
Unexpected superhard phases of niobium triborides: first-principles calculations†
Abstract
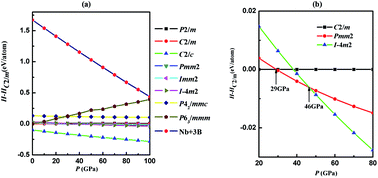
Using an unbiased structure search method based on particle-swarm optimization algorithms in combination with density functional theory calculations, we investigate the phase stability and electronic properties of NbB3 under high pressures. By structure searching as implemented in the CALYPSO code, we obtained the most stable monoclinic phase (C2/c) and four metastable phases (P63/mmm, C2/m, Pmm2 and I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) m2) for NbB3. Under high pressure, the C2/m structure transforms to Pmm2 phase and to Im2 phase at about 29 and 46 GPa, respectively. Surprisingly, the five phases of NbB3 are all dynamically and mechanically stable at ambient conditions. The high bulk and shear modulus, and low Poisson's ratio for both phases in NbB3, make it a promising low compressible material. Moreover, interestingly the hardness of the five phases of NbB3 are very close to 40 GPa, in particular, the C2/m and Pmm2 phases have sufficient hardness (45 and 44 GPa, respectively), for it to be considered as a superhard material. All phases of NbB3 with high hardness are stable due to the strong covalent bonding nature via electronic density of states and electron localization function analysis.
m2) for NbB3. Under high pressure, the C2/m structure transforms to Pmm2 phase and to Im2 phase at about 29 and 46 GPa, respectively. Surprisingly, the five phases of NbB3 are all dynamically and mechanically stable at ambient conditions. The high bulk and shear modulus, and low Poisson's ratio for both phases in NbB3, make it a promising low compressible material. Moreover, interestingly the hardness of the five phases of NbB3 are very close to 40 GPa, in particular, the C2/m and Pmm2 phases have sufficient hardness (45 and 44 GPa, respectively), for it to be considered as a superhard material. All phases of NbB3 with high hardness are stable due to the strong covalent bonding nature via electronic density of states and electron localization function analysis.
 Please wait while we load your content...
Please wait while we load your content...

