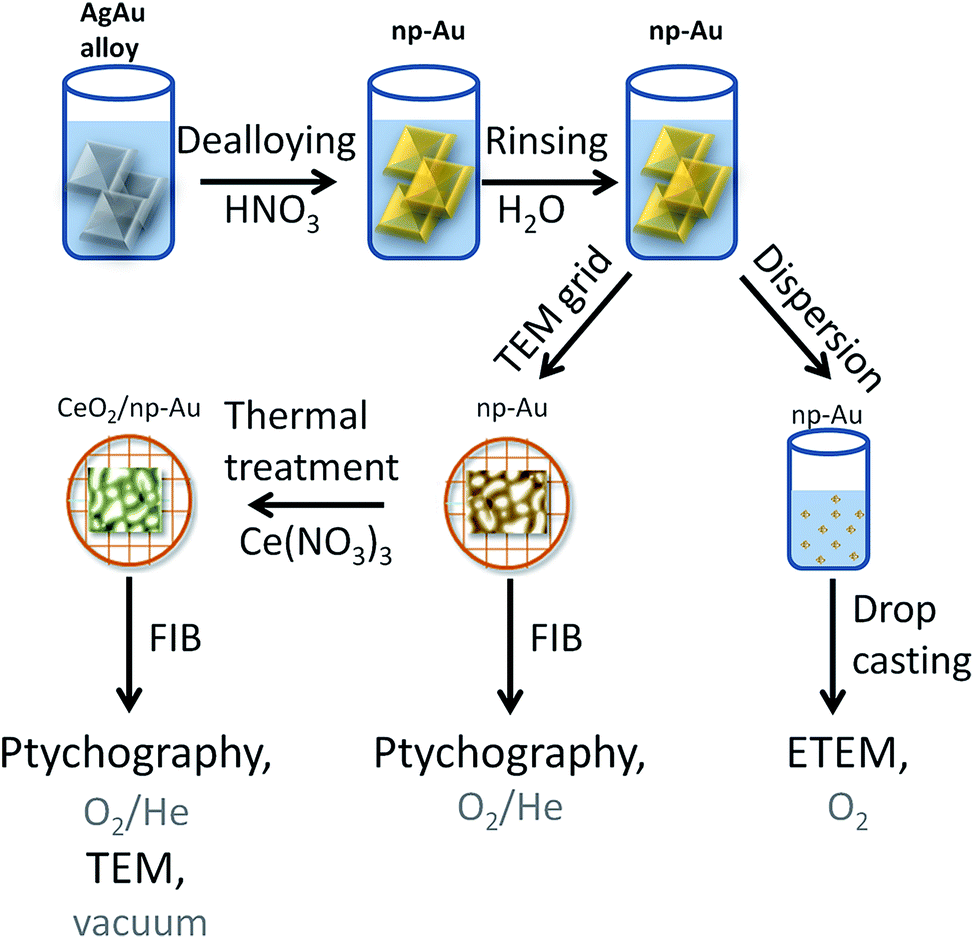 | ||
| Fig. 1 Schematic representation of the processes involved for the sample preparation. | ||
2.1 Sample preparation
Approximately 100 nm thick np-Au films were prepared by dealloying of AgAu alloy leaves (American White Gold, Au 35.4 at% and Ag 64.6 at%, 12 Karat, Noris Blattgold) using concentrated nitric acid (Sigma Aldrich, puriss p.a., ≥65 wt%). During corrosion the films were floating on the surface of the acid due to surface tension. For rinsing, the floating films were transferred onto deionized water for 30 min by lifting them onto a substrate such as a glass slide. Finally they were either transferred onto a glass layer or onto a copper TEM grid (100 mesh). The samples on the glass substrate were dried and subsequently dispersed in ethanol by carefully brushing, i.e., removal of small pieces from the substrate. The sample on the TEM grid was further processed by Focused Ion Beam (FIB) milling as described below. The CeO2/np-Au was prepared by impregnation of the np-Au on the copper TEM grid with 10 μL of an ethanolic solution of cerium nitrate (Ce(NO3)3, 1 mol L−1). After drying in air for at least 30 min, the sample was calcined at 250 °C for two hours yielding the oxide (for details about the preparation see ref. 23).2.2 Sample preparation for in situ ptychography
The as prepared samples on the copper TEM grids were transferred to a FIB microscope. Hexagonal shaped gold frames of approximately L × W × H = 25 μm × 15 μm × 500 nm for pure np-Au and quadratic gold frames L × W × H = 13 μm × 13 μm × 500 nm for CeO2/np-Au were used to attach the sample. The frame was fixed on the np-Au based samples by deposition of platinum on the outside of the frame. Afterwards, the sample was milled out and fixed on a Protochips E-Chip™25,34 by platinum deposition. In this way, one np-Au and one CeO2/np-Au sample were deposited on the same E-Chip™. FIB milling was performed using a FEI Strata dual beam FIB system (Karlsruhe Nano Micro Facility (KNMF) at the Institute of Nanotechnology). In parallel, another set of np-Au samples was prepared by drop casting of an ethanolic dispersion of np-Au on either a copper TEM grid with lacey carbon film or on a Protochips E-Chip™.2.3 Electron microscopy
Annealing studies were performed in conventional TEM via heating the sample using either (a) a Gatan 652 Inconel® heating holder (Gatan, Pleasanton) for the np-Au sample on the TEM grid, or (b) by the Protochips Aduro heating holder (Protochips, Morrisville). TEM measurements were performed on three different instruments: (a) Tecnai T20 G2 operating at 200 kV and in Bright Field (BF) mode for the drop casted pure np-Au sample on a lacey carbon film coated copper grid. (b) FEI Titan 80–300 image aberration corrected electron microscope operated at 300 kV, acquiring Scanning Transmission Electron Microscopy (STEM) images by a Fischione model 3000 High Angle Annular Dark Field (HAADF) STEM detector, using the Protochips heating holder for annealing studies. This instrument was used for the FIB prepared CeO2/np-Au sample on an E-Chip™ and the STEM analysis of the samples treated by in situ ptychography, as well as for Energy Electron Loss Spectroscopy (EELS) on those samples. EELS analysis was performed at 200 °C to avoid any contamination on the sample using a Gatan Tridiem imaging filter. (c) FEI Titan E-Cell 80–300 aberration corrected electron microscope operated at 300 kV with a pressure of 3.2 mbar O2, acquiring images in STEM and BF mode using the Protochips heating holder (drop casted pure np-Au sample on an E-Chip™).2.4 X-ray ptychography
For in situ X-ray ptychography, a dedicated in situ cell32 was used which enables heating in a controlled gas atmosphere. A flow of 3 mL min−1 of 20% O2/He was used. Measurements were performed at the cSAXS beamline of the Swiss Light Source (SLS) at the Paul Scherrer Institute (PSI, Villigen, Switzerland) using a photon energy of 5.72 keV. The beam was defined using a coherently illuminated Fresnel zone plate (FZP) made of Au35 with a diameter of 170 μm and an outer-most zone width of 60 nm, corresponding a focal length of 47.03 mm at this energy. The FZP provided an estimated flux of 2.7 × 108 photons per s. The samples were placed at a distance of 350 μm downstream the focus, such that the illumination on the sample had a diameter of about 1.3 μm. Ptychographic scans were recorded by scanning the sample over a field of view of 2 × 2 μm2 in a grid of about 400 points in a Fermat spiral pattern,36 with an average step size of 100 nm. At each scanning position diffraction patterns of 0.2 s exposure time were recorded with a Pilatus 2M detector with 172 μm pixel size37 placed 7.255 m downstream from the specimen. The total duration of a scan was about 3 minutes and 35 s, taking into account overhead time between acquisitions. The scans were repeated at two detector positions in order to record intensities at missing regions of the detector due to module gaps. Such pairs of scans were repeated at 4 different positions covering a total field of view of 3.5 × 3.5 μm2 with an overlapping region of 0.5 μm in between. For each pair of scans, an estimated flux density of 8.6 × 109 photons per μm2 was irradiated on the samples, from which the surface dose deposited on the specimen can be estimated as described by Howells et al.38 Assuming a relative density of 30% for the np-Au samples39 (5.8 g cm−3), an estimated dose of about 3.8 × 108 Gy was applied to the specimen.In order to study samples at different temperatures, the samples were heated using the resistive heating possibility of the E-Chip™ by applying a heating power. Different temperature steps were set and the sample was kept at each temperature for ∼90 minutes, in which the temperature was determined and ptychographic datasets were acquired.
The ptychographic image reconstructions were performed using 1000 iterations of the difference map algorithm40 followed by 100 iterations of a maximum likelihood optimization used as a refinement step.41 All 8 scans recorded for each sample and for each temperature step were fed simultaneously in a ptychographic reconstruction, in which a single object was reconstructed while refining a different probe for each scan. The idea of sharing the object in ptychographic reconstructions is described by Dierolf.42 It has been introduced by Guizar-Sicairos et al.43 to account for changes in the illumination probe when scanning large field of views, while benefiting from the robustness of reconstructing the entire region of interest in the same ptychographic reconstruction. All reconstructions were applied using a region of the detector of 600 × 600 pixels containing the diffraction patterns, which corresponded to a reconstructed pixel size in the images of 15.2 nm. The resolution was estimated from a dataset on a CeO2/np-Au sample acquired at room temperature. For this purpose, reconstructions were performed from 2 pairs of scans acquired at two different positions, reconstructing for each pair a common object with two different illumination probes. The Fourier shell correlation of the two objects at the overlapping area, which had a total extension of 0.5 × 2 μm2, was then computed and compared with a threshold corresponding to the 1 bit criterion,44 obtaining an estimated resolution of 20 nm.
Reconstructed images were post-processed to remove a phase ramp and an offset which are intrinsic degrees of freedom in ptychographic reconstructions.45 Methods developed previously for the phase ramp removal could not be applied because of the very small areas of air which are typically used as a reference for constant phase.45 Instead an approach was used in which the ramp was removed visually by checking different ramps in steps of 0.02, which corresponds to an error between pixels at both extremes of the image of 0.02 × 2π = 0.12 rad. For the phase offset correction, the largest area without sample in the images was used and an error below 0.12 rad was estimated.
2.5 Infrared thermography
Infrared (IR) thermography was performed to determine the temperature during in situ ptychography using an ImageIR® 8300 camera (InfraTec, Dresden) equipped with a macro objective M = 1.0× with a field of view of 9.6 mm × 7.7 mm and a pixel size of 15 μm. Furthermore, intermediate rings were used to further improve the spatial resolution to approximately 5 μm. For the determination of the sample temperature it is assumed that the sample was integrally attached to the Si3N4 membrane, for which an emissivity of 0.9 was assumed.463. Results and discussion
3.1 Combination of in situ X-ray microscopy using ptychography and IR thermography
In order to demonstrate the improved capabilities of the hard X-ray ptychography cell, which was presented in detail previously,32 results on both the temperature measurement and in situ ptychography are reported. As the temperature of the sample is very important during the measurements, IR thermography was used directly during annealing treatment to determine the temperature immediately prior to the acquisition of in situ ptychography datasets. A typical IR thermography image with a spatial resolution better than 10 μm is shown in Fig. 2, with the sample compartments located in the middle. For determining the temperature during in situ ptychographic measurements, which required a measurement through the window made of Kapton® foil, the temperature was calibrated by measuring empty chips beforehand. In this way the influence of the temperature determination through the 25 μm thick Kapton® foil could successfully be estimated. Fig. 2 (left) shows the calibration measurement from the front of the cell with and without Kapton® foil, compared to the measurement at the back, with the sample deposited on the silicon nitride membrane of the E-Chip™ used in the cell. A temperature difference of approximately 10% with and without the foil was detected, as shown in Fig. 2. Hence, during in situ ptychography, the temperature on the sample estimated by the IR camera behind the Kapton® foil was corrected by an extra 10% of the measured temperature. The thermogram in Fig. 2, which corresponds to the sample presented in Fig. 3 at 250 °C, shows that the sample seems to be at slightly higher temperature. As the emissivity chosen for temperature determination was set to the emissivity of the Si3N4 membrane, the higher temperature is probably due to the different emissivity of the sample and the membrane.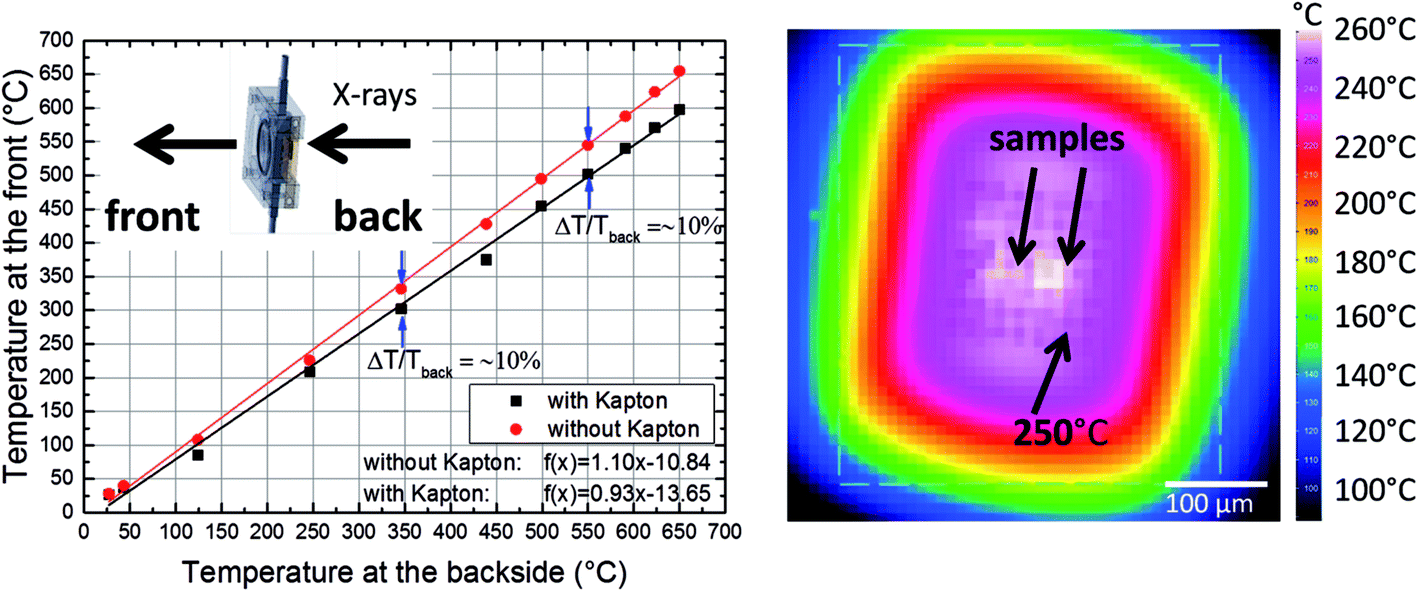 | ||
| Fig. 2 Left: IR thermography measurement with (through) and without Kapton® foil in comparison to the temperature determined at the backside of the chip. Right: IR thermography measurement of the E-Chip™ in the in situ ptychography cell at 250 °C (arrow pointing to the Si3N4 membrane) through the Kapton® foil. | ||
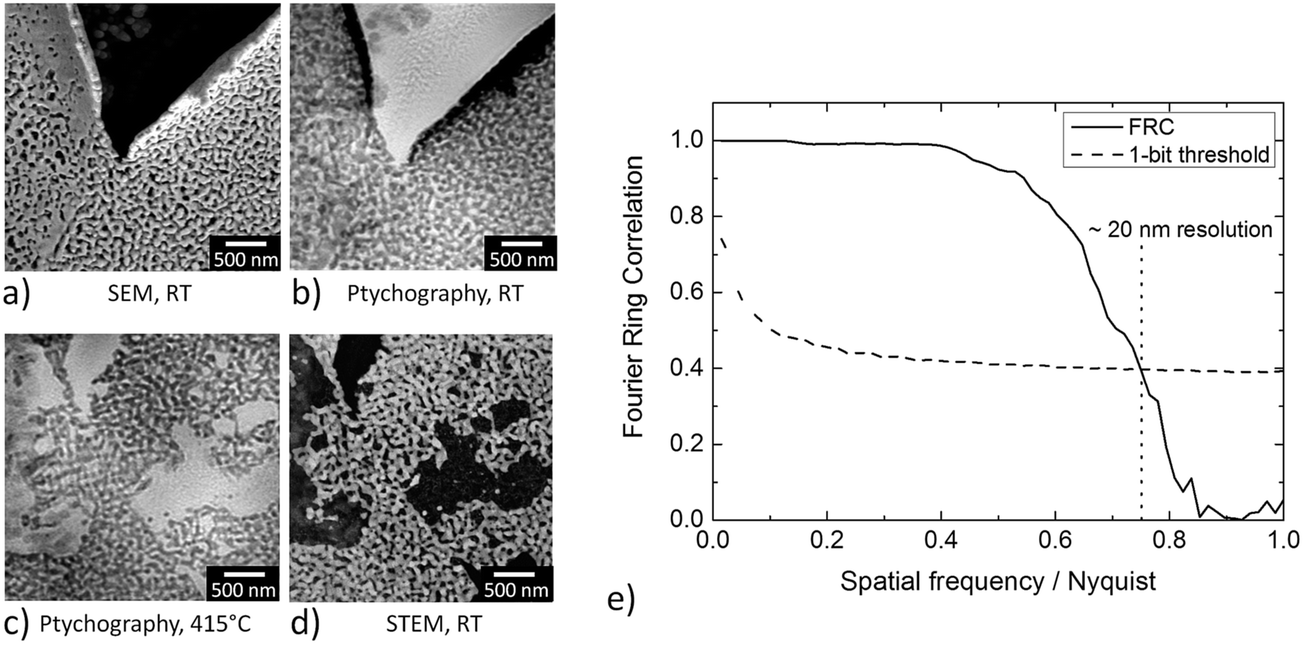 | ||
| Fig. 3 (a) SEM image of the CeO2/np-Au sample studied at room temperature under vacuum conditions before the annealing treatment, (b) ptychographic image of the same sample acquired at RT and atmospheric pressure and (c) at 415 °C, (d) STEM image under vacuum conditions at room temperature acquired after the annealing treatment of the CeO2/np-Au sample. (c) and (d) show slightly different areas of the sample. (e) Estimation of the image resolution in (b) by Fourier Ring Correlation (FRC). | ||
Fig. 3a–d shows some examples of ptychographic imaging compared to electron microscopy. SEM images of the as-prepared CeO2/np-Au sample acquired at RT are in good agreement with ptychographic measurements acquired immediately before the annealing experiment, as shown in Fig. 3a and b. Furthermore the STEM image in Fig. 3d acquired after the annealing experiment compares well with the ptychographic image acquired during the last annealing step at 415 °C, shown in Fig. 3c. Fig. 3e shows a Fourier Ring Correlation (FRC) analysis which was performed on the image in Fig. 3b to derive the spatial resolution44 (see Section 2). The intersection of the FRC with the 1 bit threshold curve indicates a spatial resolution of 20 nm, which is significantly better than in previous studies.32,33
In this study, the temperature measurement was performed directly before the ptychographic one. A measurement during ptychographic data acquisition was not conducted due to geometric constraints at the beamline. However, using the same conditions applied during in situ ptychography enabled a precise knowledge of the temperature during the image acquisition. Applying IR thermography with a spatial resolution better than 10 μm offers a distinct advantage over classic temperature sensors, which are generally only capable of determining the average temperature of the heated area.11,24 Using IR thermography, imaging of the heated area can also account for temperature inhomogeneities on the microscale, which can occur when catalysts are studied during reactions.47,48 In contrast to the use of pre-calibrated heating chips in vacuum,25 the direct determination of the temperature allows its use in a gas atmosphere as well, where the calibration performed in vacuum would no longer be valid. The good agreement of electron microscopy and the high spatial resolution derived by in situ ptychography clearly underlines the potential of the latter, namely in bridging the gap between high resolution imaging under model conditions in electron microscopy and realistic conditions.
3.2 Annealing of np-Au under different atmospheres and pressures
To study the annealing of np-Au at elevated temperatures under vacuum conditions, in situ heating was performed during TEM from room temperature (RT) up to 800 °C. A sample suspended in ethanol was prepared on a copper TEM grid by drop-casting and heated to the desired temperature in 100 °C intervals. Overview bright field TEM images of the sample at different temperatures (RT to 800 °C) are presented in Fig. 4. Due to the thickness of the sample only the corner region was electron transparent. However at 300 °C, a few changes including contraction of material (marked by a red arrow in Fig. 4), were visible in the top region. Hardly any differences could be observed until 600 °C, while at 700 °C the first changes became visible in the thicker section of material as well, possibly indicating a loss or migration of material. At 700 °C, this effect was visible in a single area (indicated by the red arrow), whereas it continued steadily during temperature increase to 800 °C. Where the material was no longer visible, some residues with weak contrast may have been left behind and thick areas were formed at the same time at different positions, indicating a coarsening of the sample and material transfer. Similar observations were also made for areas not probed with the electron beam during annealing, which excludes a direct influence of the electron beam. EDX and EELS analysis of an area with a similar material loss, studied on a sample on a Si3N4 membrane, revealed the presence of Si, N and O, but excluded any trace of Au. | ||
| Fig. 4 BF TEM images during annealing of a pure np-Au flake drop casted on a copper TEM grid covered by lacey carbon film. Annealing studies were performed in vacuum. Only the corners of the flake were thin enough to be electron transparent. However, at a temperature of 700 °C, the first changes are seen in the dark area as well, which changed constantly at 800 °C. | ||
In Fig. 5, a zoom into the top region of the area studied in Fig. 4 is presented, which shows that only slight changes, like a contraction of the ligaments, can be observed at temperatures of 200 °C (marked by red arrows in Fig. 5).
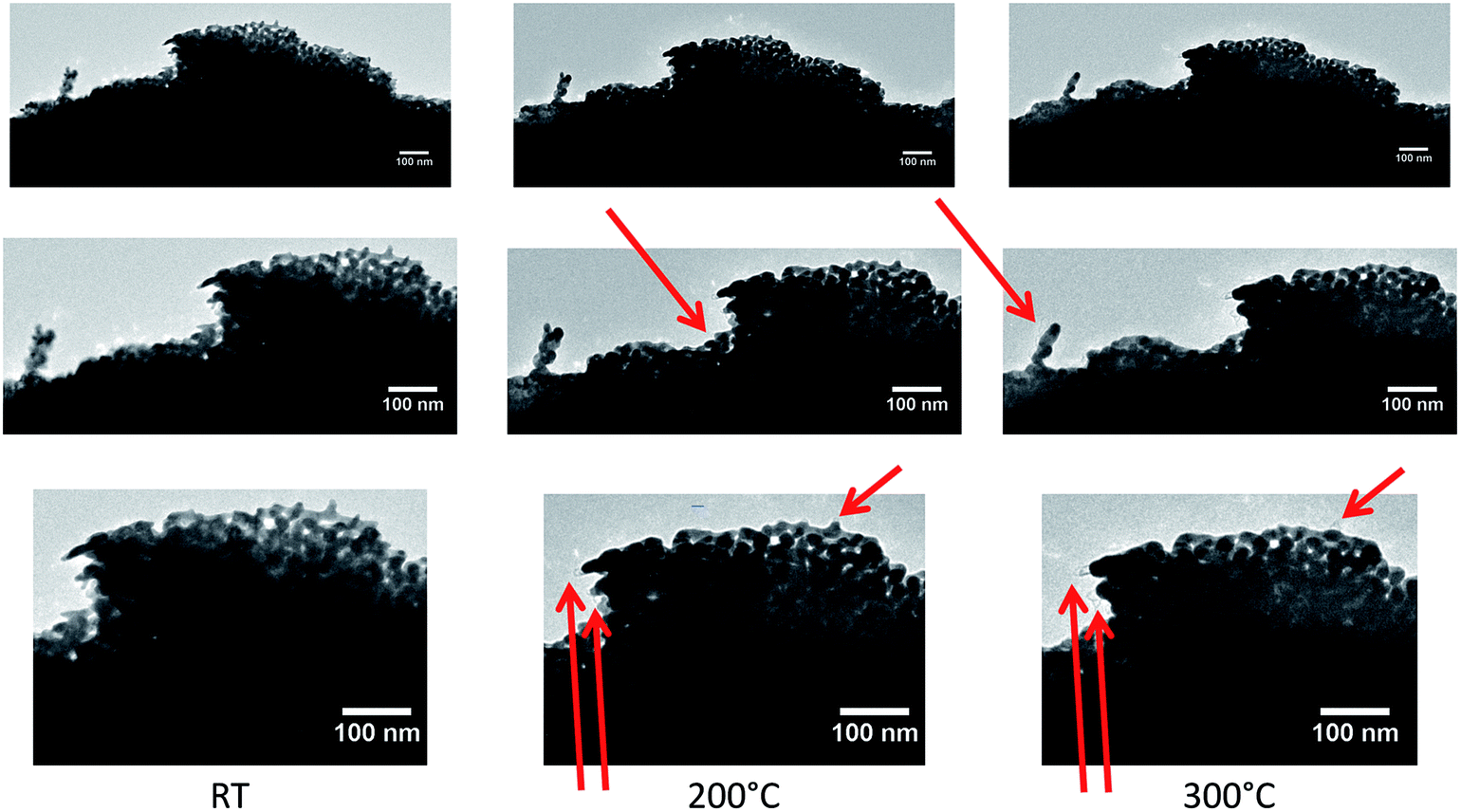 | ||
| Fig. 5 Close-up of top region in Fig. 4 showing the porous structure at the corner of the studied flake at different temperatures. From left to right, TEM images are shown at RT, 200 °C and 300 °C, respectively, with increasing magnifications (top to bottom). Already at relatively low temperatures, slight changes of the sample occurred. At 100 °C no changes were observed with respect to the measurement at RT. | ||
In the next step in situ annealing was carried out in the ETEM on a np-Au sample prepared by drop casting of dispersed flakes on a Protochips E-Chip™ in an atmosphere of 3.2 mbar of O2, in order to study the influence of an oxygen containing atmosphere. Selected images are presented in Fig. 6. By drop casting, the flakes were not placed on the thin Si3N4 windows of the E-Chip™, but were located on thicker parts of the SiC membrane (∼100–200 nm). This results in a higher background signal for the TEM images compared to the approximately 50 nm thick Si3N4 membrane. Some flakes deposited on the thicker part of the heated membrane were studied during annealing, but in comparison to the flake studied in vacuum, they were smaller and showed bigger pores and ligaments. Similar to previous measurements in vacuum, the sample was heated in 100 °C intervals and no changes could be observed up to 200 °C. Between 200 °C and 300 °C changes were detected in the sample, which can be both seen in the overview on the left in HAADF-STEM images, as well as in the image showing the top right flake in higher magnification, which is depicted as BF TEM images on the right. Both the overview and the magnified image show that the flakes start to coarsen at 300 °C, implying the collapse of some ligaments (indicated by the red arrows) and an increase in thickness. The latter becomes especially visible in the HAADF-STEM images, as the transparency of the material decreased and the contrast changed (thicker areas got darker), which can be explained by a sintering behavior. In contrast to measurements in vacuum, no material loss was observed (also at higher temperatures up to 900 °C, not shown here), but the sample showed coarsening under oxygen as also reported under air in the literature.10–14,17,49
 | ||
| Fig. 6 ETEM study of the in situ annealing of np-Au drop casted on an E-Chip™. Annealing treatment was performed at different temperatures steps in an atmosphere of 3.2 mbar O2. Both HAADF-STEM images and BF TEM images were obtained for this sample. | ||
In addition to the measurements performed under model conditions, i.e. the reduced pressure in TEM, complementary measurements were performed during in situ ptychography to examine the influence of pressure and oxygen at ambient conditions. Phase contrast images from the ptychographic measurements at a constant energy of 5.72 keV are presented in Fig. 7. The morphological changes with respect to the previous image at lower temperature are marked with red arrows. Despite attempting to monitor the same area of interest during the annealing treatment, a slight shift in sample position is visible especially from RT to 275 °C, which is due to sample drift with increasing temperature. The positions were manually corrected between measurements to overcome this issue and the same positions were marked by blue circles whenever they were not obvious. At a temperature of around 300 °C, the first changes were observed, which showed slight loss or migration of material similar to that observed during heating in vacuum. With increasing temperature, the loss of material continued but with no detectable coarsening behavior. This is also supported by quantitative comparison of the ligament diameter before and after the annealing treatment (see ESI, Fig. S1†). For comparison of the local studies performed by electron microscopy with those performed during in situ ptychography, a smaller field of view of comparable size from the ptychographic reconstructions is also presented (see ESI, Fig. S2†).
 | ||
| Fig. 7 Phase contrast images of np-Au at different temperatures performed in the in situ cell for X-ray ptychography at the cSAXS beamline at SLS using a constant energy of 5.72 keV and a flow of 3 mL min−1 of 20% O2/He. Changes are marked by red arrows, whereas the same sample position is marked by a blue circle. | ||
To gain quantitative information on the material loss, the normalized grayscale values which correspond to the phase shift of X-rays after propagation through the sample, have been estimated between 275 °C and 355 °C (Fig. 8). As the phase shift depends on the thickness of the material, the change in grayscale values can be related to a change in thickness. In Fig. 8a, the area where the material loss was studied is marked by a red box. The area in the center of the box became slightly brighter with increasing temperature, in agreement with the higher grayscale value. For quantitative analysis, the average grayscale value measured in a 200 × 200 nm area located in the center of the red box, was divided by the average grayscale value within the non-changing area marked by the blue box. Fig. 8b shows a steady increase in relative mean grayscale value with increasing temperature. However, this is only true assuming that changes in the material occur in the area used for calibration and should therefore be taken as an additional information. No absolute calibration can be performed due to the application of the manual phase ramp correction, (compare Section 2).
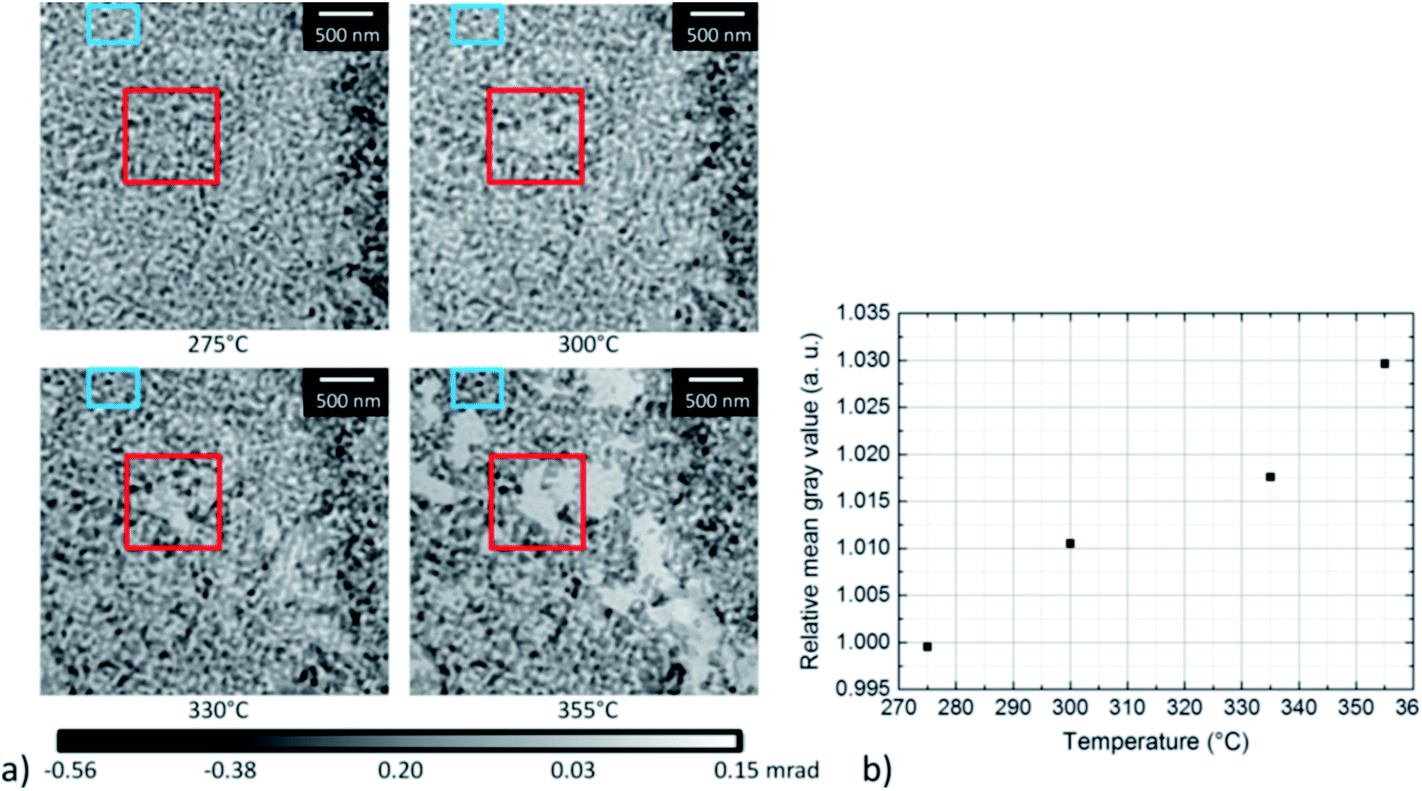 | ||
| Fig. 8 (a) Selected phase contrast images of np-Au at different temperatures during annealing treatment, showing a material loss. The area where the loss starts to be visible is marked by a red box, whereas a non-changing area is marked by a blue box. (b) Plot of the relative mean grayscale values with respect to temperature. The grayscale values were normalized by correcting the grayscale values measured in the center of the red box by the grayscale values measured in the blue box. | ||
The results presented for the annealing of np-Au in different atmospheres show that, during annealing of np-Au in vacuum and in 20% O2/He, a material loss was observed, in contrast to measurements in 20% O2/N2 (ref. 32) or pure O2 in the mbar regime. Taking the high X-ray dose for the measurements performed in 20% O2/He into account, the differences might be explained by beam damage. However, the loss was also observed in areas which were not probed by the X-ray beam, as determined by TEM after the ptychography treatment (compare Section 3.3). Nevertheless, systematic investigations of the influence of the X-ray dose should be performed in future. In contrast to previous studies,18,24 it could be shown that np-Au samples did not start to change in vacuum at temperatures below 700 °C. At those high temperatures, a simultaneous material loss and sintering were observed. Interestingly, the loss observed during in situ ptychography in 20% O2/He at ambient pressure started at much lower temperature than under vacuum conditions, but at a comparable temperature to the onset of coarsening during ETEM analysis in oxygen atmosphere. These new findings indicate that the atmosphere strongly influences the coarsening and its underlying mechanisms. Furthermore, it shows that measurements at ambient pressure are possible if techniques like in situ ptychography are applied. Such measurements are considered necessary to accurately study processes occurring under realistic (non-model) conditions.
3.3 Stabilization of np-Au by supported CeO2 – annealing of CeO2/np-Au under different atmospheres and pressures
Next, the influence of deposition of a CeO2 layer on the np-Au surface was studied. Often such an additional layer has a stabilization effect.50,51 Hence, CeO2/np-Au samples were studied under vacuum conditions during in situ annealing in TEM and at atmospheric pressure during in situ ptychography in 20% O2/He to compare the behavior under model and realistic conditions. The corresponding samples were prepared by FIB micromanipulation like the np-Au samples used for ptychography. Furthermore, the CeO2/np-Au sample studied during in situ ptychography was placed next to the np-Au to mimimize differences in temperature or atmosphere between the CeO2-stabilized and the pure np-Au.Ptychography was thus carried out following the same experimental conditions as for the pure np-Au and the phase contrast reconstructions are presented in Fig. 9. As before, structural changes with respect to the previous image at lower temperature are marked with red arrows. The reconstructions obtained at RT and at 275 °C show a different part of the sample due to a drift. Additionally, the crack in the center had a slightly different shape at temperatures above RT. In fact, this change already occurred before the measurement around 50 °C (not shown here) and might be explained by decreased stress due to enhanced temperature. Furthermore, for the measurement performed at RT, reconstruction artifacts are visible in the center of the crack, indicated by a noisy area in the center. A magnified part of the top right area from 300 °C to 385 °C is depicted in the ESI (Fig. S3†).
 | ||
| Fig. 9 Phase contrast images of CeO2/np-Au at different temperatures performed in the in situ cell for X-ray ptychography at the cSAXS beamline at SLS using a constant energy of 5.72 keV and a flow of 3 mL min−1 20% O2/He. Changes are marked by red arrows, whereas the same position is marked by a blue circle. | ||
Similar to the unmodified pure np-Au sample a material loss could be detected. Once the gold ligaments severed, a residue with weak contrast was observed in the STEM images. This residue can be attributed to CeO2 containing material for the stabilized sample, concluded from EELS analysis shown (ESI Fig. S4†). A coarsening could not be found (cf. Fig. S1b†). By comparing the onset temperature when the loss appeared, it becomes obvious that changes occur first on the pure np-Au sample around 300 °C (Fig. 7) whereas the first changes for the CeO2/np-Au sample started around 355 °C (Fig. 9). This indicates an enhanced thermal stability due to CeO2 by about 50 °C. Furthermore, it shows that the observed effect of material loss or migration occurs regardless of the presence of stabilizing oxide, although the process starts at a slightly higher temperature.
To study the annealing behavior of the stabilized sample under vacuum conditions, in situ heating was performed during TEM in a conventional microscope (vacuum conditions around the sample). A selection of STEM images from the annealing of CeO2/np-Au on a Protochips E-Chip™ at 840 °C are given in Fig. 10. Note that the bright area on the bottom left corner of Fig. 10a–d was not there before the annealing treatment, but might result from redeposition as the electron beam was kept in the lower left corner. Comparably to the previous experiment of the unstabilized sample in vacuum, a material loss could be observed. At lower temperatures, no changes could be seen. An indication for the stabilizing effect of the CeO2 in vacuum can be evaluated from the comparison of the onset of changes (e.g. material loss) in the unstabilized material versus the stabilized np-Au. For the pure np-Au sample, the onset started at 700 °C (see Fig. 4), but continued steadily at 800 °C. In contrast, the onset started at 840 °C for the CeO2/np-Au sample annealed in vacuum and continued at this temperature (see Fig. 10). The stabilization effect in vacuum is therefore comparable to the stabilizing effect in O2/He under atmospheric pressure, where the onset of material loss was shifted to about 50 °C higher temperatures.
 | ||
Fig. 10 STEM images on low resolution (top, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000× magnified) and higher resolution (bottom, 57000× magnified) at 840 °C in vacuum, showing the material loss. (a) Represents the sample after being heated at 840 °C for 11 minutes, (b) and (c) after 12 minutes, and (d) after 33 minutes at 840 °C. (e)–(h) were recorded within one minute directly after reaching 840 °C, indicating that material loss started immediately. Red arrows indicate examples of areas where changes occurred in comparison to the image before. Additionally, in (d) green boxes indicate the area for the zoomed in regions from Fig. 11. 000× magnified) and higher resolution (bottom, 57000× magnified) at 840 °C in vacuum, showing the material loss. (a) Represents the sample after being heated at 840 °C for 11 minutes, (b) and (c) after 12 minutes, and (d) after 33 minutes at 840 °C. (e)–(h) were recorded within one minute directly after reaching 840 °C, indicating that material loss started immediately. Red arrows indicate examples of areas where changes occurred in comparison to the image before. Additionally, in (d) green boxes indicate the area for the zoomed in regions from Fig. 11. | ||
Apart from material loss on larger length scale, changes were also observed on a smaller length scale. Examples of such changes are given in Fig. 11 and seem to result from contractions of the gold ligaments, which appear similar to the expected coarsening resulting from ligament pinch-off20,52 (Fig. 11a and b) or sintering (Fig. 11c and d) of the np-Au samples. The positions of the studied areas are marked with green boxes in Fig. 10d. Interestingly, the material loss started at one area and continued outgoing from the same area, as it becomes visible by comparing Fig. 10a–f. Fig. 10e indicates that not only very small ligaments pinch off, but also some neighboring ligaments sever, whereas others stay stable and break off later (Fig. 10h). Additionally, Fig. 10f–h shows (marked positions) that parts of the ligament can pinch off, while small single isolated particles, which would be expected to be unstable at elevated temperature, remain.
 | ||
| Fig. 11 Zoomed in region (indicated by green boxes in Fig. 10d) in the STEM images on the top row of Fig. 10. The images are acquired at different times and changes symbolized by a black arrow. (a) and (c) represent the structure derived after 11 minutes at 840 °C while the change to (b) occurred immediately after 33 minutes and to (d) after 25 minutes. | ||
In Fig. 12, a comparison between the SEM image of the CeO2/np-Au sample used for in situ ptychography before the annealing (Fig. 12a), and two STEM images after annealing (Fig. 12b and c) are shown. The depicted area shows a position which was not imaged by ptychography, indicating that potential interaction of the sample by the X-ray beam did not significantly influence the observed annealing behavior resulting in material loss. Comparing Fig. 12a and b reveals that at the marked position (red arrows) next to a cut prepared by Ga FIB milling, one part of the gold ligaments pinched off, while the other part was unchanged. However, some areas also changed their structure and the original ligaments cannot be recognized anymore, which might be explained by slight coarsening or restructuring. Additionally to the obvious material loss in the bottom right corner of Fig. 12b, the remaining ligaments seem to be of the same size (cf. Fig. S1b†), as far as conclusions can be drawn on the comparison of SEM and STEM images. In contrast, it seems as if the sample showed a contraction, as observed in Fig. 5 previously, which also corresponds to images shown by Kuwano-Nakatani et al.24 These contractions suggest that the gold tends to be mobile at the annealing temperature, but the ligament seems to be hindered from the usual coarsening route, which appears to be the case for the annealing in vacuum and in O2/He.
 | ||
| Fig. 12 (a) SEM image of the FIB prepared CeO2/np-Au sample before the annealing with red arrows as markers for comparison, (b) STEM-HAADF image of the prepared CeO2/np-Au sample after the annealing at the same area from (a), (c) STEM-HAADF image of the prepared CeO2/np-Au sample after the annealing at another area showing the contraction of the gold ligaments. | ||
The most intuitive way of describing coarsening, i.e., the displacement of atoms resulting in larger structures well below the melting point is by diffusion. While at high temperature diffusion can take place through the bulk, interfacial, as the case may be, surface diffusion is the dominant mechanism at lower temperatures.10,11,13,14,19,50 According to Peale and Cooper,53 the presence of chemisorbed gas molecules promotes the generation of gold adatoms and edge vacancies facilitating surface diffusion. In case of surface contaminations, i.e., strongly adsorbed molecules coarsening was found to be prevented or delayed.55,56 While most studies in the past performed the annealing experiments under gas atmosphere we observe a stabilization of the material when heating in vacuum, likely induced by a hindered formation of adatoms or vacancies at step edges of the surface. However, at high temperatures well above the hüttig temperature (∼550 °C), changes in the material can be observed also in vacuum (compare Fig. 4), indicating the onset of bulk diffusion.54
At even higher temperatures close to 800 °C in the TEM loss of the gold ligaments was observed. Reasons to consider are (a) surface tension driven viscous flow above the melting point,13 (b) sublimation of the material or (c) mechanical loss, e.g., by stress. The vapor pressure of gold at around 800 °C is about 10−8 mbar, rendering sublimation a viable explanation under vacuum conditions.57 However, for the in situ ptychography, material loss at temperatures (Treached = 375 °C) far away from the melting point, even for nanoparticulate materials,8 was observed, indicating that sublimation cannot be the only explanation for this behavior. From mechanical tests, it is known that np-Au undergoes a ductile–brittle transition due to microstructural changes58 and that stress enhancement is observed at defects.59 Additionally, in a narrow ligament-strength distribution a rupture of the weakest ligament can initiate a catastrophic failure of the material.60 While our experiments show that the mechanism for coarsening of the material strongly depends on the conditions of heating (gas atmosphere, vacuum), at this stage, the material loss in some of the experiments cannot be unequivocally attributed to one of the mechanisms detailed above.
Furthermore, it could be shown by the combination of ETEM and in situ ptychography that pressure differences between the mbar and 1 bar regime in the present study did not influence the onset of the changes due to annealing. Similar temperature ranges were observed for annealing in O2 in the mbar regime and in O2/He or O2/N2 (ref. 32) at ambient pressure. If the samples were annealed in vacuum, which leads to a different surface coverage, the onset of changes differed strongly compared to the measurements in oxygen containing atmosphere. While the temperature was measured locally by IR thermography during the in situ ptychography experiments, the temperature was controlled by a thermocouple in the TEM heating holder (Gatan Inconel® heating holder) or by external calibration (Protochips E-Chip™) during the TEM experiments. As the temperature ranges are similar for the stabilized and unstabilized sample in vacuum or in oxygen containing atmosphere, respectively, it can be assumed that inaccuracies in temperature determination could be minimized. Moreover, due to similar temperature ranges for the different sample preparation techniques, namely FIB micromanipulation versus drop casting, influences of FIB preparation and possible contamination introduced by Ga ion milling and Pt deposition can also be neglected. Therefore the different behavior of the samples studied at the various atmospheres is mainly attributed to those atmospheres. The results show that further studies on the coarsening behavior of np-Au have to be performed under realistic conditions to further unravel the underlying mechanisms.
4. Conclusions
Combined (E)TEM and in situ X-ray ptychography measurements were performed to study the thermal annealing of np-Au. This allows exploiting the advantage of more realistic reaction conditions in case of X-ray microscopy with the higher spatial resolution obtainable by (E)TEM. For the first time, the temperature applied during in situ ptychography was precisely determined using parallel IR thermography, which is crucial when varying the gas atmosphere. Strikingly, we obtained an unparalleled 20 nm resolution by in situ hard X-ray ptychography. This demonstrates the potential for studying other functional nanoscale materials during heating and gas treatment.The present study demonstrates that annealing of np-Au based samples is strongly dependent on the gas atmosphere: np-Au was found to be stable up to 700–800 °C in vacuum, but at higher temperatures a material loss occurred. In contrast, coarsening was observed in oxygen atmosphere around 200–300 °C in the mbar regime. Notably, a similar material loss to that observed in vacuum at ca. 800 °C was already found around 300 °C at ambient pressure in O2/He atmosphere. Furthermore, the present analysis points to the presence of additional coarsening mechanisms than just surface diffusion. This effect may be similar to the different growth mechanisms for metallic nanoparticles in classical catalysts, such as particle migration, coalescence or Ostwald ripening that also depend strongly on the support, contaminants/structural promoters and the gas atmosphere. Finally, it could be shown that np-Au and CeO2/np-Au show a comparable behavior in the different atmospheres, but stability was improved by the presence of CeO2, as indicated by the onset of changes shifted towards higher temperatures.
The necessity of complementary techniques, as presented by this work, shows the potential for the study of heterogeneous catalysts under real conditions, which can be furthered by i.e. three dimensional high-resolution structural information by ptychographic tomography,29,61,62 or by elemental and chemical contrast offered by X-ray absorption spectroscopy.63,64
Acknowledgements
We thank the “Stiftung der Deutschen Wirtschaft” (S. B.) and the China Scholarship Council (J. S.) for PhD grants. Moreover, we acknowledge the BMBF projects “X-ray microscopy” (05K10VK1/05K10OD1) and “Nanoscopy” (05K13VK2/05K13OD4), the virtual institute VI-403 “In situ Nano Imaging of Biological and Chemical Processes” and the Helmholtz Research Program “Science and Technology of Nanosystems” (STN) as well as the Danish National Research Foundation's Center for Individual Nanoparticle Functionality (DNRF54) for financial support. The A. P. Møller and Chastine Mc-kinney Møller Foundation is gratefully acknowledged for its contribution towards establishment of the Center for Electron Nanoscopy at the Technical University of Denmark. The authors acknowledge financial support from the European Commission under the Seventh Framework Program by means of the grant agreement for the Integrated Infrastructure Initiative N.262348 European Soft Matter Infrastructure (ESMI). In addition, we acknowledge the Karlsruhe Nano Micro Facility (KNMF), a Helmholtz Research Infrastructure at KIT, for the opportunity to use the FIB-SEM. Finally, we thank the cSAXS beamline team, Lorenz Bäni and Mirko Holler for their help during the beamtime.References
- T. Fujita, P. Guan, K. McKenna, X. Lang, A. Hirata, L. Zhang, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka, Y. Ishikawa, N. Asao, Y. Yamamoto, J. Erlebacher and M. Chen, Nat. Mater., 2012, 11, 775–780 CrossRef CAS PubMed.
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Perez, Science, 2007, 318, 1757–1760 CrossRef CAS PubMed.
- V. Zielasek, B. Jürgens, C. Schulz, J. Biener, M. M. Biener, A. V. Hamza and M. Bäumer, Angew. Chem., Int. Ed., 2006, 45, 8241–8244 CrossRef CAS PubMed.
- A. Wittstock, J. Biener and M. Bäumer, Phys. Chem. Chem. Phys., 2010, 12, 12919–12930 RSC.
- L. Y. Chen, X. Y. Lang, T. Fujita and M. W. Chen, Scr. Mater., 2011, 65, 17–20 CrossRef CAS.
- D. Hung, Z. Liu, Y. W. Hao and P. C. Searson, Abstr. Pap. Am. Chem. Soc., 2005, 230, U1113–U1114 Search PubMed.
- C. A. R. Chapman, H. Chen, M. Stamou, J. Biener, M. M. Biener, P. J. Lein and E. Seker, ACS Appl. Mater. Interfaces, 2015, 7, 7093–7100 CAS.
- G. Schmid and B. Corain, Eur. J. Inorg. Chem., 2003, 3081–3098 CrossRef CAS.
- A. J. Forty and P. Durkin, Philos. Mag. A, 1980, 42, 295–318 CrossRef CAS.
- A. Y. Chen, S. S. Shi, F. Liu, Y. Wang, X. Li, J. F. Gu and X. F. Xie, Appl. Surf. Sci., 2015, 355, 133–138 CrossRef CAS.
- L. Schade, S. Franzka, M. Mathieu, M. M. Biener, J. Biener and N. Hartmann, Langmuir, 2014, 30, 7190–7197 CrossRef CAS PubMed.
- J. Wang, R. Xia, J. Zhu, Y. Ding, X. Zhang and Y. Chen, J. Mater. Sci., 2012, 47, 5013–5018 CrossRef CAS.
- C. A. R. Chapman, L. Wang, J. Biener, E. Seker, M. M. Biener and M. J. Matthews, Nanoscale, 2015, 785–795 Search PubMed.
- Y.-C. K. Chen-Wiegart, S. Wang, Y. S. Chu, W. Liu, I. McNulty, P. W. Voorhees and D. C. Dunand, Acta Mater., 2012, 60, 4972–4981 CrossRef CAS.
- Z. Li, S. Zhou, H. Li, W. Si and Y. Ding, J. Nanosci. Nanotechnol., 2009, 9, 1651–1654 CrossRef CAS PubMed.
- J. Biener, A. Wittstock, M. M. Biener, T. Nowitzki, A. V. Hamza and M. Bäumer, Langmuir, 2010, 26, 13736–13740 CrossRef CAS PubMed.
- E. Seker, J. T. Gaskins, H. Bart-Smith, J. Zhu, M. L. Reed, G. Zangari, R. Kelly and M. R. Begley, Acta Mater., 2007, 55, 4593–4602 CrossRef CAS.
- Y. Sun, S. A. Burger and T. J. Balk, Philos. Mag., 2014, 94, 1001–1011 CrossRef CAS.
- J. Erlebacher, Phys. Rev. Lett., 2011, 106 Search PubMed.
- K. Kolluri and M. J. Demkowicz, Acta Mater., 2011, 59, 7645–7653 CrossRef CAS.
- M. P. Klein, B. W. Jacobs, M. D. Ong, S. J. Fares, D. B. Robinson, V. Stavila, G. J. Wagner and I. Arslan, J. Am. Chem. Soc., 2011, 133, 9144–9147 CrossRef CAS PubMed.
- A. Wichmann, A. Wittstock, K. Frank, M. M. Biener, B. Neumann, L. Madler, J. Biener, A. Rosenauer and M. Bäumer, ChemCatChem, 2013, 5, 2037–2043 CrossRef CAS.
- J. Shi, C. Mahr, M. M. Murshed, V. Zielasek, A. Rosenauer, T. M. Gesing, M. Bäumer and A. Wittstock, Catal. Sci. Technol., 2016, 6, 5311–5319 CAS.
- S. Kuwano-Nakatani, T. Fujita, K. Uchisawa, D. Umetsu, Y. Kase, Y. Kowata, K. Chiba, T. Tokunaga, S. Arai, Y. Yamamoto, N. Tanaka and M. Chen, Mater. Trans., 2015, 56, 468–472 CrossRef CAS.
- L. F. Allard, M. Flytzani-Stephanopoulos and S. H. Overbury, Microsc. Today, 2009, 17, 50–55 CrossRef.
- N. P. Young, M. A. van Huis, H. W. Zandbergen, H. Xu and A. I. Kirkland, Ultramicroscopy, 2010, 110, 506–516 CrossRef CAS PubMed.
- J. M. Thomas and J.-C. Hernandez-Garrido, Angew. Chem., Int. Ed., 2009, 48, 3904–3907 CrossRef CAS PubMed.
- J. M. Rodenburg and H. M. L. Faulkner, Appl. Phys. Lett., 2004, 85, 4795–4797 CrossRef CAS.
- M. Dierolf, A. Menzel, P. Thibault, P. Schneider, C. M. Kewish, R. Wepf, O. Bunk and F. Pfeiffer, Nature, 2010, 467, 436–439 CrossRef CAS PubMed.
- J. Vila-Comamala, A. Diaz, M. Guizar-Sicairos, A. Mantion, C. M. Kewish, A. Menzel, O. Bunk and C. David, Opt. Express, 2011, 19, 21333–21344 CrossRef PubMed.
- A. Schropp, R. Hoppe, J. Patommel, D. Samberg, F. Seiboth, S. Stephan, G. Wellenreuther, G. Falkenberg and C. Schroer, Appl. Phys. Lett., 2012, 100, 253112 CrossRef.
- S. Baier, C. D. Damsgaard, M. Scholz, F. Benzi, A. Rochet, R. Hoppe, T. Scherer, J. Shi, A. Wittstock, B. Weinhausen, J. B. Wagner, C. G. Schroer and J.-D. Grunwaldt, Microsc. Microanal., 2016, 22, 178–188 CrossRef CAS PubMed.
- K. Høydalsvik, J. B. Floystad, T. Zhao, M. Esmaeili, A. Diaz, J. W. Andreasen, R. H. Mathiesen, M. Ronning and D. W. Breiby, Appl. Phys. Lett., 2014, 104, 241909 CrossRef.
- L. F. Allard, A. Borisevich, W. Deng, R. Si, M. Flytzani-Stephanopoulos and S. H. Overbury, J. Electron Microsc., 2009, 58, 199–212 CrossRef CAS PubMed.
- S. Gorelick, J. Vila-Comamala, V. A. Guzenko, R. Barrett, M. Salomé and C. David, J. Synchrotron Radiat., 2011, 18, 442–446 CrossRef PubMed.
- X. Huang, H. Yan, R. Harder, Y. Hwu, I. K. Robinson and Y. S. Chu, Opt. Express, 2014, 22, 12634–12644 CrossRef PubMed.
- B. Henrich, A. Bergamaschi, C. Broennimann, R. Dinapoli, E. Eikenberry, I. Johnson, M. Kobas, P. Kraft, A. Mozzanica and B. Schmitt, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 607, 247–249 CrossRef CAS.
- M. R. Howells, T. Beetz, H. N. Chapman, C. Cui, J. Holton, C. Jacobsen, J. Kirz, E. Lima, S. Marchesini and H. Miao, J. Electron Spectrosc. Relat. Phenom., 2009, 170, 4–12 CrossRef CAS PubMed.
- A. M. Hodge, J. R. Hayes, J. A. Caro, J. Biener and A. V. Hamza, Adv. Eng. Mater., 2006, 8, 853–857 CrossRef CAS.
- P. Thibault, M. Dierolf, A. Menzel, O. Bunk, C. David and F. Pfeiffer, Science, 2008, 321, 379–382 CrossRef CAS PubMed.
- P. Thibault and M. Guizar-Sicairos, New J. Phys., 2012, 14, 063004 CrossRef.
- M. Dierolf, Ptychographic X-ray microscopy and Tomography, Technische Universität München, 2015, ch. 7 Search PubMed.
- M. Guizar-Sicairos, I. Johnson, A. Diaz, M. Holler, P. Karvinen, H.-C. Stadler, R. Dinapoli, O. Bunk and A. Menzel, Opt. Express, 2014, 22, 14859–14870 CrossRef PubMed.
- M. van Heel and M. Schatz, J. Struct. Biol., 2005, 151, 250–262 CrossRef CAS PubMed.
- M. Guizar-Sicairos, A. Diaz, M. Holler, M. S. Lucas, A. Menzel, R. A. Wepf and O. Bunk, Opt. Express, 2011, 19, 21345–21357 Search PubMed.
- N. M. Ravindra, S. Abedrabbo, W. Chen, F. M. Tong, A. K. Nanda and A. C. Speranza, IEEE Trans. Semicond. Manuf., 1998, 11, 30–39 CrossRef.
- S. Hannemann, J.-D. Grunwaldt, B. Kimmerle, A. Baiker, P. Boye and C. Schroer, Top. Catal., 2009, 52, 1360–1370 CrossRef CAS.
- A. M. Gänzler, M. Casapu, A. Boubnov, O. Müller, S. Conrad, H. Lichtenberg, R. Frahm and J.-D. Grunwaldt, J. Catal., 2015, 328, 216–224 CrossRef.
- T. S. Dorofeeva and E. Seker, Nano Res., 2015, 8, 2188–2198 CrossRef CAS.
- J. Shi, A. Schaefer, A. Wichmann, M. M. Murshed, T. M. Gesing, A. Wittstock and M. Bäumer, J. Phys. Chem. C, 2014, 118, 29270–29277 CAS.
- A. Wichmann, A. Wittstock, K. Frank, M. M. Biener, B. Neumann, L. Mädler, J. Biener, A. Rosenauer and M. Bäumer, ChemCatChem, 2013, 5, 2037–2043 CrossRef CAS.
- R. N. Viswanath, V. A. Chirayath, R. Rajaraman, G. Amarendra and C. S. Sundar, Appl. Phys. Lett., 2013, 102, 253101 CrossRef.
- D. R. Peale and B. H. Cooper, J. Vac. Sci. Technol., A, 1992, 10, 2210–2215 CAS.
- T. W. Hansen, A. T. Delariva, S. R. Challa and A. K. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef CAS PubMed.
- F. Kertis, J. Snyder, L. Govada, S. Khurshid, N. Chayen and J. Erlebacher, JOM, 2010, 62, 50–56 CrossRef CAS.
- D. L. Olson, J. M. Blakely and H. R. Patil, Scr. Metall., 1972, 6, 229–240 CrossRef CAS.
- R. E. Honig and D. A. Kramer, RCA Rev., 1969, 30, 285–305 CAS.
- R. Li and K. Sieradzki, Phys. Rev. Lett., 1992, 68, 1168–1171 CrossRef CAS PubMed.
- J. Biener, A. M. Hodge and A. V. Hamza, Appl. Phys. Lett., 2005, 87, 121908 CrossRef.
- B. Kahng, G. G. Batrouni, S. Redner, L. de Arcangelis and H. J. Herrmann, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 7625–7637 CrossRef.
- M. Holler, A. Diaz, M. Guizar-Sicairos, P. Karvinen, E. Farm, E. Harkonen, M. Ritala, A. Menzel, J. Raabe and O. Bunk, Sci. Rep., 2014, 4, 3857 CAS.
- J. C. da Silva, K. Mader, M. Holler, D. Haberthür, A. Diaz, M. Guizar-Sicairos, W. C. Cheng, Y. Shu, J. Raabe and A. Menzel, ChemCatChem, 2015, 7, 413–416 CrossRef CAS PubMed.
- M. Beckers, T. Senkbeil, T. Gorniak, M. Reese, K. Giewekemeyer, S.-C. Gleber, T. Salditt and A. Rosenhahn, Phys. Rev. Lett., 2011, 107, 208101 CrossRef PubMed.
- R. Hoppe, J. Reinhardt, G. Hofmann, J. Patommel, J.-D. Grunwaldt, C. D. Damsgaard, G. Wellenreuther, G. Falkenberg and C. G. Schroer, Appl. Phys. Lett., 2013, 102, 203104 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra12853j |
| This journal is © The Royal Society of Chemistry 2016 |