
Effect of variations of CuIIX2/L, surface area of Cu0, solvent, and temperature on atom transfer radical polyaddition of 4-vinylbenzyl 2-bromo-2-isobutyrate inimers†
Chih-Feng Huang*a,
Shiao-Wei Kuob,
Daniela Moravčíkovác,
Jyun-Ci Liaoa,
Yu-Min Hana,
Ting-Han Leea,
Po-Hung Wanga,
Rong-Ho Leea,
Raymond Chien-Chao Tsiangd and
Jaroslav Mosnáček*c
aDepartment of Chemical Engineering, National Chung Hsing University, 250 Kuo Kuang Road, South District, Taichung 40227, Taiwan. E-mail: HuangCF@dragon.nchu.edu.tw
bDepartment of Materials and Optoelectronic Science, Centre for Nanoscience and Nanotechnology, National Sun Yat-Sen University, Kaohsiung 80424, Taiwan
cPolymer Institute, Slovak Academy of Sciences, Dúbravská cesta 9, Bratislava 84541, Slovakia. E-mail: Jaroslav.Mosnacek@savba.sk
dDepartment of Chemical Engineering, National Chung Cheng University, 168 University Road, Minhsiung Township, Chiayi 62102, Taiwan
First published on 12th May 2016
Abstract
An asymmetrical AB type inimer (4-vinylbenzyl 2-bromo-2-isobutyrate (VBBiB)) was synthesized and a variety of conditions for atom transfer radical polyaddition (ATRPA) of VBBiB was investigated regarding a CuX2/Cu0/ligand catalytic system (where, X = Br or Cl; ligand = 4,4′-dinonyl-2,2′-bipyridine (dNBpy), N,N,N′,N′′,N′′-pentamethyl diethylenetriamine (PMDETA) or 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA)). We focused on investigation of the effect of four reaction factors, i.e. type of ligand, Cu0 surface area, solvent and temperature on the final structure and molecular characteristics of the polyesters. The VBBiB/CuBr2/Cu0(wire)/PMDETA system showed formation of branched polymers due to very high activity of the catalyst complex. Replacing Cu0 wire with Cu0 powder, possessing higher surface area, resulted in only slight increase of the rate of inimer consumption. The polyaddition was more significantly affected by reaction temperature. Decreasing reaction temperature down to 0 °C increased significantly self-degradation by-products formation as a result of longer reaction times. Optimization of reaction conditions using commercially available catalytic systems allowed obtaining control over the polyester architecture and functionality, as well as ready achievement of functional linear polyesters with high molecular weight (Mw = 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200).
200).
1. Introduction
With the evolution of controlled/living polymerisations in previous decades, reversible-deactivation radical polymerisations (RDRPs) have been developed as a robust and versatile tool for the creation of unique macromolecules for application in many aspects, for example, electronic devices, medicine, pharmacy, biomedical, as well as agriculture and environmentally-friendly materials. To most of the RDRPs belong various variations of atom transfer radical polymerisation (ATRP),1–7 degenerative transfer polymerisation [including reversible addition–fragmentation chain transfer (RAFT) polymerisation8–10 and reversible iodine transfer polymerisation (RITP)],11 and stable free radical polymerisation [including nitroxide-mediated polymerisation (NMP)12,13 and organometallic radical polymerisation (OMRP)],14–16 as well as the recently developed organo-stilbene/-tellurium/-bismuthine radical polymerisations (SBRP/TERP/BIRP).17–19Nowadays, linear aliphatic polyesters are an important group of biocompatible and biodegradable materials.20–23 In addition to polyesterification and ring-opening polymerisations of lactones, the polyesters have also been alternatively synthesized by an innovative approach of radical ring-opening polymerisations (RROP) of particular cyclic monomers. Bailey et al. have extensively studied a family of cyclic ketene acetal (CKA) monomers to obtain polyesters through conventional free radical polymerisation.24–26 Various RDRP techniques were applied for RROP as well. Detailed mechanisms and characterization have been investigated using the ATRP technique to initiate RROP (i.e., ATRROP).27,28 Accordingly, Pan et al.29,30 and Matyjaszewski et al.20,31 have addressed the capability of ATRROP to afford polyesters efficiently. Hawker et al. elegantly introduced different tuneable biologically and chemically degradable structures to obtain linear aliphatic polyesters with controlled molecular weights.32 Moreover, some examples of synthesis of aliphatic polyesters by nitroxide-mediated radical ring-opening polymerisation (NMRROP)33–35 have been reported. Although the synthesis of the degradable polyesters by this promising method has attracted great interest, the RROP approach still faces difficulties in the preparation of functional monomers.
Taking advantage of mechanistic studies of atom transfer radical reactions, an emerging polymerisation technique, namely, atom transfer radical polyaddition (ATRPA),36,37 demonstrated success in providing a polyadduct through atom transfer radical addition (ATRA).38,39 This method provides a novel strategy for the synthesis of functional aliphatic polyesters. Furthermore, this technique can be extended to copolymerize some common vinyl monomers to proceed simultaneous chain- and step-growth radical polymerization.40–44 Accordingly, production of readily degradable analogues of most commodities becomes facile. Regarding an ATRA process, it generally occurs through the following mechanism (Scheme 1) to yield a 1:1 monoadduct: (i) an AB-type inimer with organic halide is activated (ka) by a lower-oxidation-state transition metal catalyst to produce a radical (1) and a higher-oxidation-state transition metal catalyst; (ii) the produced radical adds to a vinyl bond (kadd) to attain a monoadduct in radical form (2); and (iii) the monoadduct radical is deactivated (k′da) by the higher-oxidation-state metal complex to obtain a new organic halide species (3) and a lower-oxidation-state transition metal catalyst. The key for switching such a monoadduct of ATRA to a polyadduct of ATRPA is through the proper design of a set of A and B groups (i.e., A: = –R1– and B: –R2-X) to form a different type of alkyl halide (i.e., B′: –C(X)-R1–), which is inactive towards the metal catalyst. Kamigaito et al. have successfully achieved this radical polyaddition to synthesize aliphatic polyesters using AB type inimers with a 2-chloropropanoate-based initiating site and an allyl group36,45,46 while most of the chloro groups on the polyester backbone of the resulting polymers remained inactive. Nevertheless, the reaction times were in a range of days to weeks. An efficient ATRPA example was then depicted by Li et al. through the polyadduct between an α-bromoisobutyrate-based initiating site and a styrene group while the reaction was carried out at low temperature in the presence of a CuBr2/Cu0/BPMOA catalytic complex system.37,47 Methacrylate radicals that were efficiently generated from an α-bromoisobutyrate-based initiating site not only reacted with a styrene, but also formed a dormant benzyl bromide group at 0 °C.
 | ||
| Scheme 1 Mechanism of ATRPA for aliphatic polyesters (i.e., R2 = ester group). | ||
Based on mechanistic understanding, our previous study indicated that the ATRPA of an AB type inimer through proper manipulation of “freezing/adding” processes can provide linear polyesters even at high temperature using a commercially obtainable copper complex.48 Accordingly, a new catalogue of functional aliphatic polyesters can be obtained through control of the various activation/deactivation balances of the initiator structures when using ATRPA. In this context, an asymmetrical AB type inimer (4-vinylbenzyl 2-bromo-2-isobutyrate (VBBiB)) was investigated under a variety of ATRPA conditions using a commercially available copper complex system (Scheme 2) to optimize the conditions for the preparation of linear functionalized degradable aliphatic polyesters and to enrich the scope of this newly developed technique.
 | ||
| Scheme 2 Preparation of aliphatic polyester PVBBiB by ATRPA. | ||
2. Experimental
2.1. Materials
Copper(II) bromide (CuBr2, 99%), copper(II) chloride (CuCl2, 99%), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA, 99%), 4,4′-dinonyl-2,2′-bipyridine (dNBpy, 97%), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 97%), alumina (neutral), Dowex 50WX8 (H form/200–400 mesh) and 4-vinylbenzyl chloride (VBC, 90%) were purchased from Sigma-Aldrich. Copper wire (d = 0.5 mm) and copper powder (150 mesh, 99.5%) were purchased from Alfa. 4-Vinylbenzyl 2-bromo-2-isobutryrate (VBBiB) was synthesized from VBC as described previously.47,48 All solvents were distilled prior to use.2.2. ATRPA of VBBiB
An example for ATRPA of VBBiB: A Schlenk flask was charged with anisole (4 mL), VBBiB (16.0 mmol), CuBr2 (0.32 mmol), and PMDETA (0.96 mmol) and the mixture was degassed through freeze–pump–thaw cycles three times. The flask was backfilled with N2 and copper powder (0.32 mmol) was added to the frozen solution. The flask was fastened and degassed through two additional freeze–pump–thaw cycles (VBBiB/CuBr2/Cu0(p)/dNBpy = 50/1/2/6; [VBBiB]0 = 1.8 M). A t0 sample was collected and the reaction mixture was stirred in an ice bath at 0 °C. Samples were taken in defined time intervals via syringes to monitor inimer conversion by GC and changes in molecular weights by GPC. The polymerisation was stopped by cooling the flask in an ice bath, exposing the sample to air and diluting it with THF. The mixture was stirred with Dowex 50WX8 for a period of time, passed through a fresh alumina column, and poured into cold petroleum ether. The white polyesters were collected, dried overnight under vacuum, and stored in the fridge.2.3. Characterization
1H NMR spectra were obtained with a Varian Inova 400 in CDCl3, which was also utilized as a chemical shift standard (7.26 ppm). A Nicolet Avatar 320 FT-IR spectrometer was used to obtain FT-IR spectra (32 scans at a resolution of 1 cm−1). The sample solution was drop-casted onto a KBr disk and dried under vacuum; the measuring chamber was purged with nitrogen. Inimer conversion was determined using anisole as an internal standard through a Hewlett Packard 5890 series II gas chromatograph equipped with an FID detector and a 30 m CNW CD-5 column. The gel permeation chromatography (GPC) instrument was equipped with a pump (Waters 515; flow rate of 1 mL min−1; eluent: tetrahydrofuran (THF)), a differential refractometer (Waters 410), an absorbance detector (Waters 486; detecting wavelength = 254 nm), and two PSS SDV columns in a series of linear S and 100 Å at 40 °C to determine the average molecular weights Mn and Mw and dispersity Mw/Mn (abbreviated as Đ in context). Calibration based on monodisperse polystyrene (PSt) standards was performed.3. Results and discussion
3.1. Ligand effects for ATRPA of VBBiB
An important factor in ATRPA is the activity of the catalytic system. When the activity is very high, both the original alkyl halide and the new formed alkyl halide can be activated and a branched polymer can be obtained. Opposite to that when the activity of the catalytic system is too low, the reaction rate of ATRPA can be too slow. Therefore, first, the effect of ligand structure, influencing the activity of the catalyst complex, on the rate of polyaddition and topology of the final polymer was investigated (Table 1, entries 1 and 2). Based on KATRP, recently determined for polymerisation systems involving PMDETA and dNBpy ligands,49 it could be anticipated that a faster polymerisation rate should be observed for a VBBiB/CuBr2/Cu wire/PMDETA system than that for VBBiB/CuBr2/Cu wire/dNBpy. However, as observed from Fig. 1A, the inimer conversion was found to be faster when dNBpy was used as a ligand. It has been reported that using excess of some multi-dentate ligands such as PMDETA with respect to a CuIIBr2 compound may lead to the formation of a poorly soluble metal complex network.50,51 Therefore, using PMDETA ligands can lead to the partial precipitation of the copper complex and thus decrease of the total concentration of copper catalyst dissolved in the polymerisation mixture. A lower concentration of CuI complex (activator) was thus obtained and provided a slower polymerisation rate (i.e., curve (a) in Fig. 1A).| Entrya | Compositiona | Catalyst | Temp. (°C) | Time (h) | Conv. (%) | Mwb | Đb |
|---|---|---|---|---|---|---|---|
| a Compositions consisted of VBBiB/CuX2/Cu0/Ligand, respectively; entries 1, 2, 4 and 8 were conducted with Cu0 wire (L = 2.9 mm; d = 0.5 mm) and entries 3, 5–7, and 9 were conducted with Cu0 powder (150 mesh).b Mw and Đ were determined by GPC using THF as an eluent and PSt standards. | |||||||
| 1 | 50/1/2/3 | CuBr2/PMDETA | 0 | 456 | 98 | 8190 | 2.58 |
| 2 | 50/1/2/6 | CuBr2/dNBpy | 0 | 125 | 88 | 1750 | 1.35 |
| 3 | 50/1/2/6 | CuBr2/dNBpy | 0 | 167 | 85 | 2070 | 2.35 |
| 4 | 50/1/2/6 | CuCl2/dNBpy | 10 | 144 | 98 | 10410 |
2.95 |
| 5 | 50/0.5/1/3 | CuCl2/dNBpy | 10 | 60 | 92 | 4200 | 1.76 |
| 6 | 50/1/2/6 | CuCl2/dNBpy | 0 | 219 | 91 | 2110 | 1.60 |
| 7 | 50/1/2/6 | CuCl2/dNBpy | 10 | 65 | 95 | 4500 | 1.80 |
| 8 | 50/1/2/6 | CuCl2/dNBpy | 25 | 72 | 97 | 3200 | 1.50 |
| 9 | 50/1/2/6 | CuBr2/dNBpy | 10 | 45 | 98 | 16200 |
2.47 |
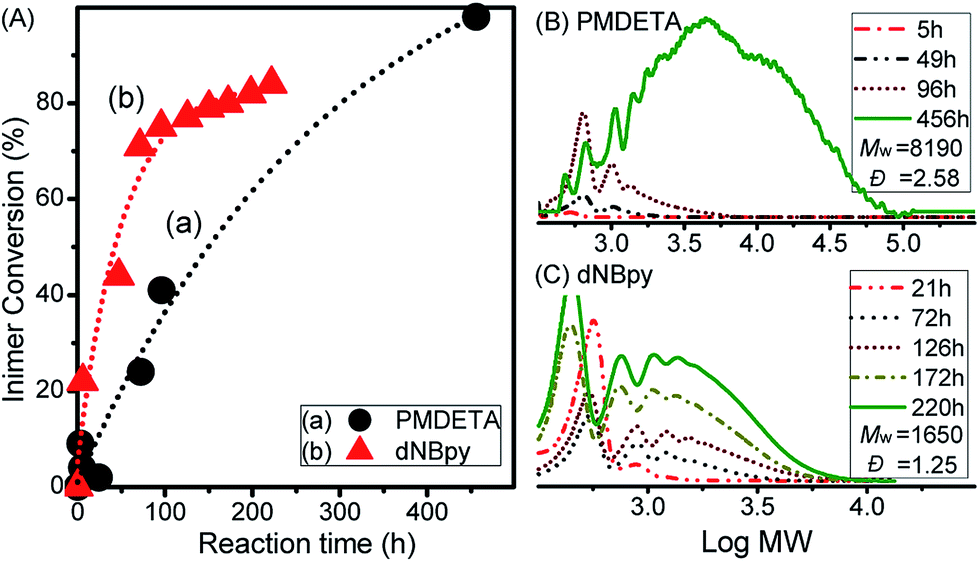 | ||
| Fig. 1 Results of ATRPA of VBBiB: (A) VBBiB conversion with the polyaddition time; (B, C) GPC traces for reactions performed using (B) PMDETA and (C) dNBpy ligand recorded at various polyaddition times. Polyaddition conditions were as follows: VBBiB/CuBr2/Cu0 wire/(PMDETA or dNBpy) = 50/1/2/(3 or 6), respectively, with ([VBBiB]0 = 1.8 M; L = 5.8 mm; d = 0.5 mm) in anisole at 0 °C. | ||
GPC evolutions during the polyadditions for both ligands are displayed in Fig. 1B and C. A clear shift towards higher molecular weight (MW) products was observed in both cases, even though some differences were obvious after longer polyaddition time. While the polymer with higher MW (Mw = 8190, Đ = 2.58) was formed after 456 h with the VBBiB/CuBr2/Cu0/PMDETA system (Fig. 1B), lower molecular weight compounds were formed in the case of polyaddition with the VBBiB/CuBr2/Cu0/dNBpy system (Fig. 1C) after reaction time above 130 h.
The newly formed B′ moiety (i.e., phenyl ethyl bromide (PEBr)) in the backbone might be re-activated, depending on the activity of CuI/L complex (e.g., ka,PEBr(PMDETA)/ka,PEBr(dNBpy) = 4.6 at 25 °C).49 To obtain information about the product structures, both of the obtained polymers were analyzed by 1H NMR spectroscopy. Unlike the case for a linear aliphatic polyester structure, peak broadenings were observed using PMDETA as a ligand in Fig. 2A (i.e., the peaks of d at 5.16 ppm, e/e′ at 5.06 ppm, d′/d′′ at 4.75 ppm, and f at 2.4–2.8 ppm). As mentioned above, the formation of a poorly soluble metal complex network when using excess of multi-dentate ligands (herein, PMDETA) with respect to CuIIBr2 compound leads to decrease of dissolved deactivators. Thus, the lower concentration of deactivators combined with the known higher activity of the CuI/PMDETA complex compared to the dNBpy one resulted in higher probability of reaction of radicals, which formed along the polymer chains, with vinyl groups to form a branched structure. According to previous literature,4,52 the degree of branching (DB) can be further estimated from Fig. 2A (i.e., DB = [2(number of branched units)]/[(total number of units) − 1] = [2(d/b − 1)]/[(d + d′ + d′′)/b − 1]. DB of 0.35 was obtained, implying a highly branched structure.
 | ||
| Fig. 2 1H NMR spectra (400 MHz, CDCl3) of the reaction mixtures from: (A) after 456 h of ATRPA using PMDETA as a ligand and (B) after 220 h of ATRPA using dNBpy as a ligand. ATRPA conditions were as follows: VBBiB/CuBr2/Cu0 wire/(PMDETA or dNBpy) = 50/1/2/(3 or 6), respectively, with ([VBBiB]0 = 1.8 M; L = 5.8 mm; d = 0.5 mm) in anisole at 0 °C. | ||
When dNBpy was used as a ligand, formation of an aliphatic polyester structure can be observed, however, without formation of branched structures (Fig. 2B). Therefore, the balance of metal complex solubility and activity are crucial for obtaining aliphatic polyester structures in a controlled manner. In this case, however, some peaks other than those from the polyester chain were observable in the NMR spectra. We have recently reported that different backbone structures of aliphatic polyesters showed differing tendency to undergo self-degrading side reactions.48 Thus, the slightly visible main peaks of 1, 3, 5, and 7 in Fig. 2B can be assigned to by-products formed after cleavage of the polyester chain accompanied with formation of lactones. The side reaction of lactonization was outlined in Scheme 3.48
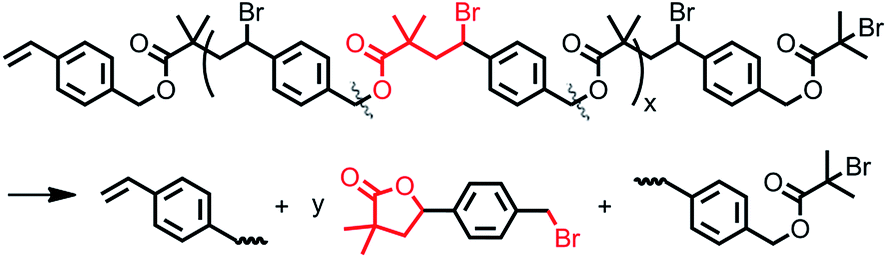 | ||
| Scheme 3 Self-degrading reactions of aliphatic polyesters (i.e., lactonization). | ||
3.2. Influence of Cu0 surface area and solvent on ATRPA of VBBiB
To vary the overall catalytic activity, our next attempt focused on investigation of the effect of Cu0 surface area by using wire (Cu(w)) or powder (Cu(p)) and type of copper salt using CuBr2 (Fig. 3). By comparing the influence of the surface area of Cu0 on the rate of inimer conversion during polyadditions performed at 0 °C, it could be observed that the rate of inimer conversion was only slightly affected by change in the Cu0 surface area. This might be due to the rate of comproportionation reaction between Cu0 (wire/powder) and CuII being faster than the addition reaction, leading to insignificant influence regarding Cu0 surface area. As shown in Fig. 3B and C, obvious differences in MW evolution were observed between experiments with Cu0 wire and Cu0 powder, showing that more products with low MW (=600) were observed for the system with Cu0 wire. This might be ascribed to low inimer conversion (curve a in Fig. 3A) or high extent of degradation of the polyester chain. When the polyadditions were performed at 10 °C using CuCl2, the conversions for both experiments, i.e. performed with Cu0 wire and Cu0 powder, were similar after 76 h and 60 h, respectively (curves a and b in Fig. 4A). In general, at 10 °C (Fig. 4B–D), polymers with higher MW in a range from 4200 to 16200 were obtained in comparison to polymers with Mw ≤ 2000 prepared at 0 °C.
 | ||
| Fig. 3 (A) Inimer conversion vs. reaction time and (B, C) GPC traces for ATRPA in anisole performed with CuBr2 and different surface areas of Cu0 at 0 °C. ([VBBiB]0 = 1.8 M; w = wire, p = powder). | ||
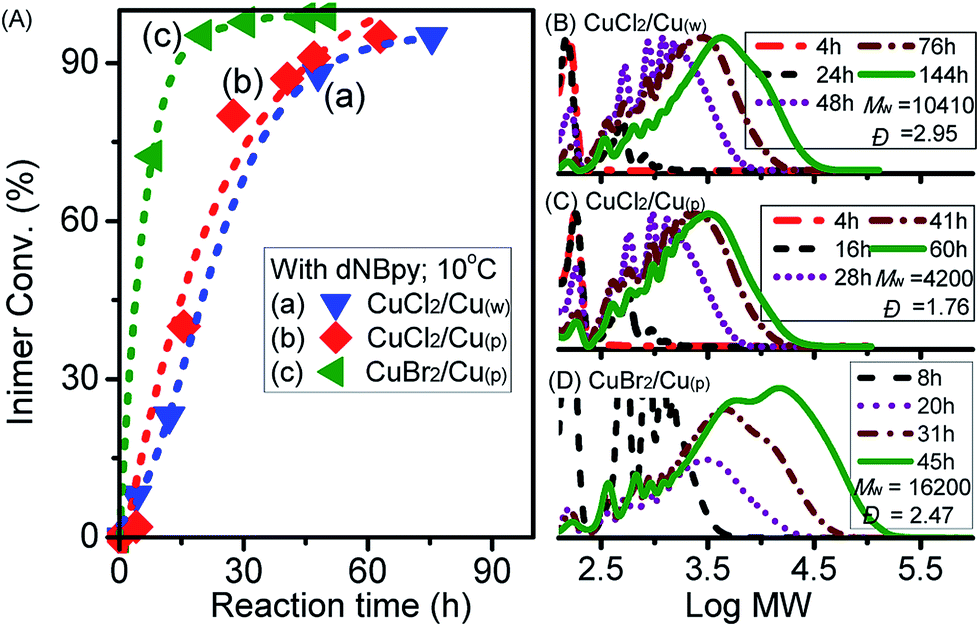 | ||
| Fig. 4 (A) Inimer conversion vs. reaction time and (B–D) GPC traces for ATRPA in anisole performed with different catalysts at 10 °C. ([VBBiB]0 = 1.8 M; w = wire, p = powder). | ||
Chemical structural details of the resulted polymers prepared using either Cu0 wire or Cu0 powder at various temperatures were investigated by 1H NMR spectroscopy (Fig. 5). Fig. 5A and C displayed the structural assignments of the corresponding aliphatic polyesters. In all cases, the characteristic peaks (d′), (f), and (e/e′) from polyester chains were mainly observed. In addition, peaks indicating self-degradation by-products could be observed. The amount of these by-products, however, depended on the polyaddition conditions. In compliance with the GPC traces, the highest amount of degradation by-products could also be observed from 1H NMR spectra when the polyaddition was performed at 0 °C using Cu0 wire. A longer polyaddition time at a lower temperature can result in occurrences of side reactions such as degradation of the polyesters accompanied with lactonization of the cleaved compounds. It can be noted that signals from chain end functional groups elucidated information about the choice of halogen.48,53 The g′/g peaks intensity ratio in Fig. 5C was higher than that in Fig. 5D, consistent with the added amounts of CuCl2 for the ATRPA. The reaction conditions and results of product characterization are summarized in Table 1 (entries 2–5).
 | ||
| Fig. 5 1H NMR spectra (400 MHz, CDCl3) of products from the ATRPA in anisole performed with Cu0 characterized by different surface areas, copper catalysts and temperatures ([VBBiB]0 = 1.8 M; w = wire, p = powder; VBBiB/CuX2 = 50/1 for A–C and 50/0.5 for D). | ||
Possible further optimization of the best system (i.e., ATRPA with CuBr2/Cu0 powder/L at 10 °C) by changing the ligand or solvent was investigated as well (Table S1†). Performing ATRPA with Cu0 powder at 10 °C, but using the PMDETA ligand, yielded a polymer with higher MW (Mw = 30800, Đ = 8.86). However, similarly to ATRPA performed at 0 °C, branching with DB = 0.21 was observed after 45 h of polymerization (see the ESI: Fig. S1A (GPC) and S2† (1H NMR)). On the other hand, using HMTETA, which is known to be a slightly less active ligand in ATRP than dNBpy, yielded after 168 h a polymer with lower MW (Mw = 4950, Đ = 2.45) (Fig. S1B (GPC)†). As shown in Fig. S3,† no branching was observed in the NMR spectra. In addition, the rate of polymerization was significantly lower in the case of HMTETA compared to those of dNBpy and PMDETA (Fig. S1C†).
Replacement of the anisole with more polar solvents (herein, DMF and DMSO) was further proceeded. As shown in Fig. S3,† only oligomers can be observed because the polyadditions stopped at about 50% conversion. 1H NMR spectra from ATRPA performed in DMF (Fig. S5†) and DMSO (Fig. S6†) showed that decomposition products were formed in both cases and their formation was already observed after a few hours of polyaddition. The extent of the decomposition increased with the polarity of the solvent. Moreover, additional peaks were observed in the DMSO case due to branching structure (DB = 0.75). In DMSO, the copper comproportionation can occur fast, leading to low concentration of CuII deactivators and thus increased probability of branching.
3.3. Temperature effects on ATRPA of VBBiB/CuCl2/dNBpy system
Finally, we looked more in detail into the ATRPA of VBBiB under different temperatures using the CuCl2/dNBpy complex. This system showed good stability against self-degradation reactions at 10 °C. Fig. 6A displays kinetic plots for ATRPA of VBBiB performed under different temperatures. The reaction rate of inimer conversion increased with temperature increase confirming that temperature has a significant effect on the reaction rate. Fig. 6B–D present the MW evolutions of the corresponding reactions. The MWs showed obvious changes at higher inimer conversions (>80%) and the Đ vales of the resulting polymers were above 1.50. These results indicated step-growth polymerisation during ATRPA. It can be noted that low MW signals (highlighted in yellow in Fig. 6D) arising from the self-degradation side reaction were relatively obvious at 0 °C. This could be ascribed to the longer reaction times, which enhanced the occurrences of lactonization.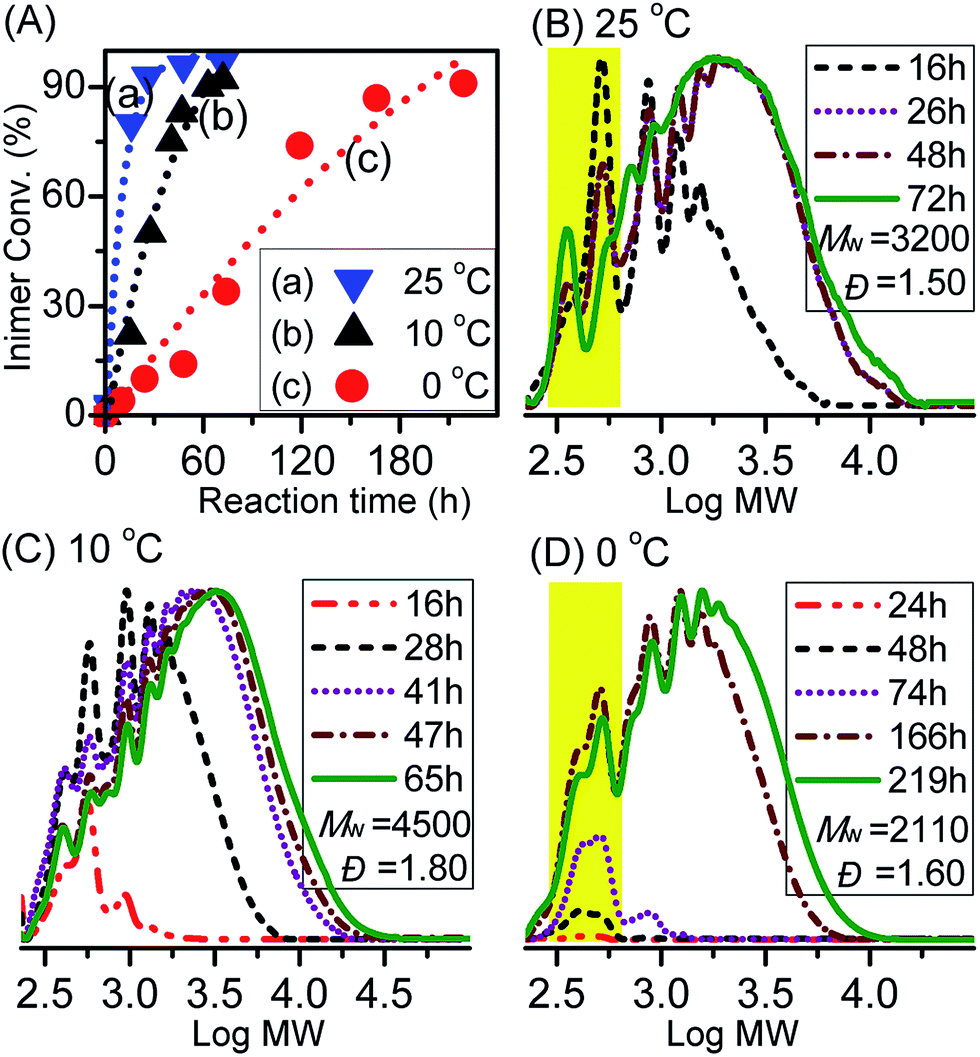 | ||
| Fig. 6 (A) Inimer conversion vs. reaction time for ATRPA under different temperatures. (B–D) GPC traces of ATRPA performed under different temperatures (for 0 and 10 °C: VBBiB/CuCl2/Cu0 powder/dNBpy = 50/1/2/6; for 25 °C: Cu0 wire ([VBBiB]0 = 1.8 M in anisole; L = 2.9 mm and d = 0.5 mm)). | ||
The resulting aliphatic polyesters were characterized by 1H NMR (Fig. 7). Significant intensities arising from self-degradation were observable for samples polymerized at 25 °C and 0 °C (peaks 1–8 in Fig. 7a and c, respectively). This was consistent with the observation in GPC traces for polyaddition performed at 0 °C due to the longer reaction time. Some extent of self-degradation observed for polyesters prepared at 25 °C indicates that in addition to the reaction time, the higher temperature resulted in even faster self-degradation of the aliphatic polyesters. The reaction conditions and characterization of the products are summarized in Table 1 (entries 6–8).
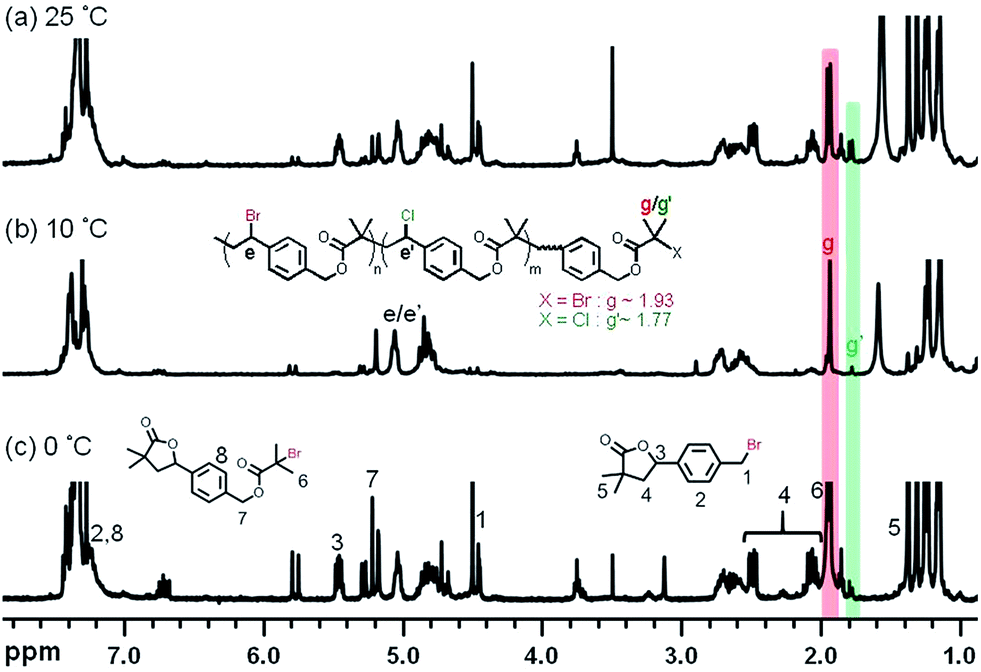 | ||
| Fig. 7 1H NMR spectra (400 MHz, CDCl3) of the ATRPA products in Fig. 6: (a) after 72 h at 25 °C, (b) after 65 h at 10 °C and (c) after 219 h at 0 °C. | ||
4. Conclusions
When reaction performed at 0 °C, first, the kinetic results using PMDETA and dNBpy as ligands were opposite to the mechanistic viewpoint. We deduced that the homogeneity of the CuII complex (deactivator) in solution was decreased in the system of VBBiB/CuBr2/Cu0/PMDETA at low reaction temperature due to the formation of an insoluble metal complex network. GPC and 1H NMR analysis showed that in the system of VBBiB/CuBr2/Cu0/PMDETA, a side reaction led to formation of highly branched polyesters. In further investigations of polymerization with various ligands (i.e., PMDETA, dNBpy, and HMTETA), the dNBpy system showed good kinetics and chemical structures than those of PMDETA and HMTETA systems. The surface area of Cu0 (wire vs. powder) had only a slight influence on the rate of inimer consumption. Proper activity of the metal complex and temperature served as a more important reaction factor than that of the Cu0 surface area for the ATRPA procedure. Differing the temperature had significant influence on the self-degradation behaviour of the polyesters. The longer reaction time needed at 0 °C led to the higher extent of self-degradation than that observed when the polyaddition was performed at 10 °C for a shorter amount of time. In addition, performing polyaddition at 25 °C led, again, to higher extent of self-degradation. Polar solvents of DMF or DMSO caused higher extent of degradation, thus preventing formation of polymers with reasonable MW. To sum up, we studied the crucial balance of metal complex activity, solubility, and reaction temperature to readily (reaction time < 48 h) obtain a new functional aliphatic polyesters with high molecular weight (Mw = 16200).
Acknowledgements
Ministry of Science and Technology, Taiwan (MOST102-2221-E-005-089 and MOST104-2923-E-194-001), Slovak Academy of Sciences (SAS-MOST JRP 2014-9), and Industrial Technology Research Institute were appreciated for financial support. JM also thanks to grant agency VEGA through the grant VEGA-2/142/14.Notes and references
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689–3745 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- Z. C. Chen, C. L. Chiu and C. F. Huang, Polymers, 2014, 6, 2552–2572 CrossRef.
- C. F. Huang, Y. A. Hsieh, S. C. Hsu and K. Matyjaszewski, Polymer, 2014, 55, 6051–6057 CrossRef CAS.
- C. F. Huang, R. Nicolay, Y. Kwak, F. C. Chang and K. Matyjaszewski, Macromolecules, 2009, 42, 8198–8210 CrossRef CAS.
- J. Mosnacek, A. Eckstein-Andicsova and K. Borska, Polym. Chem., 2015, 6, 2523–2530 RSC.
- C. Barner-Kowollik, T. P. Davis, J. P. A. Heuts, M. H. Stenzel, P. Vana and M. Whittaker, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 365–375 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- S. G. Gaynor, J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 8051–8056 CrossRef CAS.
- D. Benoit, V. Chaplinski, R. Braslau and C. J. Hawker, J. Am. Chem. Soc., 1999, 121, 3904–3920 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- K. Matyjaszewski, M. L. Wei, J. H. Xia and N. E. McDermott, Macromolecules, 1997, 30, 8161–8164 CrossRef CAS.
- B. B. Wayland, L. Basickes, S. Mukerjee, M. L. Wei and M. Fryd, Macromolecules, 1997, 30, 8109–8112 CrossRef CAS.
- B. B. Wayland, G. Poszmik, S. L. Mukerjee and M. Fryd, J. Am. Chem. Soc., 1994, 116, 7943–7944 CrossRef CAS.
- S. Yamago, E. Kayahara, M. Kotani, B. Ray, Y. Kwak, A. Goto and T. Fukuda, Angew. Chem., Int. Ed., 2007, 46, 1304–1306 CrossRef CAS PubMed.
- S. Yamago, B. Ray, K. Iida, J. Yoshida, T. Tada, K. Yoshizawa, Y. Kwak, A. Goto and T. Fukuda, J. Am. Chem. Soc., 2004, 126, 13908–13909 CrossRef CAS PubMed.
- S. Yusa, S. Yamago, M. Sugahara, S. Morikawa, T. Yamamoto and Y. Morishima, Macromolecules, 2007, 40, 5907–5915 CrossRef CAS.
- D. J. Siegwart, S. A. Bencherif, A. Srinivasan, J. O. Hollinger and K. Matyjaszewski, J. Biomed. Mater. Res., Part A, 2008, 87, 345–358 CrossRef PubMed.
- J. F. Lutz, J. Andrieu, S. Uzgun, C. Rudolph and S. Agarwal, Macromolecules, 2007, 40, 8540–8543 CrossRef CAS.
- V. Delplace, A. Tardy, S. Harrisson, S. Mura, D. Gigmes, Y. Guillaneuf and J. Nicolas, Biomacromolecules, 2013, 14, 3769–3779 CrossRef CAS PubMed.
- C. Riachi, N. Schuwer and H. A. Klok, Macromolecules, 2009, 42, 8076–8081 CrossRef CAS.
- W. J. Bailey, Z. Ni and S. R. Wu, Macromolecules, 1982, 15, 711–714 CrossRef CAS.
- W. J. Bailey, S. R. Wu and Z. Ni, Makromol. Chem., 1982, 183, 1913–1920 CrossRef CAS.
- T. Endo and W. J. Bailey, J. Polym. Sci., Part A: Polym. Chem., 1975, 13, 2525–2530 CrossRef CAS.
- Q. Smith, J. Y. Huang, K. Matyjaszewski and Y. L. Loo, Macromolecules, 2005, 38, 5581–5586 CrossRef CAS.
- S. Agarwal, Polym. Chem., 2010, 1, 953–964 RSC.
- J. Y. Yuan, C. Y. Pan and B. Z. Tang, Macromolecules, 2001, 34, 211–214 CrossRef CAS.
- T. He, Y. F. Zou and C. Y. Pan, Polym. J., 2002, 34, 138–143 CrossRef CAS.
- I. S. Chung and K. Matyjaszewski, Macromolecules, 2003, 36, 2995–2998 CrossRef CAS.
- J. M. J. Paulusse, R. J. Amir, R. A. Evans and C. J. Hawker, J. Am. Chem. Soc., 2009, 131, 9805–9812 CrossRef CAS PubMed.
- Y. Wei, E. J. Connors, X. R. Jia and C. Wang, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 761–771 CrossRef CAS.
- A. Tardy, V. Delplace, D. Siri, C. Lefay, S. Harrisson, B. D. A. Pereira, L. Charles, D. Gigmes, J. Nicolas and Y. Guillaneuf, Polym. Chem., 2013, 4, 4776–4787 RSC.
- V. Delplace, S. Harrisson, A. Tardy, D. Gigmes, Y. Guillaneuf and J. Nicolas, Macromol. Rapid Commun., 2014, 35, 484–491 CrossRef CAS PubMed.
- K. Satoh, M. Mizutani and M. Kamigaito, Chem. Commun., 2007, 1260–1262 RSC.
- B. T. Dong, Z. L. Li, L. J. Zhang, F. S. Du and Z. C. Li, Polym. Chem., 2012, 3, 2523–2530 RSC.
- T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087–1097 RSC.
- D. P. Curran, Synthesis, 1988, 489 CrossRef CAS.
- M. Mizutani, K. Satoh and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 7498–7507 CrossRef CAS PubMed.
- M. Mizuntani, K. Satoh and M. Kamigaito, Macromolecules, 2011, 44, 2382–2386 CrossRef CAS.
- M. Mizutani, E. F. Palermo, L. M. Thoma, K. Satoh, M. Kamigaito and K. Kuroda, Biomacromolecules, 2012, 13, 1554–1563 CrossRef CAS PubMed.
- M. Mizutani, K. Satoh and M. Kamigaito, Aust. J. Chem., 2014, 67, 544–554 CrossRef CAS.
- X. M. Zhang, H. Q. Dou, Z. B. Zhang, W. Zhang, X. L. Zhu and J. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3907–3916 CrossRef CAS.
- K. Satoh, S. Ozawa, M. Mizutani, K. Nagai and M. Kamigaito, Nat. Commun., 2010, 1, 1–6 CrossRef CAS PubMed.
- M. Mizutani, K. Satoh and M. Kamigaito, Macromolecules, 2009, 42, 472–480 CrossRef CAS.
- B.-T. Dong, Y.-Q. Dong, F.-S. Du and Z.-C. Li, Macromolecules, 2010, 43, 8790–8798 CrossRef CAS.
- Y. M. Han, H. H. Chen and C. F. Huang, Polym. Chem., 2015, 6, 4565–4574 RSC.
- W. Tang, Y. Kwak, W. Braunecker, N. V. Tsarevsky, M. L. Coote and K. Matyjaszewski, J. Am. Chem. Soc., 2008, 130, 10702–10713 CrossRef CAS PubMed.
- S. Faucher, P. Okrutny and S. Zhu, Ind. Eng. Chem. Res., 2007, 46, 2726–2734 CrossRef CAS.
- S. Faucher, P. Okrutny and S. P. Zhu, Macromolecules, 2006, 39, 3–5 CrossRef CAS.
- D. Y. Yan, A. H. E. Muller and K. Matyjaszewski, Macromolecules, 1997, 30, 7024–7033 CrossRef CAS.
- K. Matyjaszewski, D. A. Shipp, J. L. Wang, T. Grimaud and T. E. Patten, Macromolecules, 1998, 31, 6836–6840 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Kinetics, GPC traces and NMR spectra – Table S1 and Fig. S1–S6. See DOI: 10.1039/c6ra06186a |
| This journal is © The Royal Society of Chemistry 2016 |