DOI:
10.1039/C6RA08582B
(Paper)
RSC Adv., 2016,
6, 57780-57792
Photophysical properties of di-Schiff bases: evaluating the synergistic effect of non-covalent interactions and alkyl spacers in enhanced emissions of solids†
Received
4th April 2016
, Accepted 2nd June 2016
First published on 6th June 2016
Abstract
Photophysical properties of di-Schiff base compounds in the solution and solid states are explored. The di-Schiff bases with alkyl spacers (ethyl, butyl and hexyl) showed enhanced light emitting properties in the solid state, while quenching was observed for di-Schiff bases with a hydrazine spacer. The photoluminescence spectra and the crystal structure were compared with bis-pyridyl-ethyl-di-imine and bis-pyridyl-butyl-di-imine. The crystal structure analysis of the compounds rationalizes the observed results. The concentration dependent 1H-NMR and NOESY spectra showed the aggregation behaviour of the compounds.
Introduction
The potential application1 of light emitting materials includes organic light-emitting diodes (OLEDs), sensors and biological imaging, which requires them to have enhanced luminescence efficiency in the aggregated state. Tang and coworkers in 2001 first explored the aggregation based emission (AIE) and reported a series of tetraphenylethene derivatives.2 Park and co-workers,3 Tian and coworkers4 have reported several material systems with AIE properties. The AIE behavior is rationalized by considering the restriction of Intramolecular Rotation, formation of J-aggregates and intramolecular planarization.5
The solid state arrangement of the molecules, in general, decides the properties of a compound, so the crystal structure analysis will provide information on arrangement of the molecules. The knowledge on supramolecular packing of the molecules and its relation with the photophysical property will pave the way to develop the strategies to design new materials targeting the light emitting properties. The synthesis of materials by utilizing non covalent interaction is an efficient route and is a handy method to tailor made light emitting solids. Draper and coworkers have used supramolecular synthesis and tuned the solid state luminescence of 2-cyano-3(4-(diphenylamino)phenyl)acrylic acid by reacting them with substituted pyridines and amines.6 The review on non-covalent methods to synthesize solid state emitting molecular materials and their applications is highlighted by Varughese.7
The Schiff base based compounds are explored widely because of their easy synthetic procedures, photophysical properties and biological activity.8 Kawasaki et al. have reported the solid state PL spectra for a series of Salen compounds (Schiff bases) with varying alkyl chain length. They have observed a relation of photo physical property with the alkyl chain length.9 Xiang et al. have reported AIE in salicylaldehyde azine where despite having planar geometry, no quenching was observed.10 Recently, Xiang, Liu and coworkers have reported the non conjugated Salen compounds with unique AIE, where the small π-conjugated system resulted in aggregation-induced emission (AIE) with large Stokes shifts and high fluorescence quantum yields was observed.10e
In the present work, we have studied the photophysical properties of di Schiff base molecules (Scheme 1) in the solid state and in solution. We have rationalized the observations on the following grounds: (i) effect of alkyl chain length; (ii) presence of methyl group on the methine carbon; (iii) comparison of the light emitting properties with the pyridyl based Schiff base compounds. Recently our group has reported the photo physical properties along with the structural description of pyridyl based Schiff bases, where we have concluded that the flexible spacer (alkyl bridge) in these molecules helped in aggregating the molecules which results in enhanced emission.11
 |
| | Scheme 1 Di Schiff base compounds. | |
There are reports on the crystal structure analysis of L1a, L1b and L2a along with their property studies.12 The absorption spectra of azines and dianils, which included L1a, L1b, L2a and L2b, were reported by Ferguson and Goodwin in 1949.12a Sinha reported the crystal structure of L1a and reported the effect of conjugation on the bond lengths.12b Glaser et al. have reported the solid state structures of series of acetophenone azines including L1b12c while the structure of L2b is reported by the research group of Tiekink.12d The electrochemical behavior of the acetophenone azines were studied by Workentin et al., where the effect of substituents on aryl group on the electrochemical potential was studied.12e Research group of Ding reported the high pressure spectral analysis of L1a and observed pressure induced phase transformation.12f Xiang et al. have studied the emission properties of L1a and reported that no AIE properties was observed.10a
Experimental section
General
Infra Red Spectra were taken in FTIR ABB Bomen MB-3000. UV-Visible Spectra and Fluorescence Spectra were taken in Shimadzu spectrophotometer model UV-2450 and Fluorimax-4 0426C0809, respectively. Proton and Carbon-13 Nuclear Magnetic Resonance (1H NMR and 13C NMR) spectra were recorded on a 400 MHz spectrophotometer (Bruker). Powder X-ray Diffraction (XRD) were recorded with a Rigaku miniflex II, λ = 1.54, Cu Kα. Fluorescence quantum yields are determined by using quanta phi instrument (Jobin Horiba, Fluorimax-4).
The compounds L1–L4 were synthesized by the usual method of preparation of Schiff bases, which involved the condensation reaction of primary diamines with an aldehyde/ketone precursor in alcoholic solution under reflux conditions.
1,2-Dibenzylidenehydrazine (L1a). Hydrazine hydrate (0.5 ml, 10 mmol) was added drop wise to an ethanolic solution of benzaldehyde (2.0 ml, 20 mmol). The mixture was kept for 6 h under refluxing condition. The solvent was removed under vacuum and the yellow color solid product was recrystallized with methanol. Yield: 36%, melting point: 86.5 °C. IR (cm−1 KBr pellet): 3418(w), 3171(w), 3047(w), 3001(w), 2947(m), 1666(m), 1628(vs), 1574(w), 489(w), 1443(m), 1304(w), 1203(w), 957(m), 856(w) (Fig. S1†). 1H-NMR (400 MHz, DMSO) δ ppm: 8.73 (s, 1H), 7.90 (dd, J = 7.4, 2.2 Hz), 7.51 (dq, J = 6.6, 3.2 Hz, 3H) (Fig. S2†). 13C-NMR (101 MHz, DMSO) δ ppm: 161.99, 134.24, 131.84, 129.38, 128.84 (Fig. S3†).
1,2-Bis(1-phenylethylidene)hydrazine (L1b). 0.5 ml (10 mmol) hydrazine hydrate was added dropwise to a solution of acetophenone (2.3 ml, 20 mmol) in ethanol (40 ml). The mixture was refluxed at 80 °C for 6 h. The solvent was removed under vacuum and the yellow color solid product was recrystallized with methanol. Yield: 46%, melting point: 123 °C. IR (cm−1 KBr pellet): 3418(m), 3055(m), 2962(m), 2916(w), 1643(vs), 1566(vs), 1489(w), 1443(vs), 1358(vs), 1281(s), 1072(m), 1018(m), 756(vs) (Fig. S4†). 1H-NMR (400 MHz, DMSO) δ ppm: 7.92 (dd, J = 6.6, 3.1 Hz, 2H), 7.50–7.44 (m, 3H), 2.28 (s, 3H) (Fig. S5†). 13C-NMR (101 MHz, DMSO) δ ppm: 157.77, 138.77, 130.54, 128.79, 126.80, 15.19 (Fig. S6†).
N1,N2-Dibenzylideneethane-1,2-diamine (L2a). 0.5 ml (10 mmol) ethylene diamine was added dropwise to a solution of benzaldehyde (2.0 ml, 20 mmol) in ethanol (40 ml). The mixture was refluxed at 80 °C for 6 h. The solvent was removed under vacuum and the orange semi-solid product was crystallized with hexane. Yield: 38%, melting point: 60 °C. IR (cm−1 KBr pellet): 3418(m), 3271(w), 3055(w), 3032(w), 2908(w), 2843(m), 1674(w), 1643(vs), 1574(w), 1450(m), 1373(m), 1281(w), 157(w), 1018(s), 972(w), 918(w) (Fig. S7†). 1H-NMR (400 MHz, DMSO) δ ppm: 8.34 (s, 1H), 7.71 (dd, J = 7.2, 2.5 Hz, 2H), 7.48–7.36 (m, 3H), 3.54–3.31 (m, 2H) (Fig. S8†). 13C-NMR (101 MHz, DMSO) δ ppm: 162.42, 136.56, 131.07, 129.09, 128.27, 61.17 (Fig. S9†).
N1,N2-Bis(1-phenylethylidene)ethane-1,2-diamine (L2b). 0.5 ml (10 mmol) ethylene diamine was added dropwise to a solution of acetophenone (2.3 ml, 20 mmol) in ethanol (40 ml). The mixture was refluxed at 80 °C for 6 h. The solvent was removed under vacuum and brown semi-solid product was recrystallized with hexane. Yield: 36%, melting point: 107.5 °C. IR (cm−1 KBr pellet): 3418(m), 3248(w), 3063(w), 3016(w), 2885(m), 2824(m), 1628(vs), 1574(w), 1489(w), 1443(m), 1373(m), 1265(vs), 1180(w), 1080(m), 1041(m), 910(m), 756(vs) (Fig. S10†). 1H-NMR (400 MHz, DMSO) δ ppm: 7.92 (dd, J = 6.6, 3.1 Hz, 2H), 7.51–7.44 (m, 3H), 3.48–3.30 (m, 2H), 2.28 (s, 3H) (Fig. S11†). 13C-NMR (101 MHz, DMSO) δ ppm: 165.09, 140.97, 129.84, 128.45, 126.89, 52.74, 15.17 (Fig. S12†).
N1,N2-Dibenzylidenebutane-1,4-diamine (L3a). 0.5 ml (10 mmol) diaminobutane was added dropwise to a solution of benzaldehyde (2.0 ml, 20 mmol) in ethanol (40 ml). The mixture was refluxed at 80 °C 6 h. The solvent was removed under vacuum and the brown color semi-solid product was recrystallized with hexane. Yield: 49%, melting point: 50 °C. IR (cm−1 KBr pellet): 3394(w), 3271(w), 3055(w), 3024(w), 2932(m), 2847(m), 2646(w), 1643(vs), 1605(w), 1574(w), 1443(s), 1381(w), 1288(w), 965(w), 756(vs) (Fig. S13†). 1H-NMR (400 MHz, DMSO) δ ppm: 8.34 (s, 1H), 7.73 (dd, J = 6.6, 3.1 Hz, 2H), 7.49–7.39 (m, 3H), 3.60 (d, J = 1.1 Hz, 2H), 1.67 (Dt, J = 5.6, 3.0 Hz, 2H) (Fig. S14†). 13C-NMR (101 MHz, DMSO) δ ppm: 161.05, 136.70, 130.96, 128.97, 128.18, 60.31, 28.58 (Fig. S15†).
N1,N2-Bis(1-phenylethylidene)butane-1,4-diamine (L3b). 0.5 ml (10 mmol) diaminobutane was added dropwise to a solution of acetophenone (2.3 ml, 20 mmol) in ethanol (40 ml). The mixture was refluxed at 80 °C for 6 h. The solvent was removed under vacuum and the green color semi-solid product was recrystallized with hexane. Yield: 50%, melting point: 62 °C. IR (cm−1 KBr pellet): 3418(m), 3240(w), 3086(w), 3047(w), 2932(m), 2878(m), 1628(vs), 1574(w), 1489(w), 1443(w), 1350(m), 1281(m), 1180(w), 1080(w), 918(w), 764(vs) (Fig. S16†). 1H-NMR (400 MHz, DMSO) δ ppm: 7.82 (dd, J = 6.8, 2.9 Hz, 2H), 7.43–7.35 (m, 3H), 3.52–3.45 (m, 2H), 2.20 (s, 3H), 1.84–1.75 (m, 2H) (Fig. S17†). 13C-NMR (101 MHz, DMSO) δ ppm: 163.85, 141.02, 129.73, 128.09, 127.33, 51.52, 28.98, 15.06 (Fig. S18†).
N1,N2-Dibenzylidenehexane-1,6-diamine (L4a). 0.5 ml (10 mmol) hexamethylene diamine was added dropwise to a solution of benzaldehyde (2.0 ml, 20 mmol) in ethanol (40 ml). The mixture was refluxed at 80 °C for 6 h. The solvent was removed under vacuum and the brown semi-solid product was recrystallized with hexane. Yield: 48%, melting point: 170 °C. IR (cm−1 KBr pellet): 3394(w), 3148(m), 3001(w), 2939(w), 2854(w), 2716(w), 2106(w), 1643(s), 1589(w), 1551(vs), 1379(vs), 1234(w), 1173(w), 1065(w), 949(w), 825(s) (Fig. S19†). 1H-NMR (400 MHz, DMSO) δ ppm: 7.89 (s, 1H), 7.44–7.23 (m, 3H), 2.70 (dd, J = 59.6 Hz, 2H), 1.63–1.45 (m, 2H), 1.40–1.18 (m, 2H) (Fig. S20†). 13C-NMR (101 MHz, DMSO) δ ppm: 170.46, 139.62, 129.80, 129.42, 127.83, 27.84, 25.92 (Fig. S21†).
N1,N2-Bis(1-phenylethylidene)hexane-1,6-diamine (L4b). 0.5 ml (10 mmol) hexamethylene diamine was added dropwise to a solution of acetophenone (2.3 ml, 20 mmol) in ethanol (40 ml). The mixture was refluxed at 80 °C for 6 h. The solvent was removed under vacuum and the brown semi-solid product was recrystallized with hexane. Yield: 47%, melting point: 47.5 °C. IR (cm−1 KBr pellet): 3811(w), 3433(m), 3256(w), 3055(w), 2924(m), 2854(w), 1628(vs), 1582(w), 1443(w), 1412(m), 1381(m), 1281(w), 794(m), 694(s), 571(w) (Fig. S22†). 1H-NMR (400 MHz, DMSO) δ ppm: 7.84–7.76 (m, 2H), 7.45–7.29 (m, 3H), 3.39 (d, J = 8.9 Hz, 2H), 2.17 (s, 3H), 1.64 (dt, J = 33.6, 6.9 Hz, 2H), 1.54–1.27 (m, 2H) (Fig. S23†). 13C-NMR (101 MHz, DMSO) δ ppm: 164.09, 141.13, 129.71, 128.26, 126.75, 51.40, 31.07, 27.55, 15.40 (Fig. S24†).
Results and discussion
In order to analyze the relation of the photophysical properties of the compounds with the geometry and supramolecular arrangement of the molecules, the crystal structure description of some compounds were studied. The crystal structure of L1a,12b L1b,12c L2b12d and L2c12g were reported by other groups while our groups had reported the crystal structure description of L1d, L2d and L3c.11 The following features were observed while analyzing the structural aspects of the compounds.
Crystal structure analysis of L1a12b
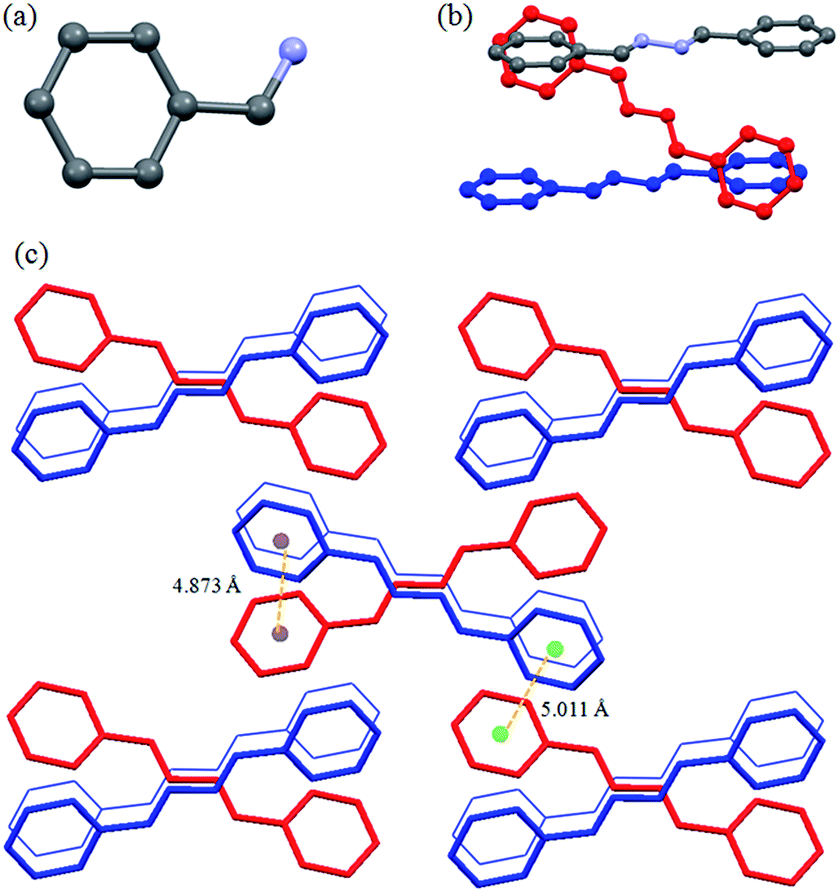
The compound L1a had orthorhombic Pbcn space group and only half of the molecule was present in the asymmetric unit (Fig. 1a). The molecules in L1a were packed in such a way that the N–N bonds of the adjacent molecules were arranged in parallel fashion with a distance of 3.810 Å between the two nitrogen of the neighboring molecules (Fig. 1b). The aromatic moieties of the two adjacent molecules have inclined/tilted arrangements with centroid-to-centroid distance of 4.873 Å (Fig. 1c). This results in a 1D arrangement of the molecules, which were further assembled in 3D via aromatic interactions between the inclined aromatic rings (centroid-to-centroid distance of 5.011 Å) (Fig. 1c).
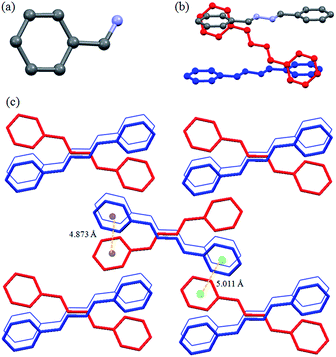 |
| | Fig. 1 Illustration of crystal structure of L1a:12b (a) asymmetric unit of L1a; (b) arrangement of adjacent molecules; (c) packing of the molecules (figures were generated from the data obtained from CCDC no. 1118104). | |
Crystal structure analysis of L1b12c
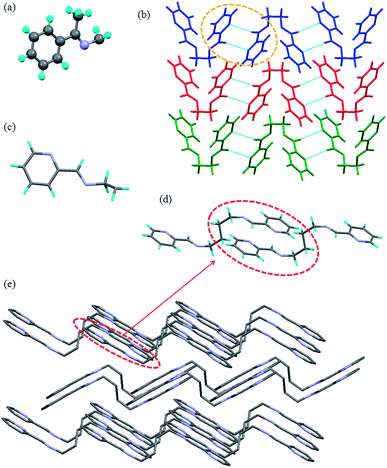
The compound L1b was crystallized in monoclinic P21/n space group with one molecule present in the asymmetric unit. The molecule of L1b had a non planar geometry, where the C–N–N–C torsion angle was 138.72° and the two phenyl rings of a molecule were inclined at an angle of 64.62° (Fig. 2a). Although inclined face-to-face stacking of the molecules were observed in this case due to the non planar geometry of the molecules, but the significant number of π⋯π interactions resulted in packing of the molecules (Fig. 2c). The phenyl ring of a molecule of L1b was in the vicinity of neighboring phenyl rings such that the centroid-to-centroid distance between the aromatic rings was in the range of 4.6–4.9 Å (Fig. 2b).
 |
| | Fig. 2 Illustration of crystal structure of L1b:12c (a) asymmetric unit of L1b: notice the non planar geometry of the molecule; (b) centroid-to-centroid distance between the adjacent aromatic rings; (c) packing of the molecules (figures were generated from the data obtained from CCDC no. 1207284). | |
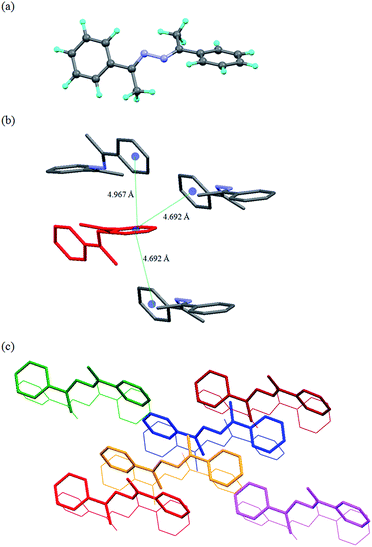
Crystal structure analysis of L1d11
The crystal structure analysis of L1d showed that there are three molecules in the asymmetric unit, which differed in the orientation of the pyridyl and imine moieties. The two molecules were arranged parallel such that they form dimer (Fig. 3). The distance between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N of the two molecules were 3.906 and 3.943 Å, while the centroid to centroid distance between the two pyridyl rings were 3.888 Å and 4.009 Å. The third type of molecule interacted with the other two molecules via C–H⋯N and aromatic π⋯π interactions and helped in the formation of dimers.
N of the two molecules were 3.906 and 3.943 Å, while the centroid to centroid distance between the two pyridyl rings were 3.888 Å and 4.009 Å. The third type of molecule interacted with the other two molecules via C–H⋯N and aromatic π⋯π interactions and helped in the formation of dimers.
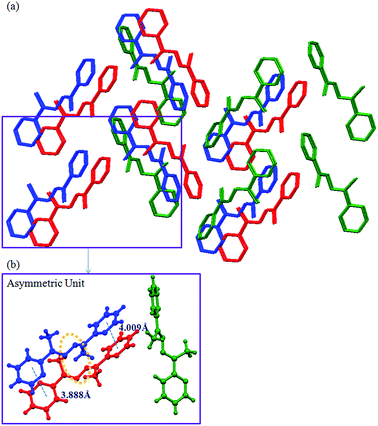 |
| | Fig. 3 Illustration of crystal structure of L1d:11 (a) packing of the molecules (hydrogen atoms are removed for clarity); (b) three types of molecules of L1d are present in the asymmetric unit, which are shown in different colors (figures were generated from the data obtained from CCDC no. 963345). | |
Crystal structure analysis of L2b12d
The compound L2b was crystallized in P21/n space group with half of the molecule in the asymmetric unit (Fig. 4a). The ethyl group in the molecule adopted all anti conformation. The molecules were stacked in parallel manner to form a one-dimensional network (Fig. 4b). The 3D arranged of the molecules can be viewed as herringbone arrangement of the one dimensional networks (Fig. 4c).
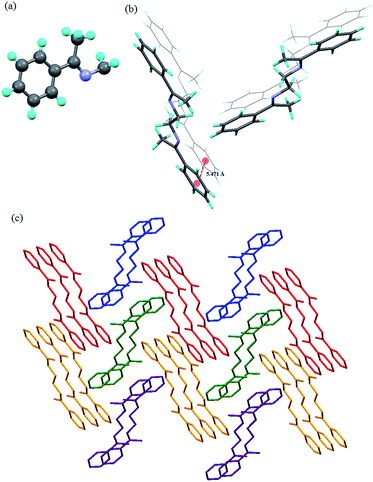 |
| | Fig. 4 Illustration of crystal structure of L2b:12d (a) asymmetric unit; (b) arrangement of the molecules: notice that the aromatic centroid-to-centroid distance is more than 5 Å; (c) herringbone arrangement of the molecules of L2b (figures were generated from the data obtained from CCDC no. 608471). | |
Crystal structure analysis of L2d11
The crystal structure of L2d was reported by our group11 and it showed that there are two molecules in the asymmetric unit (Fig. 5a). The ethyl spacer of L2d adopted anti conformation thereby resulting in linear geometry of the molecule. The neighboring molecules were arranged in a offset manner to form a one dimensional network (Fig. 5b). The overall supramolecular network can be viewed as herringbone arrangement of these one dimensional network (Fig. 5c).
 |
| | Fig. 5 Illustration of crystal structure of L2d:11 (a) asymmetric unit; (b) offset arrangement of the molecules to form 1D network; (c) herringbone arrangement of the molecules of L2d (hydrogen atoms were removed for clarity) (figures were generated from the data obtained from CCDC no. 962823). | |
Crystal structure analysis of L2c12g and L3c11
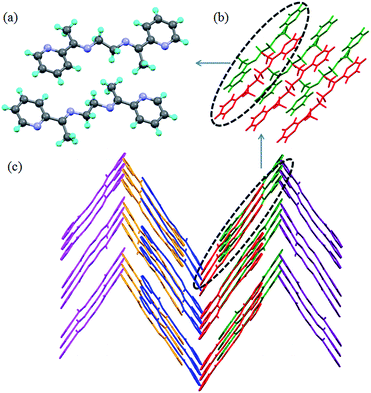
The asymmetric unit in L2c has half of the molecule and the ethyl spacer adopts gauche conformation. The C–H⋯N hydrogen bond interactions between the pyridyl N and C–H of methine resulted in the formation of “non-covalently bonded chromophore” unit (Fig. 6a and b). The crystal structure analysis of L3c showed that it has half of the molecule in the asymmetric unit (Fig. 6c). Further it was observed that the butyl spacer of L3c adopted gauche–anti–gauche conformation and not expected all anti conformation. The pyridyl-N⋯HC-(methine) interactions resulted in formation of a non covalently bonded “macrocyclic” moiety (Fig. 6d). The butyl chain conformation resulted in formation of corrugated layers and offset packing of the layers results in the overall 3D arrangement of the L3c (Fig. 6e).
 |
| | Fig. 6 Illustration of crystal structure of L2c12g and L3c:11 (a) asymmetric unit in L2c; (b) packing of the molecules of L2c via C–H⋯N interactions to form non covalent macrocyclic moiety; (c) asymmetric unit in L3c; (d) non covalent “macrocyclic” moiety in L3c; (e) offset packing of the corrugated layers in L3c (hydrogen atoms are removed for clarity) (figures were generated from the data obtained from CCDC no. 721559 (L2c) and 930055 (L3c)). | |
Powder XRD spectra of the compounds
Powder XRD spectra were recorded for L1a, L1b, L2a, L2b, L3a, L3b, L4a and L4b. The experimental powder XRD spectra of the compounds L1a, L1b, and L2b were compared with the simulated powder XRD spectra from the single crystal XRD data. Fig. 7 shows the phase purity of the compounds L1a, L1b, and L2b. The experimental spectra of other compounds are shown in Fig. S25–S29.†
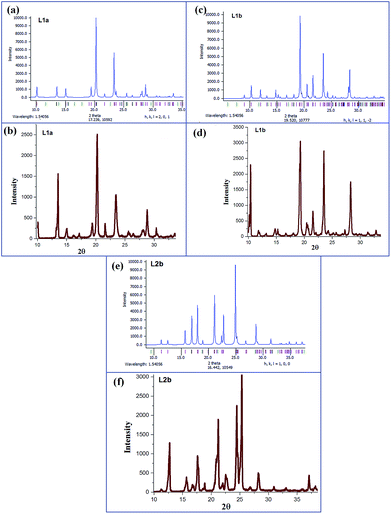 |
| | Fig. 7 (a) Calculated PXRD of L1a; generated from crystal data CCDC no. 1118104; (b) experimental PXRD of L1a; (c) calculated PXRD of L1b; generated from crystal data CCDC no. 1207284; (d) experimental PXRD of L1b; (e) calculated PXRD of L2b; generated from crystal data CCDC no. 608471; (f) experimental PXRD of L2b. | |
UV-Visible absorption spectra
The solid state UV-Visible spectra of L1a showed absorption maxima at 196 nm (π → π*) and 300 nm (n → π*) while L1b shows λmax at 206 nm (π → π*) and 266 nm (n → π*) (Fig. 8a and b). The absorption maxima for L1d11 were at 253 nm (π → π*) and 286 nm (n → π*); which clearly indicated the effect of changing phenyl group with pyridyl group. The solid state UV-Visible absorption maxima for L2a, L2b, L3a, L3b, L4a and L4b were in the range 192–194 nm (π → π*) and 223–237 nm (n → π*) (Table 1, Fig. 8c–h), whereas for the L2 and L3 compounds with pyridyl group, the λmax was observed in the range of 232–235 nm (π → π*) and 269–271 nm (n → π*).11
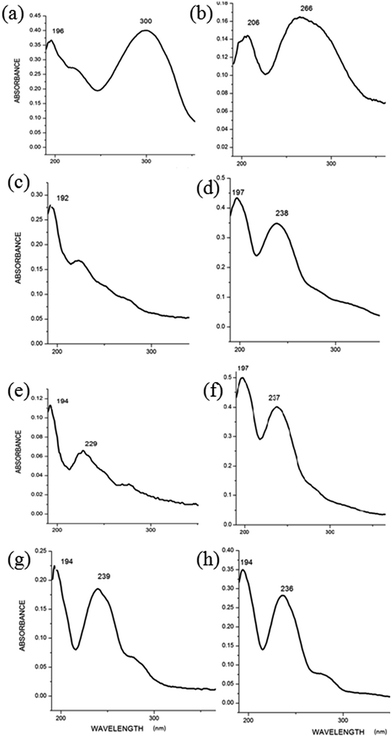 |
| | Fig. 8 UV-Visible absorption spectra of compounds in solid state (a) L1a; (b) L1b; (c) L2a; (d) L2b; (e) L3a; (f) L3b; (g) L4a; (h) L4b. | |
Table 1 UV-Visible absorption maxima for compounds L1a, L1b, L2a, L2b, L3a, L3b, L4a and L4b
| Compd |
Solid state (λmax in nm) |
Compds in MeOH (conc. 10−4 M) (λmax in nm) |
| L1a |
196, 300 |
206, 299 |
| L1b |
206, 266 |
205, 265 |
| L2a |
192, 223 |
209, 247 |
| L2b |
197, 238 |
207, 241 |
| L3a |
194, 229 |
204, 223 |
| L3b |
197, 237 |
205, 237 |
| L4a |
194, 239 |
203, 223 |
| L4b |
194, 236 |
206, 237 |
The UV-Visible absorption spectra in methanolic solution for the compounds L1a, L1b, L2a, L2b, L3a, L3b, L4a and L4b showed red shift (7–12 nm) in the π → π* peak position compared to their solid state spectra (Fig. S30–S37†); which indicates the possibility of parallel stacking of aromatic rings in solid state to form face-to-face stacking of the aromatic moieties/H-aggregates.13
Photoluminescence spectra of L2a and L2b
The PL spectra of L2a and L2b in solid state as well as in methanolic solutions showed the enhanced emission properties with increase in concentration. The observation from the concentration dependent PL spectra of L2a showed the appearance of a new peak on increasing concentration.
At 10−4 M concentration, by fixing the excitation wavelength at 300 nm, structured bands in PL spectra was observed with λmax at 330 and 364 nm, whereas at 10−3 M concentration, an extra shoulder peak appeared at 426 nm and at 5 × 10−1 M, band at around 359 nm disappeared completely and the only peak at 426 nm was observed. But when the excitation wavelength was fixed at 450 nm, concentration lower than 10−3 M didn't show any emission and on increasing the concentration to 10−1 M, an intense emission peak appeared at 518 nm (Fig. 9). The PL spectra of L2b showed a structured emission spectra (λmax at 330 and 365 nm) at a concentration of 10−4 M, when the excitation wavelength was kept at 300 nm while a shoulder peak at 424 nm appeared at 10−3 M. On changing the excitation wavelength to 450 nm, 10−1 M concentration showed an intense peak at 500 nm (Fig. 10). The PL spectra of L2c and L2d was reported previously by our group. In L2d, the PL spectra showed an increase in emission intensity along with a red shift of the peaks and the solid state PL showed a λmax at 453 nm when excitation wavelength is at 400 nm. The PL spectra of L2c showed the appearance of new peak on increasing concentration. It was suggested that the presence of pyridyl group resulted in C–H⋯N interaction and hence a non covalently bonded species appeared at high concentration.11
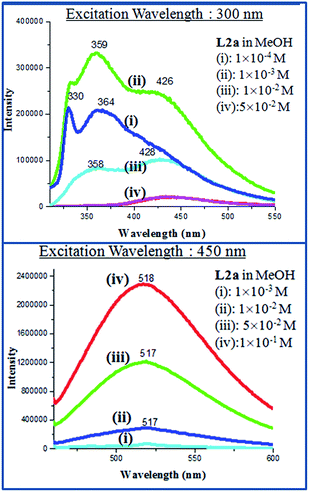 |
| | Fig. 9 PL spectra of L2a in different concentrations. | |
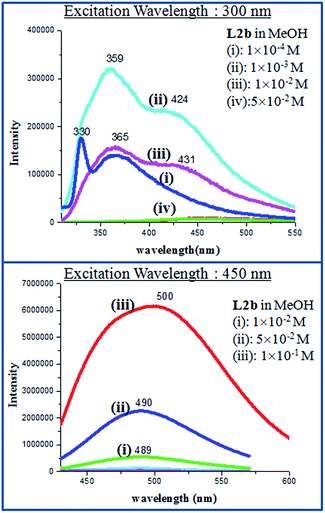 |
| | Fig. 10 PL spectra of L2b in different concentrations. | |
Photoluminescence spectra of L3a, L3b, L4a and L4b
The PL spectra of L3a, L3b, L4a and L4b in methanolic solution also showed enhanced emission on increasing the concentration. The compound L3a showed a strong emission spectra at λmax 514 nm (excitation wavelength at 440 nm) with a 10−1 M concentration, while L3b showed the emission maxima at 508 nm (excitation wavelength at 450 nm) with a 10−1 M concentration (Fig. 11, 12 and S38†). The compounds L4a and L4b at concentration of 10−1 M also showed emission maxima at 519 nm (excitation wavelength at 450 nm) and 420 nm (excitation wavelength at 493 nm), respectively (Fig. S39†). The PL spectra of L3c showed appearance of new peak at high concentration while L3d showed intensity increase along with red shift as reported previously by us.
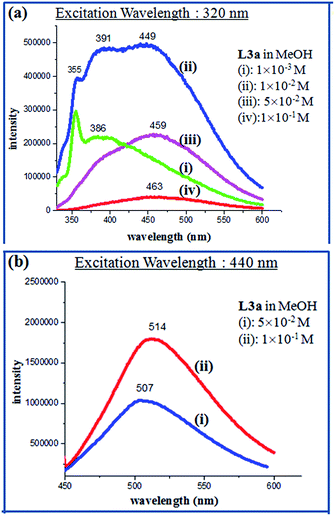 |
| | Fig. 11 PL spectra of L3a in different concentrations. | |
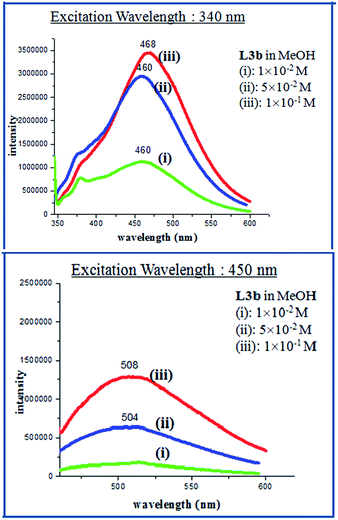 |
| | Fig. 12 PL spectra of L3b in different concentrations. | |
Solid state PL spectra
The PL spectra of compounds L1a, L1b, L2a, L2b, L3a, L3b, L4a and L4b were recorded in their powder form and crystalline form to get a comparative analysis of the emissions in different states. Compounds L1a and L1b didn't show any PL spectra in powder as well as in crystalline state. The solid state PL of L2a showed an intense peak at 531 nm when the excitation wavelength was fixed at 350 nm, while L2b showed an intense peak at 549 nm. The solid state PL of L2a, L2b, L3a, L3b, L4a and L4b are shown in Fig. S40–S44.† Tables 2 and S1† shows the comparison of λmax. For the compounds in solid state and in different concentrations in MeOH.
Table 2 λmax in the PL spectra of the compounds
| |
λmax in methanolic solution of different concentration |
λmax in solid |
| L2a |
330, 364 (Excitation 300 nm; 10−4 M) |
439 (Excitation 300 nm; 10−1 M) |
531 (Excitation 350 nm) |
| 518 (Excitation 450 nm; 10−4 M) |
518 (Excitation 450 nm; 10−1 M) |
| L2b |
330, 366 (Excitation 300 nm; 10−4 M) |
464 (Excitation 300 nm; 10−1 M) |
549 (Excitation 350 nm) |
| 489 (Excitation 450 nm; 10−4 M) |
500 (Excitation 450 nm; 10−1 M) |
| L3a |
365, 386 (Excitation 320 nm; 10−3 M) |
463 (Excitation 320 nm; 10−1 M) |
524 (Excitation 440 nm) |
| 479 (Excitation 420 nm; 10−4 M) |
509 (Excitation 420 nm; 10−1 M) |
| L3b |
376, 460 (Excitation 340 nm; 10−2 M) |
382 (Excitation 320 nm; 10−1 M) |
510 (Excitation 450 nm) |
| 478 (Excitation 420 nm; 10−4 M) |
504 (Excitation 450 nm; 10−2 M) |
| L4a |
300, 370 (Excitation 280 nm; 10−4 M) |
514 (Excitation 280 nm; 10−1 M) |
515 (Excitation 440 nm) |
| 517 (Excitation 450 nm; 10−4 M) |
517 (Excitation 450 nm; 10−2 M) |
| L4b |
350, 420 (Excitation 320 nm; 10−3 M) |
465 (Excitation 320 nm; 10−1 M) |
534 (Excitation 460 nm) |
| 484 (Excitation 420 nm; 10−4 M) |
490 (Excitation 420 nm; 10−2 M) |
Concentration and excitation dependence of PL
The steady state PL measurements have shown that the emission of the compounds depends on concentration as well as excitation wavelength. On increasing concentration, at an excitation wavelength of around 300 nm, a red shift of emission maxima is observed along with the disappearance of peak in the region of 300 nm (Table 2). Fig. 13 shows the absorption and emission spectra of compound L2a. The absorption and emission wavelengths have very less overlap in the 300 nm range. Table 3 shows the molar extinction coefficients of UV-Vis spectra of L2a in wavelengths near 300 nm. Although the molar extinction coefficient values are very less in the 300 nm region, but reabsorption at higher concentration can be expected and to some extent may be reason for disappearance of peak at high concentration. Fig. S45 and Table S2† shows the overlap of absorption and emission spectra (at an excitation ∼300 nm) and molar extinction coefficient of UV-Vis spectra in wavelength region 300 nm, respectively of the compounds L2b, L3a, L3b, L4a and L4b.
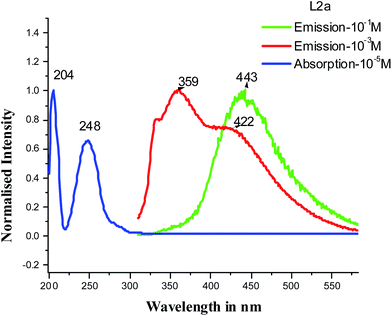 |
| | Fig. 13 Absorption and emission spectra (excitation 300 nm) of L2a. | |
Table 3 Molar extinction coefficient of UV-Vis spectra of L2a in wavelengths near 300 nm
| Compounds |
Molar extinction coefficient (ε) (M−1 cm−1) |
Wavelength in nm |
| L2a |
10−3 M |
195.2 |
330 |
| 10−4 M |
829 |
330 |
| 1095 |
300 |
| 3533 |
289 |
| 4793 |
280 |
| 10−5 M |
3190 |
330 |
| 4510 |
300 |
| 7970 |
289 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 610 610 |
280 |
| 61690 |
248 |
| 91310 |
205 |
Further, the dependence of emission wavelength on the excitation wavelength is observed from Tables 2 and S1.† The dependence is more pronounced at lower concentration. In L2a, at 10−4 M concentration, emission maxima are observed at 330 nm, 364 nm (when the excitation wavelength is 300 nm) while emission maxima is observed at 518 nm (when the excitation wavelength is 450 nm). This dependence clearly shows the formation of aggregate species, which are responsible for different emission peaks at different excitation.
Time-resolved fluorescence technique for lifetime analysis
With the help of the time-correlated single-photon counting (TCSPC) technique, we have measured the excited-state decay curve of the compounds (Fig. S46–S51†). The compounds showed a tri-exponential decay, with three lifetimes in the range of 1–2 ns, 4–7 ns and 0.1–0.2 ns at concentration more than 10−4 M. The shortest lifetime may be attributed to the deactivation (quenching) of excited aromatic groups while the longer lifetime is associated with the unquenched decay of the compounds. Tables 4 and S3† summarize the lifetime measurements of the compounds.
Table 4 Fluorescence lifetime data for L2a, L2b, L3a, L3b, L4a and 4b
| λexcitation = 375 nm |
τ1 (ns) |
τ2 (ns) |
τ3 (ns) |
τavg (ns) |
| L2a |
| 10−1 λemission 520 nm |
1.55 (α1 0.30) |
5.66 (α2 0.10) |
0.23 (α3 0.60) |
1.153 (χ2 1.24) |
| 10−4 λemission 449 nm |
1.23 (α1 0.35) |
0.26 (α2 0.54) |
4.69 (α3 0.11) |
1.076 (χ2 1.05) |
|
| L2b |
| 10−1 λemission 457 nm |
1.48 (α1 0.38) |
4.17 (α2 0.13) |
0.23 (α3 0.49) |
1.209 (χ2 1.15) |
| 10−4 λemission 457 nm |
1.51 (α1 0.37) |
5.92 (α2 0.08) |
0.21 (α3 0.55) |
1.172 (χ2 1.11) |
| 10−6 λemission 457 nm |
0.79 (α1 0.97) |
4.56 (α2 0.03) |
— |
0.90 (χ2 1.40) |
|
| L3a |
| 10−1 λemission 500 nm |
1.39 (α1 0.21) |
5.43 (α2 0.11) |
0.17 (α3 0.0.68) |
1.00 (χ2 1.23) |
| 10−2 λemission 463 nm |
0.99 (α1 0.19) |
4.53 (α2 0.09) |
0.12 (α3 0.72) |
0.682 (χ2 1.11) |
|
| L3b |
| 10−1 λemission 477 nm |
2.50 (α1 0.19) |
7.43 (α2 0.24) |
0.24 (α3 0.57) |
2.416 (χ2 1.14) |
| 10−5 λemission 473 nm |
6.24 (α1 0.93) |
4.83 (α2 0.07) |
— |
0.903 (χ2 1.34) |
|
| L4a |
| 10−1 λemission 464 nm |
1.93 (α1 0.17) |
6.90 (α2 0.11) |
0.15 (α3 0.72) |
1.201 (χ2 1.20) |
| 10−4 λemission 460 nm |
0.86 (α1 0.43) |
5.29 (α2 0.11) |
0.19 (α3 0.46) |
1.021 (χ2 1.21) |
|
| L4b |
| 10−1 λemission 540 nm |
1.42 (α1 0.11) |
5.52 (α2 0.37) |
0.19 (α3 0.52) |
2.29 (χ2 1.18) |
| 10−2 λemission 530 nm |
1.56 (α1 0.09) |
6.02 (α2 0.36) |
0.17 (α2 0.36) |
2.39 (χ2 1.29) |
Methanolic solution of L2b of concentrations 10−1 M, and 10−4 M showed a tri exponential decay when excited at 375 nm and emission was monitored at 457 nm, while a bi exponential decay was observed at a concentration of 10−6 M. The average lifetime of 10−1 M was 1.209 ns, 10−4 was 1.17 and 10−6 M was 0.90 ns (Fig. 14). The concentration dependence of the TCSPC profile of the L2b suggested the aggregation of the molecules at higher concentration. The increase in the average lifetime with concentration suggests that with aggregation in concentrated solution, the unquenched decay processes increases, which may be considered to be aggregation induced emission.
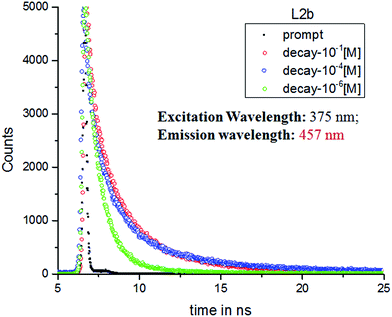 |
| | Fig. 14 TCSPC decay profiles of L2b in different concentration in MeOH with excitation at 375 nm. | |
Fluorescence quantum yield
Fluorescence quantum yield was determined for the compounds L2a, L2b, L3a, L3b, L4a and L4b in spectroscopic grade methanol using optically matching solutions of quinine sulfate (Φf = 0.546 in 0.5H2SO4 at an excitation wavelength of 345 nm) as standard. In solid state, sample was mixed with BaSO4 to measure the absolute quantum yield.
It was observed that the quantum yield of the compounds in dilute solution (10−4 M in MeOH) was in the range of 0.89–1.98, while on increasing the concentration to 10−1 M, Φf were observed in range of 1.52–7.58. The absolute quantum yield in solid state showed a further increase and values were in the range 2.28–18.68. An interesting trend was observed in the quantum yield for these compounds (Table 5). The compounds L2a, L3a and L4a showed a higher quantum yield than that of L2b, L3b and L4b, respectively. Further, on increasing the alkyl spacer length, the quantum yield was observed to increase.
Table 5 Absolute quantum yield in solution and in solid
| Compd |
10−4 M |
10−1 M |
Solid |
Compd |
10−4 M |
10−1 M |
Solid |
| L2a (Φf) |
1.06 |
4.04 |
5.81 |
L2b (Φf) |
0.89 |
1.52 |
2.28 |
| Excitation (nm) |
300 |
450 |
350 |
Excitation (nm) |
300 |
450 |
350 |
| Emission (nm) |
365 |
515 |
530 |
Emission (nm) |
365 |
515 |
545 |
| L3a (Φf) |
1.24 |
5.48 |
13.57 |
L3b (Φf) |
1.09 |
2.68 |
9.98 |
| Excitation (nm) |
320 |
440 |
440 |
Excitation (nm) |
320 |
450 |
420 |
| Emission (nm) |
380 |
505 |
524 |
Emission (nm) |
390 |
505 |
492 |
| L4a (Φf) |
1.98 |
7.58 |
18.68 |
L4b (Φf) |
1.32 |
4.48 |
14.46 |
| Excitation (nm) |
340 |
450 |
450 |
Excitation (nm) |
340 |
420 |
460 |
| Emission (nm) |
370 |
515 |
515 |
Emission (nm) |
370 |
490 |
530 |
Correlating the enhanced emission of the compounds with their crystal structure
The absence of fluorescence in L1a, L1b, L1c and L1d can be explained by observing their crystal structures. In the solid state of L1a, the molecule has planar geometry. The aromatic rings of the neighboring molecules are tilted and inclined face-to-face stacking is observed. The UV-Visible absorption maxima of crystalline solid L1a showed a blue shift compared to that in MeOH (10−4 M). Both the crystal structure as well as the absorption spectra indicated that in L1a, the formation of H-aggregates in the solid state and in concentrated solution, which may be attributed to the absence of fluorescence.
In L1b, the non planar geometry of the molecule along with face-to-face stacking of the molecules has resulted in quenching of fluorescence, while in L1d, quenching of fluorescence is explained by the dimer formation in the solid state (Scheme 2). Although the crystal structure of L1c is not reported, we can speculate that non radiative decay due to the head-to-head arrangement of the molecules.
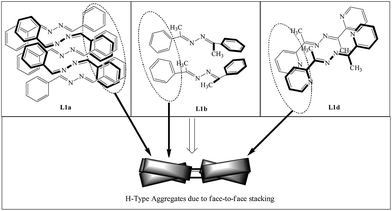 |
| | Scheme 2 Arrangement of the molecules of L1a, L1b and L1d in the solid state; notice the face-to-face stacking of the molecules. Although it is inclined in case of L1a and L1b, no fluorescence is observed in solid state. | |
In L2b, the molecules attain planarity in the solid state and face-to-face stacking of the molecules is observed. This is similar to the formation of H-aggregates, which is defined as the parallel alignment of the molecules and blue shifted absorption bands. The blue shifted UV-Visible bands (about 8 nm) and the crystal structure of L2b shows that they form H-aggregates. The appearance of the new peak in the PL spectra at higher concentration and red shift of that peak indicates that a new emissive species has formed in the high concentration. The crystal structure analysis shows that although H-aggregates are formed in L2b, presence of ethyl spacer is preventing the non radiative decay processes. The crystal structures of L2c, L3a, L3b, L4a and L4b are not reported, but the similar trend in UV-Visible absorption spectra and PL spectra indicates that the solid state arrangement of the molecules may involve parallel face-to-face stacking of the molecules (or H-aggregates) (Scheme 3).
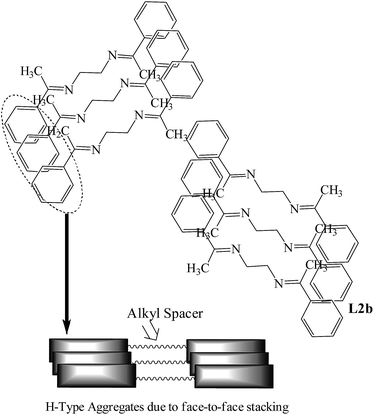 |
| | Scheme 3 Arrangement of the molecules of L2b; L2a, L3a, L3b, L4a and L4b are also expected to have similar arrangements in the solid state. Notice the face-to-face stacking of the molecules. | |
In L2d, the attainment of the planar geometry and offset arrangement of molecules resulted in increased emission in PL spectra on increasing concentration and in the solid state and similar behavior is expected for L3d in solid state (Scheme 4).
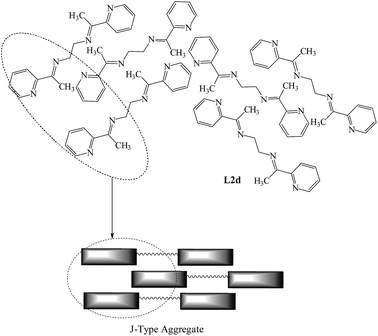 |
| | Scheme 4 Arrangement of the molecules of L2d and L3d in the solid state; notice the offset stacking of molecules. | |
In L2c and L3c, it was reported previously by our group, that the formation of a new “chromophore” species at concentrated solution and in solid state resulted in increased emission spectra. The crystal structure analysis of L3c showed that the non covalent interactions led to the arrangement of the molecules in “non covalently bonded macrocycles” (Scheme 5).
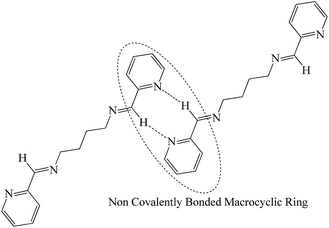 |
| | Scheme 5 Arrangement of the molecules of L2c and L3c in the solid state; notice the formation of new “chromophore” on aggregation and in the solid state. | |
NMR assay to detect the aggregation in concentrated solution
The effect of changing the concentration on the NMR spectra of aggregating compounds include changes in the chemical shifts, shape and intensity of the peaks on changing the concentration.14 The 1H-NMR of compounds L1a, L1b, L2b and L3b were recorded in different concentrations in CDCl3. The indication of the possible molecular arrangement on aggregation in concentrated solution may be obtained from final arrangement of the molecules in the solid state.
The concentration dependent 1H-NMR of L1a showed that the aromatic protons and the methine protons resonated in deshielded region when the concentration was increased from 0.01 M to 1 M (Fig. 15). The aggregation of molecules at higher concentration has resulted in the change in chemical shift of the protons. From the crystal structure analysis of L1a, it was observed that the molecules have tilted face-to-face arrangement and the aromatic moieties are clustered together.
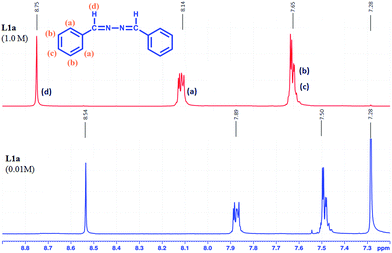 |
| | Fig. 15 1H NMR spectra of L1a in different concentrations in CDCl3. | |
The concentration dependent 1H NMR spectra of L2b and L3b was recorded in CDCl3 (Fig. 16 and S52†). In both the cases, it was observed that the protons in the aromatic region resonated in the deshielded region when the concentration was increased from 10−2 M to 1 M, whereas the methyl protons resonated in the shielded region on increasing the concentration. The crystal structure analysis of L2b showed the parallel stacking of the aromatic rings, which may be associated with the deshielding effects of aromatic protons on increasing concentrations, while on closely observing the structure, it was seen that the herringbone arrangement of the parallel stacked layer resulted in positioning of the methyl groups in such a way that they fall just above the aromatic rings (Scheme 6), which explains the shielding effect of the methyl protons on increasing concentration.
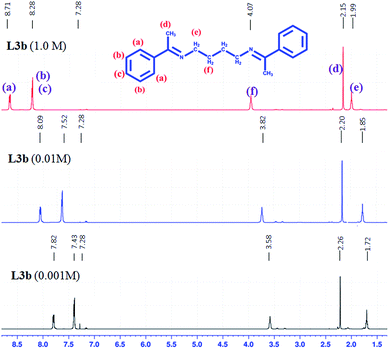 |
| | Fig. 16 1H NMR spectra of L3b in different concentrations in CDCl3. | |
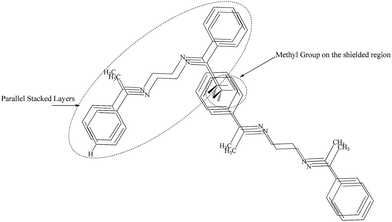 |
| | Scheme 6 Arrangement of the molecules in L2b and L3b resulted in deshielded aromatic proton and shielded methyl protons. | |
The concentration dependent 1H-NMR of L2d in CDCl3 showed that the increase in concentration from 10−2 M to 1 M resulted the aromatic proton to resonate in shielded region (Fig. 17 and S53†). The shielded aromatic proton in concentrated solution of L2d may be due to the offset arrangement of the molecules. So face-to-face stacking of aromatic rings are absent in L2d, which in case of L1a, L2b and L3b has resulted in deshielding effects.
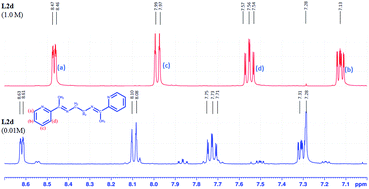 |
| | Fig. 17 1H NMR spectra of L2d in different concentrations in CDCl3. | |
The NOESY of L2b at 2 M concentration in CDCl3 was taken to observe the interaction between different types of protons at higher concentration due to aggregation. The spectrum showed interaction between the aromatic protons ‘a’ and aromatic protons ‘b’, ‘c’ & between aromatic protons (‘a’, ‘b’, ‘c’) and the methyl proton (‘d’) (Fig. 18). This suggested that at high concentration, the methyl protons interact with the aromatic protons and a structure similar to Scheme 6 may be possible. Further the interaction between the aromatic protons also suggest a face-to-face stacking of the molecules at higher concentration.
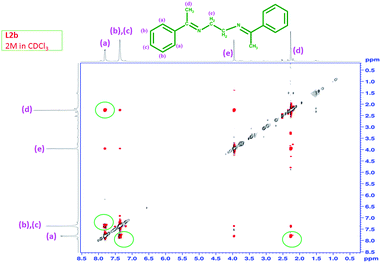 |
| | Fig. 18 NOESY of L2b in CDCl3 at a concentration of 2 M. | |
Conclusions
Systematic study of unique AIE properties of di Schiff base compounds were carried out. The crystal structure analysis of some of these compounds have given us the idea about interplay of non covalent interactions and crystal packing on the photo physical properties of the compounds. Apart from the compounds with hydrazine spacer i.e. L1, all the other compounds have small π-conjugated systems linked with alkyl spacers. In L1, where extended conjugation is present throughout the molecule didn't show any fluorescence, while other compounds with alkyl spacer showed good emission properties. The emission properties of these compounds are related to the arrangement of the molecules in aggregated states. In L1, the H-aggregate formation/dimer formation resulted in quenching of fluorescence. In L2b, the H-aggregate formation was evident from crystal structure and UV-Visible absorption spectra, but AIE properties were observed. The alkyl spacer in L2b is preventing the non-radiative decay processes usually observed for H-aggregated. The compounds L2a, L3a, L3b, L4a and L4b were also expected to show similar packing as that of L2b and the enhanced emission properties of these compounds are attributed to the presence of the alkyl spacers.
Acknowledgements
We gratefully acknowledge the financial support from DST (SR/FT/CS-24/2011) and Instrumentation facility from the DST FIST & UGC-SAP to the Department of Chemistry, BITS Pilani, Pilani Campus. We also acknowledge the DST FIST supported Powder XRD facility of the Department of Physics, BITS Pilani, Pilani Campus.
Notes and references
-
(a) T. W. Kelley, P. F. Baude, C. Gerlach, D. E. Ender, D. Muyres, M. A. Haase, D. E. Vogel and S. D. Theiss, Chem. Mater., 2004, 16, 4413 CrossRef CAS;
(b) C.-T. Chen, Chem. Mater., 2004, 16, 4389 CrossRef CAS;
(c) H. Yersin, Highly Efficient OLEDs with Phosphorescent Materials, Wiley-VCH, Weinheim, 2008 Search PubMed;
(d) H. N. Kim, Z. Q. Guo, W. H. Zhu, J. Yoon and H. Tian, Chem. Soc. Rev., 2011, 40, 79 RSC;
(e) J. S. Wu, W. M. Liu, J. C. Ge, H. Y. Zhang and P. F. Wang, Chem. Soc. Rev., 2011, 40, 3483 RSC;
(f) Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943 RSC;
(g) H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621 CrossRef CAS;
(h) M. Schaferling, Angew. Chem., Int. Ed., 2012, 51, 3532 CrossRef PubMed;
(i) M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963 RSC;
(j) A. M. Breul, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 5366 RSC.
-
(a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC;
(b) B. Z. Tang, X. Zhan, G. Yu, P. P. S. Lee, Y. Liu and D. Zhu, J. Mater. Chem., 2001, 11, 2974 RSC;
(c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC;
(d) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494 RSC.
-
(a) B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed;
(b) S. Y. Ryu, S. Kim, J. Seo, Y. W. Kim, O. H. Kwon, D. J. Jang and S. Y. Park, Chem. Commun., 2004, 70 RSC;
(c) Y. You, H. S. Huh, K. S. Kim, S. W. Lee, D. Kim and S. Y. Park, Chem. Commun., 2008, 3998 RSC;
(d) B. K. An, S. H. Gihm, J. W. Chung, C. R. Park, S. K. Kwon and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 3950 CrossRef CAS PubMed;
(e) J. W. Chung, B. K. An and S. Y. Park, Chem. Mater., 2008, 20, 6750 CrossRef CAS.
-
(a) Z. J. Ning, Z. Chen, Q. Zhang, Y. L. Yan, S. X. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799 CrossRef CAS;
(b) Z. Ning and H. Tian, Chem. Commun., 2009, 5483 RSC;
(c) B. Wang, Y. C. Wang, J. L. Hua, Y. H. Jiang, J. H. Huang, S. X. Qian and H. Tian, Chem.–Eur. J., 2011, 17, 2647 CrossRef CAS PubMed.
-
(a) Y. Hong, J. W. Y. Lama and B. Z. Tang, Chem. Commun., 2009, 4332 RSC;
(b) M. Levitus, K. Schmieder, H. Ricks, K. D. Shimizu, U. H. F. Bunz and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2001, 123, 4259 CrossRef CAS PubMed.
-
(a) S. P. Anthony, S. Varughese and S. M. Draper, J. Phys. Org. Chem., 2010, 23, 1074 CrossRef CAS;
(b) S. P. Anthony, S. Varughese and S. M. Draper, Chem. Commun., 2009, 7500 RSC.
- S. Varughese, J. Mater. Chem. C, 2014, 2, 3499 RSC.
-
(a) E. Hadjoudis and I. M. Mavridis, Chem. Soc. Rev., 2004, 33, 579 CAS;
(b) P. G. Cozz, Chem. Soc. Rev., 2004, 33, 410 RSC;
(c) M. Andruh, Chem. Commun., 2011, 47, 3025 RSC.
- T. Kawasaki, T. Kamata, H. Ushijima, S. Murata, F. Mizukami, Y. Fujii and Y. Usui, Mol. Cryst. Liq. Cryst., 1996, 286, 257 CrossRef.
-
(a) W. Tang, Y. Xiang and A. Tong, J. Org. Chem., 2009, 74, 2163 CrossRef CAS PubMed;
(b) R. Wei, P. Song and A. Tong, J. Phys. Chem. C, 2013, 117, 3467 CrossRef CAS;
(c) X. Chen and A. Tong, J. Lumin., 2014, 145, 737 CrossRef CAS;
(d) X. F. Ma, J. H. Cheng, J. Y. Liu, X. G. Zhou and H. F. Xiang, New J. Chem., 2015, 39, 492 RSC;
(e) J. Cheng, Y. Li, R. Sun, J. Liu, F. Gou, X. Zhou, H. Xiang and J. Liu, J. Mater. Chem. C, 2015, 3, 11099 RSC.
- F. Baig, Rajnikant, V. K. Gupta and M. Sarkar, RSC Adv., 2015, 5, 51220 RSC.
-
(a) L. N. Ferguson and T. C. Goodwin, J. Am. Chem. Soc., 1949, 71, 633 CrossRef CAS PubMed;
(b) U. C. Sinha, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 889 CrossRef CAS;
(c) G. S. Chen, M. Anthamatten, C. L. Barnes and R. Glaser, J. Org. Chem., 1994, 59, 4336 CrossRef CAS;
(d) R. E. Benson, T. G. Roy, B. K. Dey, K. K. Barua and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o1971 CAS;
(e) V. A. Sauro and M. S. Workentin, J. Org. Chem., 2001, 66, 831 CrossRef CAS PubMed;
(f) X. D. Tang, Z. J. Ding and Z. M. Zhang, Solid State Commun., 2009, 149, 301 CrossRef CAS;
(g) A. K. El-Qisairi, H. A. Qaseer, S. F. Alshahateet, M. K. H. Qaseer, M. H. Zaghal, W. Al-Btoush and L. N. Dawed, Croat. Chem. Acta, 2014, 87, 123 CrossRef.
-
(a) L. Wang, Y. Shen, M. Yang, X. Zhang, W. Xu, Q. Zhu, J. Wu, Y. Tiana and H. Zhou, Chem. Commun., 2014, 50, 8723 RSC;
(b) S. Basak, N. Nandi, A. Baral and A. Banerjee, Chem. Commun., 2015, 51, 780 RSC.
-
(a) A. Mitra, P. J. Seaton, R. A. Assarpour and T. Williamson, Tetrahedron, 1998, 54, 15489 CrossRef CAS;
(b) S. R. LaPlante, R. Carson, J. Gillard, N. Aubry, R. Coulombe, S. Bordeleau, P. Bonneau, M. Little, J. O'Meara and P. L. Beaulieu, J. Med. Chem., 2013, 56, 5142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR, 1H NMR, 13C NMR, UV absorption spectra, photoluminescence spectra and other supplementary material. See DOI: 10.1039/c6ra08582b |
|
| This journal is © The Royal Society of Chemistry 2016 |