
The origin of room temperature ferromagnetism mediated by Co–VZn complexes in the ZnO grain boundary
Abstract
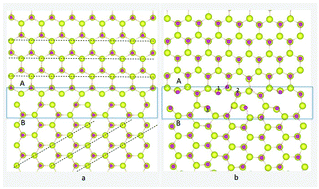
Ferromagnetism in polycrystalline ZnO doped with Co has been observed to be sustainable in recent experiments. We use first-principle calculations to show that Co impurities favorably substitute at the grain boundary (GB) rather than in the bulk. We reveal that room-temperature ferromagnetism (RTFM) at the Co-doped ZnO GB in the presence of Zn vacancies is due to ferromagnetic exchange coupling of a pair of closely associated Co atoms in the GB, with a ferromagnetic exchange coupling energy of ∼300 meV, which is in contrast to a previous study that suggested the O vacancy–Co complex induced ferromagnetism. Electronic structure analysis was used to predict the exchange coupling mechanism, showing that the hybridization of O p states with Co and Zn d states enhances the magnetic polarization originating from the GB. Our results indicate that RTFM originates from Co clusters at interfaces or in GBs.
 Please wait while we load your content...
Please wait while we load your content...

