
High dielectric constant and capacitance in ultrasmall (2.5 nm) SrHfO3 perovskite nanoparticles produced in a low temperature non-aqueous sol–gel route†
Abstract
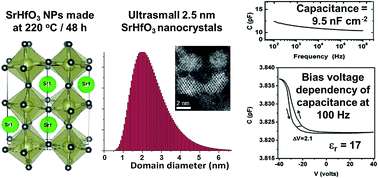
Strontium hafnium oxide (SrHfO3) has great potential as a high-k gate dielectric material, for use in memories, capacitors, CMOS and MOSFETs. We report for the first time the dielectric properties (relative permittivity and capacitance) of SrHfO3 nanoparticles (NPs), as opposed to thin films or sintered bulk ceramics. These monodisperse, ultra-small, perovskite-type SrHfO3 nanocrystals were synthesised through a non-aqueous sol–gel process under solvothermal conditions (at only 220 °C) using benzyl alcohol as a solvent, and with no other capping agents or surfactants. Advanced X-ray diffraction methods (whole powder pattern modelling, WPPM), CS-corrected high-resolution scanning transmission electron microscopy (HRSTEM), dielectric spectroscopy, and optical (UV-vis, Raman) and photoluminescent spectroscopy were used to fully characterise the NPs. These SrHfO3 NPs are the smallest reported and highly monodisperse, with a mean diameter of 2.5 nm, a mode of 2.0 nm and a small size distribution. The formation mechanism of the NPs was determined using NMR and GC-MS analysis of the species involved. Our SrHfO3 nanoparticles had a dielectric constant of 17, which is on par with literature data for bulk and thin film samples, and they also had a relatively large capacitance of 9.5 nF cm−2. As such, they would be suitable for applications as gate dielectrics for capacitors and in metal-oxide semiconductor field-effect transistor (MOSFET) technology.
 Please wait while we load your content...
Please wait while we load your content...

