
PLA-based thermogel for the sustained delivery of chemotherapeutics in a mouse model of hepatocellular carcinoma†
Abstract
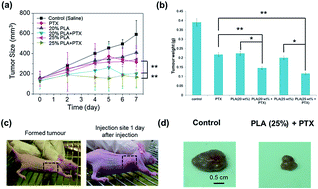
A thermogelling poly(ester urethane) comprising poly(ethylene glycol) (PEG), poly(propylene glycol) (PPG) and poly(lactic acid) (PLA) blocks was synthesized. Drug release studies of the thermogel were carried out using paclitaxel (PTX). The release rate of the drug can be achieved by changing the concentration of the gel, greatly prolonging the release and elimination times to afford long-term effects. The thermogels showed very low toxicity on HEK293 cells. A nude mice model of hepatocellular carcinoma was developed and intratumoural injection of drug-loaded thermogel showed that PTX-loaded thermogel effectively inhibited the growth of tumours.
- This article is part of the themed collection: Chemistry for Medicine: Special Collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

