
Deprotonation routes of anthocyanidins in aqueous solution, pKa values, and speciation under physiological conditions†
 *b
and
Juan Raúl
Alvarez-Idaboy
*a
*b
and
Juan Raúl
Alvarez-Idaboy
*a
Abstract
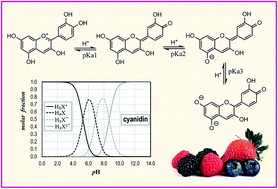
Anthocyanidins are water-soluble flavonoids that have numerous beneficial effects to human and animal health. At the same time, they present multiple acid–base equilibria that under physiological conditions may lead to a rather wide distribution of species. This particular feature might influence the activity and mechanism of action of anthocyanidins in living systems, depending on the pH of the environment. Therefore, detailed knowledge of the acid–base behavior of these compounds is crucial to fully understand their ways of action. In this work, theoretical calculations within the frame of Density Functional Theory (DFT) were carried out to investigate several aspects or the equilibria for 12 anthocyanidins. Their most likely deprotonation routes were elucidated, and most of their pKa values are reported here for the first time. Their reliability was confirmed by comparison with the available experimental data, which led to a mean unsigned error of 0.31. The obtained pKa values allowed the estimation of the populations of the different species depending on the pH, and particular attention was paid to pH = 7.4. Hopefully, the data provided here may contribute to gain better understanding on the complex processes involving anthocyanidins, under physiological conditions.
 Please wait while we load your content...
Please wait while we load your content...

