
Localized delivery and enhanced osteogenic differentiation with biodegradable galactitol polyester elastomers†
 b
and
b
and
Abstract
Given that a large fraction of the population suffers from orthopaedic diseases, the research in developing polymeric biomaterials for bone tissue regeneration applications is witnessing an exponential growth rate. We present a spectrum of novel polyesters synthesized by the reaction of galactitol with dicarboxylic acids, namely adipic acid, suberic acid and dodecanedioic acid. Fourier transform infrared spectroscopy and nuclear magnetic resonance spectroscopy confirmed the chemical structure of the polymers. Thermal characterization revealed that these polyesters were semi-crystalline. The molecular weight of the polyesters showed an increase with increase in the chain length of the diacid and the molar ratio of galactitol : diacid. Dynamic mechanical analysis showed that the polymers were elastomeric in nature with the increase in chain length and molar ratio of galactitol : diacids. Surface hydrophobicity and the swelling ratio increase with increase in the chain length and molar ratio of galactitol : diacids. Hydrolytic degradation studies demonstrated that the kinetics of the degradation followed first order. Dye release studies indicated that the rate of release followed Higuchi kinetics. In vitro studies confirmed the cytocompatible nature of these polymers. Mineralization by osteoblasts in vitro suggests that these polymers support osteogenic differentiation, thus elucidating that these polymers are promising candidate materials for bone tissue engineering. Thus, this study presents a significant advance in which the mechanical properties, degradation and release rates of the polyesters may be tuned by manipulating the process parameters.
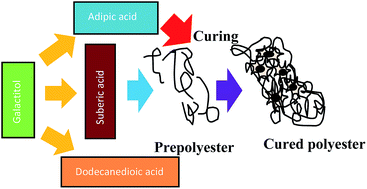
- This article is part of the themed collection: New Frontiers in Indian Research
 Please wait while we load your content...
Please wait while we load your content...

