DOI:
10.1039/C6RA03722D
(Paper)
RSC Adv., 2016,
6, 43755-43766
Multi-color solid-state emission of β-iminoenolate boron complexes tuned by methoxyl groups: aggregation-induced emission and mechanofluorochromism†
Received
10th February 2016
, Accepted 23rd April 2016
First published on 26th April 2016
Abstract
A series of new β-iminoenolate boron complexes modified by methoxyl groups in different positions of the benzene rings were synthesized. They all exhibited AIE behaviours, for example, the emission intensity of B2B in THF/H2O (v/v = 1/19) was ca. 91-fold higher than that in THF. Interestingly, the emitting color for B4B changed from green to yellow during the AIE process on account of the different aggregated states. In addition, it was found that the molecular stacking modes enormously affected the solid emission of β-iminoenolate boron complexes. B2B emitted blue light due to the weakest π–π interaction in the crystal. BB, B3B and B24B emitted blue-green light on account of their similar packing modes in crystals, in which moderate π–π interactions were involved. In the case of crystal B4B, π-aggregates were generated directed by strong π–π interactions, leading to yellow emission. On the other hand, B4B exhibited multi-color emission upon stimulation by mechanic forces. When B4B was ground into ground powder 1 in a mortar, π-aggregates were damaged and the emitting color changed from yellow to green. If a small amount of ground powder 1 was further ground on a parchment, it was turned into ground powder 2 emitting blue light derived from the isolated molecules. The fluorescence of ground powder 1 could recover to yellow upon exposure to DCM. This meant that the emitting color changes between yellow and green were reversible. Similarly, reversible mechanofluorochromic processes between ground powder 1 and 2 were observed. Although the crystal with yellow emission could be changed into ground powder 2, only green emitting ground powder 1 was gained after treating with DCM. This kind of mechanofluorochromic materials with multi-color emission might have potential applications in anti-fake labels and mechanic sensors.
Introduction
Organic fluorescent materials have gained great interest on account of their potential applications in organic light-emitting diodes (OLEDs), organic light-emitting field-effect transistors (OLEFETs) and fluorescent sensors.1 In general, the fluorescence dyes are highly emissive in dilute solutions, but the emission can usually be quenched in high concentrated solutions or in the solid state. Such aggregation-caused quenching (ACQ) may limit their applications in the solid state.2 In 2001, Tang's group prepared a new silole derivative which was non-emissive in solution but gave strong emission in an aggregated state due to the restriction of intramolecular rotations, and they termed this phenomenon as aggregation-induced emission (AIE).3 Since then, many kinds of AIE materials were developed,4 such as the derivatives of siloes,5 tetraphenylethylene,6 cyanostyrene,7 triarylamine8 and so on. Meanwhile, the AIE materials were widely used in OLED,9 fluorescent sensors,10 self-assembling,11 fluorescence switching,12 bioimaging13 and mechanofluorochromic (MFC) materials.14 As the most famous organic boron complexes,15 BODIPY derivatives were excellent fluorophores used in chemical sensors,16 solar cells17 and near-infrared emitting materials18 because of the narrow absorption and emission bands, high molar absorption coefficients, high fluorescence quantum yields and photochemical stability. However, small Stokes shift and ACQ effect of BODIPY were not beneficial to their applications. As an analogue of BODIPY, β-iminoenolate boron complexes exhibited large Stokes shift and strong emission in solid states, and their fluorescent behaviors could be easily tuned by the structures of ligands and are sensitive to external microenvironments.19 It should be noted that stimuli-responsive fluorescent materials could be used in clinical diagnosis, biochemistry and environmental science, because their photophysical properties were sensitive to pH,20 light,21 explosive compounds22 and anions,23 etc. Among them, mechanofluorochromic materials, which response to external forces gained increasing attention due to their applications in luminescent switches, mechanosensors, data storage, security inks and optoelectronic devices.24 In 2007, the first pure organic mechanofluorochromic compound was reported by Araki et al.25 They found that amide-substituted tetraphenylpyrene derivative could stack into two kinds of solid states, one ordered stable state directed by strong hydrogen bonds and another disordered metastable state in absence of hydrogen bond. The two kinds of solid states with different emission could be transformed each other by grinding and heating. Xu and coworkers developed a series of mechanofluorochromic materials containing triphenylethylene, tetraphenylethylene and divinylanthracene units with AIE feature.26 Our group has found that the moderate ICT, heavy atom effect and the steric effect of bulky groups played a key role in the performance of mechanochromic fluorescence dyes.27 In our previous work, we have synthesized carbazole, phenothiazine and triphenylamine functionalized β-iminoenolate complexes bearing a pyridine ligand, exhibiting emitting color changes in solid states.27e–g Considering the moderate electron-withdrawing ability and large conjugated degree, herein, benzoxazole was introduced to the β-iminoenolate boron complexes as a ligand (Scheme 1) to prepare new mechanofluorochromic materials. Besides, as an electron donor methoxyl group may tune the polarity of π-conjugated system to yield β-iminoenolate boron complexes with different polarity, and it might also affect the molecular packing in solid states. Therefore, different amount of methoxyls were modified to benzene rings in different positions in order to reveal the relationship between the structures of D–π–A molecules and solid-state emission. It is interesting that all the synthesized β-iminoenolate boron complexes gave strong emission in solid states although they were weak emissive or non-emissive in solutions, suggesting AIE features. In particular, compound B4B exhibited multi-color emission during mechanofluorochromic and self-assembling processes. To the best of my knowledge, the reports on the materials with multi-color emission during MFC processes are rare.28
 |
| | Scheme 1 Synthetic routes for β-iminoenolate boron complexes. | |
Experimental section
Measurement and characterization
1H NMR and 13C NMR spectra were measured with a Mercury Plus instrument at 400 MHz and 100 MHz using DMSO-d6 as solvent. FT-IR spectra were measured with a Nicolet-360 FT-IR spectrometer by incorporation of samples in KBr disks. The UV-vis absorption spectra were obtained on Shimadzu UV-3100 spectrophotometer. Fluorescent emission spectra were obtained on a Cary Eclipse fluorescence spectrophotometer. Mass spectra were obtained with Agilent 1100 MS series and AXIMA CFR MALDI-TOF (Compact) mass spectrometers. XRD patterns were obtained on an Empyrean X-ray diffraction instrument. Single crystals of all cases were selected for X-ray diffraction studies in a Rigaku RAXIS-RAPID diffractometer. Cyclic voltammograms were obtained on a CHI 604C voltammetric analyzer with a scan rate at 50 mV s−1. A three electrode configuration was used for the measurement: a platinum button as the working electrode, a platinum wire as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode. The solution of (C4H9)4NBF4 in DCM (0.1 M) was used as the supporting electrolyte.
Synthesis
THF were dried over sodium and diphenyl ketone. DCM was dried over calcium hydride. The other chemicals and reagents were used as received without further purification. Compounds 1, 2, 3, 4 and 7 were commercial available. Compounds 5 (ref. 29) and 6 (ref. 30) were synthesized according to the reported procedures.
1,1-Difluoro-3-phenyl-1H-benzo[4,5]oxazolo[3,2-c][1,3,2]oxazaborinin-10-ium-1-uide (BB)
2-Methyl benzoxazole (1.05 mL, 8.82 mmol) was dissolved in THF (30 mL) and then NaH (60%, 0.37 g, 8.82 mmol) was added. After the mixture was stirred for 0.5 h at room temperature, methyl benzoate (0.80 g, 5.88 mL) was added. The mixture was refluxed for 8 h under an atmosphere of nitrogen, and then poured into water followed by acidified with dilute HCl. The crude intermediate was precipitated and collected by suction filtration. After the intermediate was dried under vacuum, it was dissolved in DCM (50 mL) containing triethylamine (2.45 mL, 17.64 mmol). Under an atmosphere of nitrogen, boron trifluoride diethyl ether (2.23 mL, 17.64 mmol) was added in portion by injector. After that, the mixture was stirred for 12 h at room temperature. Then, water (200 mL) was added to quench the reaction. The organic layer was separated and dried over anhydrous Na2SO4. After removal of the solvent, the crude product was purified by recrystallization in petroleum ether and DCM to afford BB (1.22 g) as a light yellowish green solid. Yield: 73%; mp 244.0–246.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.12 (m, 2H), 7.94 (dd, J = 6.8 Hz, 2.2 Hz, 1H), 7.73 (m, 1H), 7.66 (m, 1H), 7.59 (m, 4H), 7.21 (s, 1H) (Fig. S20†); 13C NMR (100 MHz, DMSO-d6) δ 170.84, 165.13, 148.49, 133.20, 133.12, 130.15, 129.49, 127.53, 127.42, 126.90, 114.89, 112.51, 82.09 (Fig. S21†); FT-IR (KBr): ν = 686, 723, 738, 762, 887, 926, 979, 1018, 1013, 1033, 1118, 1131, 1164, 1177, 1214, 1275, 1295, 1338, 1373, 1404, 1465, 1495, 1559, 1594 cm−1; MALDITOF MS: m/z: calculated for C15H10BF2NO2: 285.1; found: 286.1 [M + H]+ (Fig. S22†).
1,1-Difluoro-3-(2-methoxyphenyl)-1H-benzo[4,5]oxazolo[3,2-c][1,3,2]oxazaborinin-10-ium-1-uide (B2B)
Following the synthetic procedure of BB, the intermediate was afforded by the condensation of methyl 2-methoxybenzoate (0.60 g, 3.61 mmol) and 2-methyl benzoxazole (0.64 mL, 5.41 mmol) in THF. The intermediate was complexed with boron trifluoride diethyl ether (1.37 mL, 10.83 mmol) in the presence of triethylamine (1.50 mL, 10.83 mmol) in DCM to give the crude product, which was purified by column chromatography (silica gel, petroleum/DCM, v/v = 2/1) to afford B2B as a light yellowish green solid (0.84 g). Yield: 74%; mp 208.0–210.0 °C; 1H NMR (400 MHz, DMSO-d6) 7.99 (dd, J = 7.9 Hz, 1.8H, 1H), 7.92 (m, 1H), 7.71 (dd, J = 6.5 Hz, 2.4 Hz, 1H), 7.58 (m, 3H), 7.6 (d, J = 8.2 Hz, 1H), 7.15 (m, 2H), 4.00 (s, 3H) (Fig. S23†); 13C NMR (100 MHz, DMSO-d6) δ 167.90, 165.01, 159.35, 148.48, 134.45, 130.19, 129.91, 127.36, 126.87, 121.31, 121.23, 114.88, 113.01, 112.43, 86.13, 56.47 (Fig. S24†); FT-IR (KBr): ν = 594, 616, 668, 730, 747, 762, 792, 853, 888, 982, 1009, 1085, 1115, 1138, 1166, 1179, 1246, 1268, 1280, 1295, 1344, 1362, 1399, 1466, 1488, 1547, 1633, 2845, 2948, 2976, 3013, 3085, 3112, 3167 cm−1; MALDITOF MS: m/z: calculated for C16H12BF2NO3: 315.1; found: 316.1 [M + H]+ (Fig. S25†).
1,1-Difluoro-3-(3-methoxyphenyl)-1H-benzo[4,5]oxazolo[3,2-c][1,3,2]oxazaborinin-10-ium-1-uide (B3B)
Following the synthetic procedure of BB, the intermediate was afforded by the condensation of methyl 3-methoxybenzoate (0.60 g, 3.61 mmol) and 2-methyl benzoxazole (0.64 mL, 5.41 mmol) in THF. The intermediate was complexed with boron trifluoride diethyl ether (1.37 mL, 10.83 mmol) in the presence of triethylamine (1.50 mL, 10.83 mmol) in DCM to give the crude product, which was purified by recrystallization in petroleum ether and DCM to afford B3B (0.82 g) as a light yellowish green solid. Yield: 72%; mp 235.0–237.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.95 (m, 1H), 7.72 (m, 2H), 7.58 (m, 3H), 7.50 (t, J = 8.0 Hz, 1H), 7.23 (m, 2H), 3.88 (s, 3H) (Fig. S26†); 13C NMR (100 MHz, DMSO-d6) δ 170.59, 165.10, 160.07, 148.50, 134.58, 130.63, 130.14, 127.42, 126.92, 119.92, 119.14, 114.91, 112.51, 112.28, 82.43, 55.94 (Fig. S27†); FT-IR (KBr): ν = 669, 720, 741, 769, 804, 867, 920, 980, 1014, 1033, 1050, 1082, 1117, 1128, 1178, 1207, 1247, 1272, 1295, 1367, 1400, 162, 1488, 1560; MALDITOF MS: m/z: calculated for C16H12BF2NO3: 315.1; found: 316.1 [M + H]+ (Fig. S28†).
1,1-Difluoro-3-(4-methoxyphenyl)-1H-benzo[4,5]oxazolo[3,2-c][1,3,2]oxazaborinin-10-ium-1-uide (B4B)
Following the synthetic procedure of BB, the intermediate was afforded by the condensation of methyl 4-methoxybenzoate (0.60 g, 3.61 mmol) and 2-methyl benzoxazole (0.64 mL, 5.41 mmol) in THF. The intermediate was complexed with boron trifluoride diethyl ether (1.37 mL, 10.83 mmol) in the presence of triethylamine (1.50 mL, 10.83 mmol) in DCM to give the crude product, which was purified by recrystallization in petroleum ether and DCM to afford B4B (0.79 g) as a bright yellow solid. Yield: 69%; mp 251.0–253.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.09 (d, J = 9.0 Hz, 2H), 7.90 (m, 1H), 7.68 (d, J = 7.2 Hz, 1H), 7.55 (m, 2H), 7.12 (d, J = 9.0 Hz, 2H), 7.07 (s, 1H), 3.88 (s, 3H) (Fig. S29†); 13C NMR (100 MHz, DMSO-d6) δ 171.00, 165.29, 163.49, 148.36, 130.27, 129.76, 127.27, 126.58, 125.37, 114.92, 114.65, 112.37, 80.32, 56.10 (Fig. S30†); FT-IR (KBr): ν = 597, 668, 711, 728, 745, 768, 805, 842, 891, 984, 1014, 1030, 1108, 1122, 1131, 1180, 1216, 1267, 1306, 1314, 1375, 1397, 1455, 1466, 1508, 1557, 1607, 1633, 2846, 2937 cm−1; MALDITOF MS: m/z: calculated for C16H12BF2NO3: 315.1; found: 316.0 [M + H]+ (Fig. S31†).
1,1-Difluoro-3-(2,4-dimethoxyphenyl)-1H-benzo[4,5]oxazolo[3,2-c][1,3,2]oxazaborinin-10-ium-1-uide (B24B)
Following the synthetic procedure of BB, the intermediate was afforded by the condensation of methyl 2,4-dimethoxybenzoate (0.70 g, 3.57 mmol) and 2-methyl benzoxazole (0.63 mL, 5.36 mmol) in THF. The intermediate was complexed with boron trifluoride diethyl ether (1.36 mL, 10.72 mmol) in the presence of triethylamine (1.49 mL, 10.72 mmol) in DCM to give the crude product. The crude product was purified by column chromatography (silica gel, petroleum ether/DCM, v/v = 1/1) to afford B24B (0.86 g) as a light yellowish green solid. Yield: 70%; mp 228.0–230.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.99 (m, 1H), 7.88 (dd, J = 7.2 Hz, 1.7 Hz, 1H), 7.67 (d, J = 7.2 Hz, 1H), 7.55 (m, 2H), 7.05 (s, 1H), 6.76 (m, 2H), 4.01 (s, 3H), 3.90 (s, 3H) (Fig. S32†); 13C NMR (100 MHz, DMSO-d6) δ 167.98, 165.25, 164.72, 161.38, 148.36, 131.69, 130.33, 127.20, 126.51, 114.62, 114.01, 112.27, 106.87, 99.28, 84.10, 56.58, 56.20 (Fig. S33†); FT-IR (KBr): ν = 602, 668, 733, 760, 778, 832, 890, 945, 983, 1005, 1022, 1090, 1117, 1140, 1178, 1216, 1258, 1271, 1287, 1323, 1386, 1438, 1458, 1474, 1507, 1540, 1559, 1608, 2959, 3005, 3166 cm−1; MALDITOF MS: m/z: calculated for C17H14BF2NO4: 345.1; found: 346.1 [M + H]+ (Fig. S34†).
1,1-Difluoro-3-(2,5-dimethoxyphenyl)-1H-benzo[4,5]oxazolo[3,2-c][1,3,2]oxazaborinin-10-ium-1-uide (B25B)
Following the synthetic procedure of BB, the intermediate was afforded by the condensation of methyl 2,5-dimethoxybenzoate (0.70 g, 3.57 mmol) and 2-methyl benzoxazole (0.63 mL, 5.36 mmol) in THF. The intermediate was complexed with boron trifluoride diethyl ether (1.36 mL, 10.72 mmol) in the presence of triethylamine (1.49 mL, 10.72 mmol) in DCM to give the crude product. The crude product was purified by column chromatography (silica gel, petroleum ether/DCM, v/v = 2/1) to afford B25B (0.83 g) as a light yellowish green solid. Yield: 68%; mp 179.0–181.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.91 (m, 1H), 7.71 (m, 1H), 7.58 (m, 2H), 7.56 (m, 1H), 7.21 (m, 2H), 7.16 (s, 1H), 3.94 (s, 3H), 3.80 (s, 3H) (Fig. S35†); 13C NMR (100 MHz, DMSO-d6) δ 167.45, 164.96, 153.69, 153.42, 148.50, 130.16, 127.38, 126.91, 121.74, 120.05, 114.92, 114.41, 113.91, 112.44, 86.36, 56.79, 56.10 (Fig. S36†); FT-IR (KBr): ν = 418, 428, 667, 718, 746, 756, 787, 814, 852, 885, 892, 934, 979, 1014, 1024, 1033, 1050, 1086, 1112, 1125, 1155, 1170, 1181, 1232, 1269, 1280, 1294, 1338, 1361, 1447, 1464, 1472, 1503, 1550, 1629, 2847, 2955, 2985, 3090, 3163 cm−1; MALDITOF MS: m/z: calculated for C17H14BF2NO4: 345.1; found: 346.1 [M + H]+ (Fig. S37†).
1,1-Difluoro-3-(3,4,5-trimethoxyphenyl)-1H-benzo[4,5]oxazolo[3,2-c][1,3,2]oxazaborinin-10-ium-1-uide (B345B)
Following the synthetic procedure of BB, the intermediate was afforded by the condensation of methyl 3,4,5-trimethoxybenzoate (0.80 g, 3.54 mmol) and 2-methyl benzoxazole (0.62 mL, 5.30 mmol) in THF. The intermediate was complexed with boron trifluoride diethyl ether (1.35 mL, 10.61 mmol) in the presence of triethylamine (1.47 mL, 10.61 mmol) in DCM to give the crude product, which was purified by column chromatography (silica gel, petroleum ether/DCM, v/v = 3/1) to afford B345B (0.89 g) as a light green solid. Yield: 67%; mp 218.0–220.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 7.8 Hz, 1H), 7.71 (d, J = 7.5 Hz, 1H), 7.58 (m, 2H), 7.37 (s, 2H), 7.28 (s, 1H), 3.91 (s, 6H), 3.77 (s, 3H) (Fig. S38†); 13C NMR (100 MHz, DMSO-d6) δ 170.64, 165.17, 153.52, 148.47, 141.94, 130.18, 128.39, 127.36, 126.77, 114.80, 112.45, 105.08, 81.86, 60.72, 56.70 (Fig. S39†); FT-IR (KBr): ν = 744, 766, 779, 807, 1003, 1014, 1037, 1128, 1166, 1237, 1259, 1300, 1313, 1396, 1425, 1470, 1503, 1563, 1596, 1632, 2841, 2918, 2948 cm−1; MALDITOF MS: m/z: calculated for C18H16BF2NO5: 375.1; found: 376.4 [M + H]+ (Fig. S40†).
Results and discussion
Synthesis
The synthetic routes for β-iminoenolate boron complexes (BB, B2B, B3B, B4B, B24B, B25B, B345B) were shown in Scheme 1. Compounds 1, 2, 3, 4 and 7 were commercial available. Compounds 5 (ref. 29) and 6 (ref. 30) were synthesized according to the reported procedures. The condensation reactions between the derivatives of methyl benzoates and 2-methyl benzoxazole gave the corresponding intermediates, which were complexed with boron trifluoride diethyl ether directly to afford the corresponding β-iminoenolate boron complexes. Due to the electron withdrawing ability of benzoxazole, the yields of the condensation reactions of 2-methyl benzoxazole with esters were high, and the overall yields of the two steps were around 70%. The target molecules were characterized by 1H NMR, 13C NMR, FT-IR and MALDI-TOF mass spectrometry. The single structures of BB, B2B, B3B, B4B, B24B, B25B, B345B were gained by slow evaporation of their solutions in DCM/petroleum ether, and further confirmed the molecular structures.
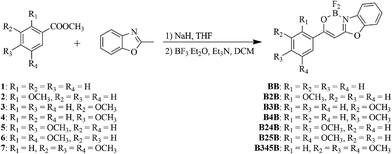
UV-vis absorption and fluorescence emission spectra in solutions
The UV-vis absorption and fluorescence emission spectra of the synthesized β-iminoenolate boron complexes in THF were shown in Fig. 1 and the corresponding data were summarized in Table S1.† The absorption bands of the β-iminoenolate boron complexes were mainly located in the range of 260–270 nm and 330–400 nm, and derived from π–π* transitions. Specifically, the maximum absorption band of BB was located at 352 nm, which was the shortest wavelength compared with others due to the absence of methoxyl. B3B with a methoxyl group in m-position of benzene ring gave a maximum absorption at 355 nm, while the absorption of B2B, B4B bearing one methoxyl in o-position or p-position of benzene, as well as the absorption of B345B with three methoxyls further red-shifted to 362–371 nm. B24B and B25B with two methoxyls, in which one was in o-position of benzene, gave the longest maximum absorption bands located at 378 nm and 381 nm, respectively. The variation of the absorption bands should be attributed to the different electron effect of methoxyl groups in different positions of benzene rings. In solutions, the complexes were almost non-emissive or gave very weak emission. As shown in Fig. 1, the maximum emission peaks of BB, B2B, B3B, B4B and B24B emerged in the range of 425–430 nm, and those for B345B and B25B were located at 442 nm and 453 nm, respectively. Their fluorescence quantum yields (ΦF) were quite low. For BB, B2B and B3B, the quantum yields were less than 0.01 using quinine sulfate as standard. As to B4B, B24B, B25B and B345B, their quantum yields were only 0.02, 0.03, 0.08 and 0.04, respectively. We suggested that the intramolecular rotation induced the increased non-radiative transition, leading to the very weak emission in solutions.
 |
| | Fig. 1 Normalized UV-vis absorption (a) and fluorescence emission spectra ((b) λex = 365 nm) of β-iminoenolate boron complexes in THF (1 × 10−5 mol L−1). | |
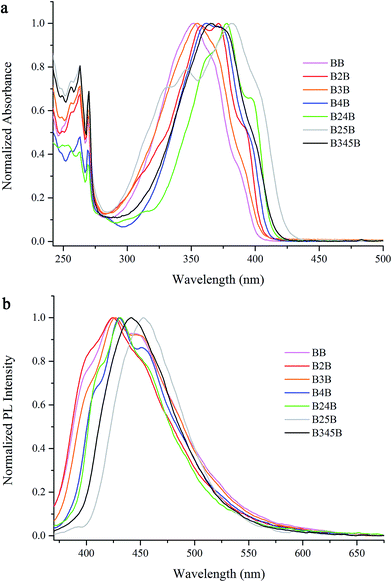
Meanwhile, we obtained the solvent-dependent UV-vis absorption and fluorescence emission spectra of β-iminoenolate boron complexes. As shown in Fig. S1,† the maximum absorption peak of BB was located at 351–355 nm in different solvents. The maximum emission peak of BB appeared at 410 nm in cyclohexane, and at 425–430 nm with vibration structures in toluene, THF, DCM and DMF. Additionally, the ΦF of BB in different solvents were also as low as 0.01 (Table S1†). The above results indicated that no intramolecular charge transfer (ICT) occurred in BB. The boron complexes of B2B, B3B, B4B, B24B and B345B exhibited similar electronic spectral features to those of BB in different solvents (Fig. S2–S6†). Therefore, ICT could not take place in these complexes. In the case of B25B, the maximum absorption band was located ca. 380 nm without evident changes in different solvents, but its emission band red-shifted obviously with increasing solvent polarity. For instance, the emission appeared at 431 nm in cyclohexane and 474 nm in DMF (Fig. 2). We deemed the ICT transition occurred in B25B, which could be further proved by theoretical calculation results. It should be noted that a negative solvatokinetic effect was found for B25B, and its ΦF was 0.02 in cyclohexane and 0.29 in DMF.31 Such negative solvatokinetic effect has been previously found in other β-iminoenolate boron complexes.19
 |
| | Fig. 2 Normalized UV-vis absorption and fluorescence emission spectra (λex = 365 nm) of B25B in different solvents (1 × 10−5 mol L−1). | |
Electrochemical properties
As we discussed above, the number and the position of methoxyl group linked to benzene affected the photophysical properties of β-iminoenolate boron complexes in solutions. In order to study whether the electrochemical properties were affected by the methoxyl group, cyclic voltammograms were measured and shown in Fig. S7.† B25B exhibited one reversible reduction process and others exhibited irreversible reduction processes. The half-wave potentials of BB, B2B, B3B, B4B, B24B, B25B and B345B were located at −1.32 V, −1.35 V, −1.34 V, −1.48 V, −1.50 V, −1.48 V and −1.39 V (vs. Fc/Fc+), respectively (Table 1). BB showed the highest reductive potential due to the absence of methoxyl group. B24B and B25B gave the lowest reductive potential because of the strong electron donating ability of methoxyl at o- and p-positions of benzene, being consistent with the fact that the maximum absorption bands of B24B and B25B emerged in low-energy region compared with other boron complexes. Furthermore, the strongest donating ability of 2,5-dimethoxyl phenyl group led to the occurrence of ICT in B25B. Therefore, the electrochemical properties of β-iminoenolate boron complexes could be tuned by methoxyl group. Meanwhile, the LUMO energy level was calculated using the equation of ELUMO = −(Ered + 4.8), and the HOMO energy level was calculated using the empirical equation of EHOMO = ELUMO − Eg, in which Eg was estimated from the onset of the absorption spectrum (Eg = 1240/λonset). The calculated results were shown in Table 1. BB showed the lowest HOMO and LUMO energy levels, which were located at −5.96 eV and −2.87 eV. B24B and B25B gave the highest HOMO/LUMO energy levels on account of the electron-donating ability of methoxyl groups.
Table 1 Electrochemical data and HOMO/LUMO energy levels
| |
Ereda (V) |
HOMOb (eV) |
LUMOb (eV) |
Egc (eV) |
HOMOd (eV) |
LUMOd (eV) |
| Ered: reduction potential; Fc/Fc+ was used as the external reference. Calculated using the empirical equation: ELUMO = −(Ered + 4.8) and EHOMO = ELUMO − Eg. Estimated from the onset of the absorption spectra (Eg = 1240/λonset). Obtained from quantum chemical calculation using DFT/B3LYP/6-31G(d). |
| BB |
−1.32 |
−5.96 |
−2.87 |
3.09 |
−5.96 |
−2.06 |
| B2B |
−1.35 |
−5.91 |
−2.84 |
3.07 |
−5.89 |
−2.09 |
| B3B |
−1.34 |
−5.92 |
−2.85 |
3.07 |
−5.95 |
−2.05 |
| B4B |
−1.48 |
−5.74 |
−2.71 |
3.03 |
−5.75 |
−1.91 |
| B24B |
−1.50 |
−5.69 |
−2.69 |
3.00 |
−5.58 |
−1.74 |
| B25B |
−1.48 |
−5.64 |
−2.71 |
2.93 |
−5.58 |
−1.82 |
| B345B |
−1.39 |
−5.79 |
−2.80 |
2.99 |
−5.77 |
−1.98 |
Theoretical calculation
We carried out the density functional theory (DFT) calculations for β-iminoenolate boron complexes by the Gaussian 09W program32 using the DFT/B3LYP/6-31G(d) method to reveal their electronic structures. The frontier orbital plots of the HOMO and LUMO were shown in Fig. S8,† and the calculated HOMO/LUMO energy levels were summarized in Table 1. It was found that the frontier orbital plots of HOMO and LUMO for β-iminoenolate boron complexes were located in the whole molecules except B25B, whose HOMO was mainly distributed in the 2,5-dimethoxyl phenyl moiety and LUMO was located at the whole molecule. It was understandable that the maximum absorption and emission bands of B25B derived from ICT transition. Moreover, the calculated HOMO energy levels were quite similar to the results obtained from electrochemical results. The LUMO energy levels were different from the experimental results, but they gave similar variation trend. The energy levels of HOMO and LUMO for B24B and B25B were the highest among the complexes.
Aggregation-induced emission
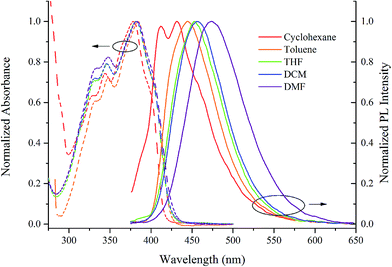
Although the fluorescence emission of the synthesized β-iminoenolate boron complexes was quite weak in solutions, we found that it could be intensified significantly in solid states, indicating the AIE activities. Therefore, the effect of content of water (%) on the emission of boron complexes was evaluated in THF/H2O. As shown in Fig. 3, BB showed very weak emission at ca. 430 nm in THF and in the mixture of THF/H2O with content of water below 70%. When the content of water was increased to 80%, the emission at 468 nm emerged, and its intensity increased significantly with increasing content of water. Compared with the emission of BB in THF, a 44-fold of emission intensity was afforded when the content of water in THF/H2O reached 90%. Although the emission decreased when the content of water was over 95% in THF/H2O, the intensity was still 21-fold higher than its THF solution. In order to confirm that the enhanced emission of BB was originated from the aggregation in THF/H2O with high content of water, the absorption spectral changes were monitored (Fig. 3c). It was clear that the absorption at 350 nm for BB was similar either in THF or in THF/H2O with the content of water below 70%. When the content of water increased to 80%, the absorbance at 350 nm decreased and was broadened significantly due to the molecular aggregation.33 Meanwhile, the appearance of tails in the visible region could be attributed to Mie scattering caused by nanoparticles.5a When the content of water increased over 90%, the absorption was further broadened and a new absorption at ca. 420 nm appeared. Thus, the bond rotations would be restricted greatly in aggregated states and the non-radiative transition would be suppressed, leading to evident enhancement of emission. In addition, the other synthesized β-iminoenolate boron complexes except B4B showed similar AIE characters (Fig. S9–S13†), and B2B exhibited the most evident AIE performance. When content of water in THF/H2O increased to 95%, the emission intensity of B2B was ca. 91-fold higher than that in THF. B3B, B24B, B25B and B345B gave ca. 48, 11, 1.2 and 2.6-fold increase of the emission in THF/H2O than that in pure THF solutions, respectively (Table S2†). In order to quantitatively evaluate the AIE performance, we measured the fluorescence quantum yields of the as-synthesized samples (ΦF,ass), and they were 0.49, 0.90, 0.86, 0.80, 0.26 and 0.27 for BB, B2B, B3B, B24B, B25B and B345B, respectively. However, the quantum yields of BB, B2B, B3B, B24B, B25B and B345B in THF (ΦF,THF) were as low as 0.01, 0.01, 0.01, 0.03, 0.08 and 0.04, respectively. Therefore, the AIE factor (aAIE = ΦF,ass/ΦF,THF) of BB, B2B, B3B, B24B, B25B and B345B were calculated to be 49, 90, 86, 27, 3.3 and 6.8, indicating evident AIE phenomenon.27d,34
 |
| | Fig. 3 Photographs of BB (a) and B4B (d) under UV light; fluorescence emission spectra of BB (b) and B4B (e) excited at 365 nm and UV-vis absorption spectra of BB (c) and B4B (f) in THF/H2O with different content of water (% volume). The concentration is maintained at 5.0 × 10−5 M. The insets in (b) and (e) showed a plot of fluorescence emission integral for BB and B4B versus content of water in THE/H2O. | |
Notably, during the aggregation process of B4B the emitting color changed with increasing content of water in THF/water. As shown in Fig. 3d, when the content of water was below 70%, B4B emitted weak blue light centered at 430 nm. If the content of water increased to 80%, B4B emitted bright bluish green light with a maximal emission peak at 480 nm. When the content of water further increased to 90% and 95%, B4B emitted yellow light with two emission peaks at 480 nm and 550 nm, meanwhile, the emission intensity exhibited 11 and 6.1-fold enhancement than that in THF. Besides, the ΦF,ass and ΦF,THF of B4B were 0.92 and 0.02, giving an AIE factor of 46. It should be noted that the emission at ca. 550 nm was not detected for other complexes. In order to reveal the origination of the emission, the fluorescence decay curves of B4B were obtained (Fig. S14†). It was found that the lifetime of the emission at 430 nm for B4B in THF was 0.48 ns, and the lifetime for the emission at 480 nm in THF/H2O with content of water of 80% increased to 18.54 ns (Table S3†). When the content of water further increased to 90%, the lifetimes for the emission at 480 nm and 550 nm were 13.28 ns and 23.68 ns, respectively. Because the lifetime for the emission at 550 nm was similar to that at 480 nm, we deemed that the emission at 550 nm was not derived from excimers but from the π-aggregates.19b We suggested that small nanoparticles formed when the content of water increased to 80%, so the emission red-shifted to 480 nm from 430 nm for isolated molecules. When the content of water further increased to 90%, the small particles would grow-up to form bigger ones, and π-aggregates were generated. In order to verify our speculation, the scanning electron microscopy (SEM) images of the aggregates gained from B4B in THF/H2O with water content of 80% and 90% were shown in Fig. S15.† From Fig. S15a and b,† we could find smaller fibres and the blocky structures in the aggregated states. When the water content was 90%, the blocky structures were more massive and concentrated. When the water content was 80%, the blocky structures separated among the fibres were much smaller and fewer. It should be noted that the fibres generated from THF/H2O with water content of 80% and 90% were both needle-like structures with similar size (Fig. S15c–f†), meanwhile, the blocky microstructures were also similar gained from THF/H2O with different water content (Fig. S15g–j†). Therefore, we deduced that when the water content was 80% in THF/H2O, most molecules aggregated into needle-like fibres and a small part of molecules grew-up into blocky structures, where π-aggregates were generated. At this moment, we could observe the emission mainly from fibres. When the water content increased to 90%, more molecules aggregated into massive blocky structures and the emission of π-aggregates were shown.
Crystal structures
Due to the occurrence of AIE, the synthesized boron complexes gave strong emission in solid states. To verify the effect of the molecular packing modes and intermolecular interactions on the solid emission, the single crystals of BB, B2B, B3B, B4B, B24B and B25B were gained by slow evaporation of their solutions in DCM/petroleum ether. As shown in Fig. 4, two molecules of BB drawn in green and in orange firstly stacked into a dimer, and the dimers further packed into a column structure in the single crystal. The distance between the two molecules in a dimer was 3.474 Å, and the distance between the adjacent dimer was 3.499 Å. Molecules overlapped partially in the crystal, so π–π interaction might take place. Hydrogen bonds parallel to the molecular plane took place between the column structures. The distance between fluorine and hydrogen atoms (F1⋯H14) in benzoxazole ring was 2.516 Å. The single crystals of B3B and B24B were similar to that of BB. In the crystal of B3B, the distances between the adjacent molecules in a dimer and between the adjacent dimers were 3.435 Å and 3.571 Å, respectively, and F1⋯H12 interaction (2.636 Å) between the columns was also found (Fig. S16†). In the crystal of B24B, the distances of the molecules in a dimer and the adjacent dimers were 3.457 Å and 3.639 Å, respectively. Besides, the overlap degree of molecules in crystal of B24B was less than those in crystals of BB and B3B. The intermolecular interactions of F1⋯C16 (3.168 Å), F1⋯H16 (2.514 Å), F2⋯H13 (2.641 Å), F2⋯H14 (2.624 Å) and B1⋯H13 (3.168 Å) were involved in crystal of B24B (Fig. S17†). In the case of crystal of B2B, the molecules drawn in red and in blue stacked into two kinds of row-liked structures, which were packed in parallel each other (Fig. 5). The π–π interaction should be very weak because of the small molecular overlap degree. In the red row, there were B1⋯H16 (3.008 Å) and F1⋯H16 (2.547 Å) interactions between the adjacent molecules. Among the adjacent rows, F2⋯H4, B1⋯C4 and H1A⋯C16 interactions were observed with distance of 2.643 Å, 3.673 Å and 2.827 Å, respectively. In the crystal of B4B, two antiparallel molecules stacked into a dimer, and then stacked into a column (Fig. 6). The distances between the adjacent molecules in a dimer and between the adjacent dimers were 3.524 Å and 3.534 Å, respectively. On the other hand, one column could interact with four neighbouring columns. The distances of F1⋯H15, F1⋯H1A, F2⋯H13 and B1⋯H13 were 2.372 Å, 2.633 Å, 2.665 Å and 3.152 Å, respectively. In the crystal of B25B, the molecules drawn in red and in blue stacked into two columns, and the adjacent green/yellow columns were almost perpendicular to the red/blue columns. Although no evident π–π interaction was found, several hydrogen bonds of H2B⋯O1, H2B⋯C15, F2⋯H1C, F2⋯H6A and C2⋯C15 with distances of 2.697 Å, 2.785 Å, 2.530 Å, 2.595 Å and 3.354 Å were involved in the crystal (Fig. S18†). The above multiple intermolecular interactions in the crystals restricted the bond rotation, and resulted in the fluorescent enhancement in solid states.
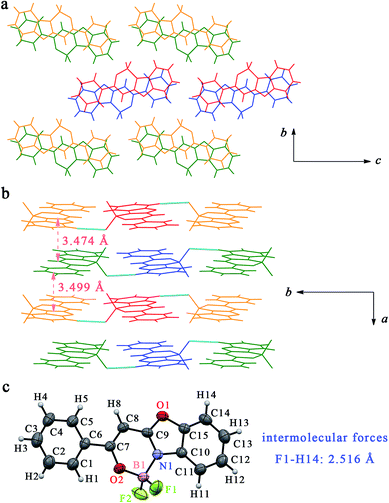 |
| | Fig. 4 (a) Crystal structure of BB viewed along a axis; (b) crystal structure of BB viewed along c axis, intermolecular forces (blue line) and distances between molecules (pink arrow); (c) ORTEP view and list of intermolecular forces. | |
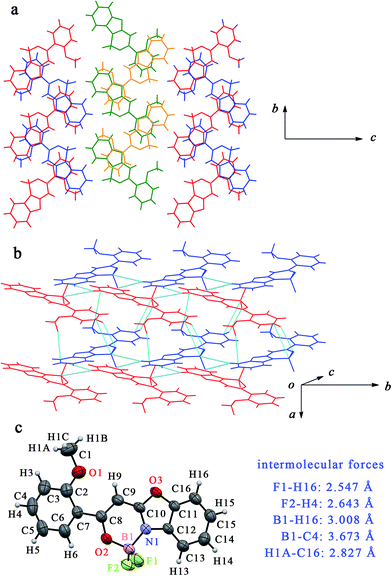 |
| | Fig. 5 (a) Crystal structure of B2B viewed along a axis; (b) crystal structure of B2B and intermolecular forces (blue line); (c) ORTEP view and list of intermolecular forces. | |
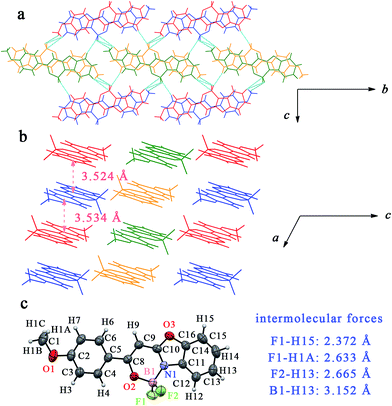 |
| | Fig. 6 (a) Crystal structure of B4B viewed along a axis; (b) crystal structure of B4B viewed along b axis, intermolecular forces (blue line) and distances between molecules (pink arrow); (c) ORTEP view and list of intermolecular forces. | |
Fluorescence emission in solid states
As shown in Fig. 7, B2B emitted blue light centred at 439 nm and 461 nm in powder and in single crystal, respectively (Table S2†). The powders and single crystals of BB, B3B and B24B emitted blue-green light located at ca. 470 nm. The powder and single crystal of B345B emitted green light at 505 nm and 512 nm, respectively. B4B emitted yellow light with two emission peaks at 480 nm and 550 nm in both powder and in crystal state, which was similar to the emission in THF/H2O with content of water of 90%. The powder and single crystal of B25B emitted blue-green light located at ca. 470 nm and 500 nm, respectively. Combined with the fluorescence emission in solutions, we suggested that the solid emission of β-iminoenolate boron complexes was mainly affected by the molecular packing modes. For example, the emission of B2B and B3B was quite similar in solutions, but their emission in single crystals was different. The reason might be that molecules in crystal of B2B stacked crossing each other, and π–π interactions would be very weak, leading to the emission of B2B appeared in high energy region. In the case of crystal B3B, the distances between the adjacent molecules in a dimer and between the adjacent dimers suggested the occurrences of π–π interactions, so that the red-shift of the emission was detected. Additionally, the stacking modes of B3B, BB and B24B in single crystals were similar, so they gave similar emission. Unexpectedly, B4B exhibited quite different emission behaviors from BB, B3B and B24B in single crystals although they packed into similar crystal structures. Since the lifetimes of the emission peaks for B4B at 480 nm and 550 nm were 10.47 ns and 16.89 ns, respectively (Table S3†), which were similar to those in the THF/H2O with content of water of 90%, the emission at 550 nm was derived from π-aggregates instead of excimers. The reason why π-aggregates could not be formed in other crystals was ascribed to weak π–π interaction.
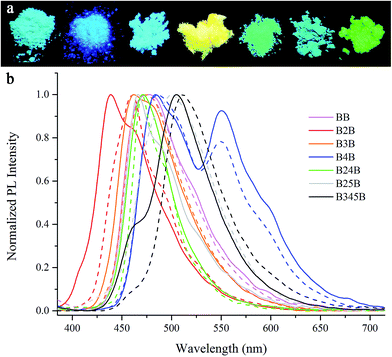 |
| | Fig. 7 (a) Photographs of the as-synthesized samples under UV (from left: BB, B2B, B3B, B4B, B24B, B25B, B345B); (b) normalized fluorescence emission spectra of as-synthesized samples (solid) and single crystals (dash) of β-iminoenolate boron complexes excited at 365 nm. | |
Mechanofluorochromic properties
From the above solid-state emission of β-iminoenolate boron complexes, we found that most of the synthesized complexes showed similar emission spectra in powders and in crystals. No obvious mechanofluorochromic behaviors of BB, B2B, B3B, B24B, B25B and B345B were observed. However, B4B exhibited interesting mechanofluorochromic phenomenon with multi-color emission (Fig. 8). The crystal of B4B emitted bright yellow light with two emission bands at ca. 480 nm and ca. 550 nm. After it was ground in a mortar, ground powder 1 emitting green light centered at 505 nm was gained. If ground powder 1 was placed at room temperature for 5–10 min, the emitting color could self-recover to yellow. Meanwhile, fumed with DCM for 3 s, the ground powder 1 could also be transformed into its initial state. Besides, when a small amount of the ground powder 1 was further ground on a vegetable parchment, it became ground powder 2 emitting blue light with the maximal emission at ca. 450 nm. The ground powder 2 could be transformed into powder 1 with green emission (ca. 480 nm) after a drop of DCM was dropped on the vegetable parchment. However, the ground power 2 could not be restored to the crystalline state with yellow emission. Meanwhile, if a small amount of crystal B4B was ground on a vegetable parchment, it could also turn to ground powder 2.
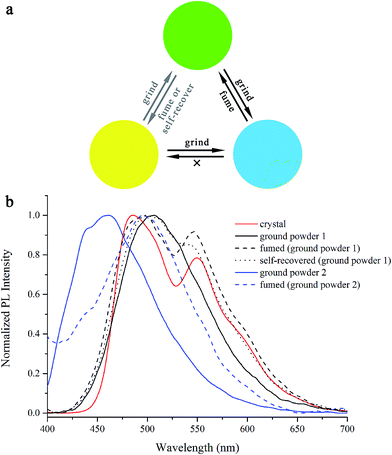 |
| | Fig. 8 (a) Schematic diagrams and (b) normalized fluorescence emission spectra of B4B in different solid states excited at 365 nm. | |
Mechanofluorochromism was often caused by the changes of molecular packing in solid states, so the XRD patterns of B4B in different solid states were measured and shown in Fig. S19.† It was clear that the as-synthesized sample of B4B gave several sharp and strong diffraction peaks, illustrating crystalline structure. After grinding the crystal in a mortar, the ground powder 1 was prepared and no evident diffraction peaks were detected due to the amorphous state. Moreover, the diffraction peaks emerged again when ground powder 1 was self-recovered or fumed with DCM. Because the ground powder 2 was obtained by grinding a small amount of ground powder 1 on a vegetable parchment, its XRD pattern could not be measured. Additionally, fluorescence lifetimes were measured to confirm the mechanofluorochromic mechanism. The lifetimes for the emission at 480 nm and 550 nm in as-synthesized crystal were 10.47 ns and 16.89 ns, respectively (Table S3†). From the above results, we knew that the lifetimes of the emission at 480 nm and 550 nm in THF/H2O (v/v = 1/9) were 13.28 ns and 23.68 ns, respectively. Therefore, we deemed that the emission at 550 nm derived from π-aggregates directed by strong π–π interactions. Moreover, the fluorescence lifetime at 505 nm for B4B in ground powder 1 was 14.87 ns, similar to that for the emission at 480 nm in crystal and in aggregates in THF/H2O (v/v = 2/8). However, the lifetime for the emission at 450 nm in ground powder 2 was as short as 3.20 ns, similar to the peak at 430 nm in THF (0.48 ns). It indicated the emission from ground powder 2 derived from isolated molecules. Therefore, we proposed the mechanofluorochromic mechanism of B4B as below. In crystal, the π-aggregates formed due to the strong π–π interactions, and emitted yellow light centered at ca. 550 nm. Meanwhile, the emission at 480 nm was also detected in crystal of B4B since parts of molecules did not packed into π-aggregates. When the crystal was ground, the crystalline structure was damaged into amorphous state so that the π-aggregates disappeared, leading to the absence of the emission at 550 nm. The green light emission of ground powder 1 was ascribed to red-shift of the emission from the partially aggregated molecules (480 nm) to 505 nm on account of the decrease of the molecular distances or the enhancement of the molecular planarity. When the ground powder 1 was fumed, the crystalline structure recovered and the π-aggregates reformed. On the other hand, when the ground powder 1 was further ground on a vegetable parchment, the molecular distances were greatly enlarged and the isolated molecules appeared, leading to blue light emission located at 450 nm. When the ground powder 2 was treated by DCM, molecules stacked partially, so the emission red-shifted to 480 nm. The reason why ground powder 2 could not convert to crystalline state was that the small amount of isolated molecules could not be packed together into π-aggregates.
It is worth noting that this kind of mechanofluorochromic materials with AIE nature was named as the piezofluorochromic AIE (PAIE) materials, and most of PAIE dyes contained typical AIE-active functional groups, such as tetraphenylethylene, triphenylethylene, cyanoethylene and 9,10-divinylanthracene.14,26,35 In our cases, no typical AIE-active groups were involved in β-iminoenolate boron complexes-based PAIE dyes. Generally, the multi-color emission was often caused by totally different molecular stacking modes in liquid crystal state or crystalline state.28 In our previous work, we also found triphenylamine functionalized β-iminoenolate boron complexes exhibiting multi-color MFC properties due to the two different crystalline states.27g To the best of our knowledge, the multi-color emission in MFC processes caused by π-aggregates was not reported. Combined with our previous work, we deduced that β-iminoenolate boron complex tended to form π-aggregates or excimers in crystal, and the crystal always exhibited two emission bands, one of which was derived from π-aggregates/excimers.27e,f Thus, the emission could be tuned by the fraction of π-aggregates/excimers in different solid states. When the crystalline structures were destroyed completely, the other emission band would emerge, and the multi-color emission could be observed.27e Therefore, it provided a new strategy for the design of new multi-color MFC materials.
Conclusions
In conclusion, we synthesized a series of β-iminoenolate boron complexes modified by methoxyl groups in different positions of benzene rings. They were almost non-emissive in solutions but highly-emissive in solid states, exhibiting evident AIE properties. For instance, the emission intensity of B2B in THF/H2O with content of water of 95% was ca. 91-fold higher than that in THF. In particular, B4B exhibited special color change in AIE process. When the content of water was increased to 80% and 90%, green emissive nanoparticles and yellow emissive π-aggregates were generated. In crystal states, due to the weakest π–π interaction, B2B emitted blue light. On account of the similar packing modes in crystals, BB, B3B, B24B emitted blue-green light. Additionally, in crystal of B4B, π-aggregates were generated directed by strong π–π interaction, and they emitted yellow light. It should be noted that no mechanofluorochromic behaviors was detected for BB, B2B, B3B, B24B and B25B. However, B4B exhibited a multi-color emission during mechanofluorochromic processes. When the as-synthesized crystal B4B was ground into ground powder 1, π-aggregates were damaged and the emitting color changed from yellow to green. If a small amount of ground powder 1 was further ground on a parchment, it became ground powder 2 emitting blue light ascribed to the electronic transition of the isolated molecules. Two kinds of reversible emitting colors changes of yellow/green and green/blue were observed. This new series of β-iminoenolate boron complexes helped us to well understand the effects of the molecular structures and the molecular packing modes on the AIE and MFC properties. This kind of novel multi-color emission of mechanofluorochromic materials might have potential applications in new anti-fake labels and in mechanic sensors, which can distinguish different strength of forces so as to well protect the fragile products.
Acknowledgements
This study is financially supported by the National Natural Science Foundation of China (21374041), the Open Project of State Key Laboratory of Supramolecular Structure and Materials (SKLSSM201615).
Notes and references
-
(a) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed;
(b) F. Cicoira and C. Santato, Adv. Funct. Mater., 2007, 17, 3421–3434 CrossRef CAS;
(c) I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS PubMed;
(d) M. D. McGehee and A. J. Heeger, Adv. Mater., 2000, 12, 1655–1668 CrossRef CAS;
(e) T. J. Dale and J. Rebek, J. Am. Chem. Soc., 2006, 128, 4500–4501 CrossRef CAS PubMed.
-
(a) J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed;
(b) J. Malkin, Photophysical and Photochemical Properties of Aromatic Compounds, CRC, Boca Raton, 1992 Search PubMed;
(c) N. J. Turro, Modern Molecular Photochemistry, University Science Books, Mill Valley, 1991 Search PubMed;
(d) X.-C. Li and S. C. Moratti, Photonic Polymer Systems, ed. D. L. Wise, G. E. Wnek, D. J. Trantolo, T. M. Cooper and J. D. Gresser, Marcel Dekker, New York, 1998 Search PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
-
(a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC;
(b) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC;
(c) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed;
(d) R. Hu, N. L. C. Leung and B. Z. Tang, Chem. Soc. Rev., 2014, 43, 4494–4562 RSC.
-
(a) Z. Li, Y. Q. Dong, J. W. Y. Lam, J. Sun, A. Qin, M. Häußler, Y. P. Dong, H. H. Y. Sung, I. D. Williams, H. S. Kwok and B. Z. Tang, Adv. Funct. Mater., 2009, 19, 905–917 CrossRef CAS;
(b) J. Mei, J. Wang, J. Z. Sun, H. Zhao, W. Yuan, C. Deng, S. Chen, H. H. Y. Sung, P. Lu, A. Qin, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, Chem. Sci., 2012, 3, 549–558 RSC;
(c) Z. Zhao, D. Liu, F. Mahtab, L. Xin, Z. Shen, Y. Yu, C. Y. K. Chan, P. Lu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, B. Yang, Y. Ma and B. Z. Tang, Chem.–Eur. J., 2011, 17, 5998–6008 CrossRef CAS PubMed.
-
(a) N. B. Shustova, T.-C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dincă, J. Am. Chem. Soc., 2012, 134, 15061–15070 CrossRef CAS PubMed;
(b) X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, S. Liu, Y. Zhang and J. Xu, J. Mater. Chem., 2011, 21, 1788–1796 RSC;
(c) Z. Zhao, C. Y. K. Chan, S. Chen, C. Deng, J. W. Y. Lam, C. K. W. Jim, Y. Hong, P. Lu, Z. Chang, X. Chen, P. Lu, H. S. Kwok, H. Qiua and B. Z. Tang, J. Mater. Chem., 2012, 22, 4527–4534 RSC;
(d) W. Wang, T. Lin, M. Wang, T.-X. Liu, L. Ren, D. Chen and S. Huang, J. Phys. Chem. B, 2010, 114, 5983–5988 CrossRef CAS PubMed.
-
(a) B.-K. An, S.-K. Kwon, S.-D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS PubMed;
(b) S.-J. Yoon, J. H. Kim, K. S. Kim, J. W. Chung, B. Heinrich, F. Mathevet, P. Kim, B. Donnio, A.-J. Attias, D. Kim and S. Y. Park, Adv. Funct. Mater., 2012, 22, 61–69 CrossRef CAS;
(c) J. W. Chung, B.-K. An and S. Y. Park, Chem. Mater., 2008, 20, 6750–6755 CrossRef CAS;
(d) S.-J. Lim, B.-K. An and S. Y. Park, Macromolecules, 2005, 38, 6236–6239 CrossRef CAS;
(e) X. Zhao, P. Xue, K. Wang, P. Chen, P. Zhang and R. Lu, New J. Chem., 2014, 38, 1045–1051 RSC.
-
(a) Z. Ning, Z. Chen, Q. Zhang, Y. Yan, S. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799–3807 CrossRef CAS;
(b) B. Wang, Y. Wang, J. Hua, Y. Jiang, J. Huang, S. Qian and H. Tian, Chem.–Eur. J., 2011, 17, 2647–2655 CrossRef CAS PubMed;
(c) Y. Liu, X. Tao, F. Wang, X. Dang, D. Zou, Y. Ren and M. Jiang, J. Phys. Chem. C, 2008, 112, 3975–3981 CrossRef CAS;
(d) T. Ishi-i, K. Ikeda, Y. Kichise and M. Ogawa, Chem.–Asian J., 2012, 7, 1553–1557 CrossRef CAS PubMed.
-
(a) Z. Zhao, C. Deng, S. Chen, J. W. Y. Lam, W. Qin, P. Lu, Z. Wang, H. S. Kwok, Y. Ma, H. Qiu and B. Z. Tang, Chem. Commun., 2011, 47, 8847–8849 RSC;
(b) P.-I. Shih, C.-Y. Chuang, C.-H. Chien, E. W.-G. Diau and C.-F. Shu, Adv. Funct. Mater., 2007, 17, 3141–3146 CrossRef CAS;
(c) L. Chen, Y. Jiang, H. Nie, P. Lu, H. H. Y. Sung, I. D. Williams, H. S. Kwok, F. Huang, A. Qin, Z. Zhao and B. Z. Tang, Adv. Funct. Mater., 2014, 24, 3621–3630 CrossRef CAS;
(d) Y. Liu, X. Tao, F. Wang, J. Shi, J. Sun, W. Yu, Y. Ren, D. Zou and M. Jiang, J. Phys. Chem. C, 2007, 111, 6544–6549 CrossRef CAS.
-
(a) H.-T. Feng and Y.-S. Zheng, Chem.–Eur. J., 2014, 20, 195–201 CrossRef CAS PubMed;
(b) L. Liu, G. Zhang, J. Xiang, D. Zhang and D. Zhu, Org. Lett., 2008, 10, 4581–4584 CrossRef CAS PubMed;
(c) F. Sun, G. Zhang, D. Zhang, L. Xue and H. Jiang, Org. Lett., 2011, 13, 6378–6381 CrossRef CAS PubMed;
(d) X. Chen, X. Y. Shen, E. Guan, Y. Liu, A. Qin, J. Z. Sun and B. Z. Tang, Chem. Commun., 2013, 49, 1503–1505 RSC.
-
(a) P. Gong, J. Sun, P. Xue, C. Qian, Z. Zhang, J. Sun and R. Lu, Dyes Pigm., 2015, 118, 27–36 CrossRef CAS;
(b) B.-K. An, D.-S. Lee, J.-S. Lee, Y.-S. Park, H.-S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS PubMed;
(c) S. Shin, S. H. Gihm, C. R. Park, S. Kim and S. Y. Park, Chem. Mater., 2013, 25, 3288–3295 CrossRef CAS;
(d) B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS PubMed.
-
(a) J. W. Chung, S.-J. Yoon, B.-K. An and S. Y. Park, J. Phys. Chem. C, 2013, 117, 11285–11291 CrossRef CAS;
(b) X. Cheng, H. Zhang, K. Ye, H. Zhang and Y. Wang, J. Mater. Chem. C, 2013, 1, 7507–7512 RSC.
-
(a) W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771–779 CrossRef CAS;
(b) E. Zhao, Y. Hong, S. Chen, C. W. T. Leung, C. Y. K. Chan, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Adv. Healthcare Mater., 2014, 3, 88–96 CrossRef CAS PubMed;
(c) D. Ding, C. C. Goh, G. Feng, Z. Zhao, J. Liu, R. Liu, N. Tomczak, J. Geng, B. Z. Tang, L. G. Ng and B. Liu, Adv. Mater., 2013, 25, 6083–6088 CrossRef CAS PubMed;
(d) Y. Yu, C. Feng, Y. N. Hong, J. Z. Liu, S. J. Chen, K. M. Ng, K. Q. Luo and B. Z. Tang, Adv. Mater., 2011, 23, 3298–3302 CrossRef CAS PubMed.
-
(a) X. Zhu, R. Liu, Y. Li, H. Huang, Q. Wang, D. Wang, X. Zhu, S. Liu and H. Zhu, Chem. Commun., 2014, 50, 12951–12954 RSC;
(b) T. Han, Y. Zhang, X. Feng, Z. Lin, B. Tong, J. Shi, J. Zhi and Y. Dong, Chem. Commun., 2013, 49, 7049–7051 RSC;
(c) S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed;
(d) J. Wang, J. Mei, R. Hu, J. Z. Sun, A. Qin and B. Z. Tang, J. Am. Chem. Soc., 2012, 134, 9956–9966 CrossRef CAS PubMed.
-
(a) D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS PubMed;
(b) D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC.
-
(a) P. Ashokkumar, H. Weißhoff, W. Kraus and K. Rurack, Angew. Chem., Int. Ed., 2014, 53, 2225–2229 CrossRef CAS PubMed;
(b) N. Sakamoto, C. Ikeda, M. Yamamura and T. Nabeshim, Chem. Commun., 2012, 48, 4818–4820 RSC;
(c) S. Erbas-Cakmak and E. U. Akkaya, Org. Lett., 2014, 16, 2946–2949 CrossRef CAS PubMed;
(d) J. J. Hu, N.-K. Wong, Q. Gu, X. Bai, S. Ye and D. Yang, Org. Lett., 2014, 16, 3544–3547 CrossRef CAS PubMed.
-
(a) T. Rousseau, A. Cravino, T. Bura, G. Ulrich, R. Ziessel and J. Roncali, Chem. Commun., 2009, 1673–1675 RSC;
(b) D. Kumaresan, R. P. Thummel, T. Bura, G. Ulrich and R. Ziessel, Chem.–Eur. J., 2009, 15, 6335–6339 CrossRef CAS PubMed;
(c) Y. Kubo, D. Eguchi, A. Matsumoto, R. Nishiyabu, H. Yakushiji, K. Shigaki and M. Kaneko, J. Mater. Chem. A, 2014, 2, 5204–5211 RSC;
(d) A. Bessette and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 3342–3405 RSC.
-
(a) T. Sarma, P. K. Panda and J.-I. Setsune, Chem. Commun., 2013, 49, 9806–9808 RSC;
(b) G. Ulrich, S. Goeb, A. D. Nicola, P. Retailleau and R. Ziessel, J. Org. Chem., 2011, 76, 4489–4505 CrossRef CAS PubMed;
(c) L. Zeng, C. Jiao, X. Huang, K.-W. Huang, W.-S. Chin and J. Wu, Org. Lett., 2011, 13, 6026–6029 CrossRef CAS PubMed.
-
(a) Y. Zhou, Y. Xiao, S. M. Chi and X. H. Qian, Org. Lett., 2008, 10, 633–636 CrossRef CAS PubMed;
(b) J. Q. Feng, B. L. Liang, D. L. Wang, L. Xue and X. Y. Li, Org. Lett., 2008, 10, 4437–4440 CrossRef CAS PubMed;
(c) Y. Kubota, Y. Ozaki, K. Funabiki and M. Matsui, J. Org. Chem., 2013, 78, 7058–7067 CrossRef CAS PubMed.
-
(a) C. Wang, J. Ren, D.-H. Qu, X.-L. Zhao, K. Chen, H. Tian and P. Erk, Dyes Pigm., 2003, 59, 143–152 CrossRef;
(b) D. Cui, X. Qian, F. Liu and R. Zhang, Org. Lett., 2004, 6, 2757–2760 CrossRef CAS PubMed;
(c) V. B. Bojinov, D. B. Simeonov and N. I. Georgiev, Dyes Pigm., 2008, 76, 41–46 CrossRef.
-
(a) A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687 CrossRef CAS PubMed;
(b) S. Yokoyama, T. Hirose and K. Matsuda, Chem. Commun., 2014, 50, 5964–5966 RSC;
(c) R. Rajaganesh, A. Gopal, T. M. Das and A. Ajayaghosh, Org. Lett., 2012, 14, 748–751 CrossRef CAS PubMed.
-
(a) Y. Che, D. E. Gross, H. Huang, D. Yang, X. Yang, E. Discekici, Z. Xue, H. Zhao, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2012, 134, 4978–4982 CrossRef CAS PubMed;
(b) T. Naddo, Y. Che, W. Zhang, K. Balakrishnan, X. Yang, M. Yen, J. Zhao, J. S. Moore and L. Zang, J. Am. Chem. Soc., 2007, 129, 6978–6979 CrossRef CAS PubMed;
(c) P. Xue, J. Sun, B. Yao, P. Gong, Z. Zhang, C. Qian, Y. Zhang and R. Lu, Chem.–Eur. J., 2015, 21, 4712–4720 CrossRef CAS PubMed.
-
(a) P. Ashokkumar, H. Weißhoff, W. Kraus and K. Rurack, Angew. Chem., Int. Ed., 2014, 53, 2225–2229 CrossRef CAS PubMed;
(b) J. F. Zhang, C. S. Lim, S. Bhuniya, B. R. Cho and J. S. Kim, Org. Lett., 2011, 13, 1190–1193 CrossRef CAS PubMed;
(c) P. Xue, J. Sun, Q. Xu, R. Lu, M. Takafuji and H. Ihara, Org. Biomol. Chem., 2013, 11, 1840–1847 RSC;
(d) P. Xue, R. Lu, J. Jia, M. Takafuji and H. Ihara, Chem.–Eur. J., 2012, 18, 3549–3558 CrossRef CAS PubMed;
(e) D. Xu, X. Liu, R. Lu, P. Xue, X. Zhang, H. Zhou and J. Jia, Org. Biomol. Chem., 2011, 9, 1523–1528 RSC.
-
(a) M. S. Kwon, J. Gierschner, J. Seo and S. Y. Park, J. Mater. Chem. C, 2014, 2, 2552–2557 RSC;
(b) X. Cheng, D. Li, Z. Y. Zhang, H. Y. Zhang and Y. Wang, Org. Lett., 2014, 16, 880–883 CrossRef CAS PubMed;
(c) A. Pucci, F. D. Cuia, F. Signoriab and G. Ruggeri, J. Mater. Chem., 2007, 17, 783–790 RSC;
(d) M. Kinami, B. R. Crenshaw and C. Weder, Chem. Mater., 2006, 18, 946–955 CrossRef CAS;
(e) S. Hirata and T. Watanabe, Adv. Mater., 2006, 18, 2725–2729 CrossRef CAS.
- Y. Sagara, T. Mutai, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2007, 129, 1520–1521 CrossRef CAS PubMed.
-
(a) X. Zhang, Z. Chi, H. Li, B. Xu, X. Li, W. Zhou, S. Liu, Y. Zhang and J. R. Xu, Chem.–Asian J., 2011, 6, 808–811 CrossRef CAS PubMed;
(b) B. Xu, Z. Chi, J. Zhang, X. Zhang, H. Li, X. Li, S. Liu, Y. Zhang and J. Xu, Chem.–Asian J., 2011, 6, 1470–1478 CrossRef CAS PubMed;
(c) H. Li, X. Zhang, Z. Chi, B. Xu, W. Zhou, S. Liu, Y. Zhang and J. Xu, Org. Lett., 2011, 13, 556–559 CrossRef CAS PubMed;
(d) H. Li, Z. Chi, B. Xu, X. Zhang, X. Li, S. Liu, Y. Zhang and J. Xu, J. Mater. Chem., 2011, 21, 3760–3767 RSC;
(e) X. Zhang, Z. Chi, J. Zhang, H. Li, B. Xu, X. Li, S. Liu, Y. Zhang and J. Xu, J. Phys. Chem. B, 2011, 115, 7606–7611 CrossRef CAS PubMed.
-
(a) P. Xue, P. Chen, J. Jia, Q. Xu, J. Sun, B. Yao, Z. Zhang and R. Lu, Chem. Commun., 2014, 50, 2569–2571 RSC;
(b) P. Xue, B. Yao, J. Sun, Q. Xu, P. Chen, Z. Zhang and R. Lu, J. Mater. Chem. C, 2014, 2, 3942–3950 RSC;
(c) P. Xue, B. Yao, P. Wang, J. Sun, Z. Zhang and R. Lu, RSC. Adv., 2014, 4, 58732–58739 RSC;
(d) G. Zhang, J. Sun, P. Xue, Z. Zhang, P. Gong, J. Peng and R. Lu, J. Mater. Chem. C, 2015, 3, 2925–2932 RSC;
(e) Z. Zhang, P. Xue, P. Gong, G. Zhang, J. Peng and R. Lu, J. Mater. Chem. C, 2014, 2, 9543–9551 RSC;
(f) Z. Zhang, Z. Wu, J. Sun, B. Yao, G. Zhang, P. Xue and R. Lu, J. Mater. Chem. C, 2015, 3, 4921–4932 RSC;
(g) Z. Zhang, Z. Wu, J. Sun, B. Yao, P. Xue and R. Lu, J. Mater. Chem. C, 2016, 4, 2854–2861 RSC.
-
(a) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef CAS PubMed;
(b) X. Luo, W. Zhao, J. Shi, C. Li, Z. Liu, Z. Bo, Y. Q. Dong and B. Z. Tang, J. Phys. Chem. C, 2012, 116, 21967–21972 CrossRef CAS;
(c) S. Yagai, S. Okamura, Y. Nakano, M. Yamauchi, K. Kishikawa, T. Karatsu, A. Kitamura, A. Ueno, D. Kuzuhara, H. Yamada, T. Seki and H. Ito, Nat. Commun., 2014, 5, 4013–4016 CAS;
(d) Y. Dong, B. Xu, J. Zhang, X. Tan, L. Wang, J. Chen, H. Lv, S. Wen, B. Li, L. Ye, B. Zou and W. Tian, Angew. Chem., Int. Ed., 2012, 51, 10782–10785 CrossRef CAS PubMed;
(e) P. Galer, R. C. Korošec, M. Vidmar and B. Šket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS PubMed.
- A. P. Skoumbourdis, R. Huang, N. Southall, W. Leister, V. Guo, M.-H. Cho, J. Inglese, M. Nirenberg, C. P. Austin, M. Xia and C. J. Thomas, Bioorg. Med. Chem. Lett., 2008, 18, 1297–1303 CrossRef CAS PubMed.
- A. P. Skoumbourdis, C. A. LeClair, E. Stefan, A. G. Turjanski, W. Maguire, S. A. Titus, R. Huang, D. S. Auld, J. Inglese, C. P. Austin, S. W. Michnick, M. Xia and C. J. Thomas, Bioorg. Med. Chem. Lett., 2009, 19, 3686–3692 CrossRef CAS PubMed.
- P. F. Wang, Z. Y. Xie, Z. R. Hong, J. X. Tang, O. Wong, C. S. Lee, N. Wong and S. Lee, J. Mater. Chem., 2003, 13, 1894–1899 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. B. Noh, R. H. Kim, W. J. Kim, S. Kim, K.-S. Lee, N. S. Cho, H.-K. Shim, H. E. Pudavar and P. N. Prasad, J. Mater. Chem., 2010, 20, 7422–7429 RSC.
- W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. H.-Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed.
- Z. Chi, X. Zhang, B. Xu, X. Zhou, C. Ma, Y. Zhang, S. Liu and J. Xu, Chem. Soc. Rev., 2012, 41, 3878–3896 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR, MALDI/TOF MS and CV spectra; fluorescence lifetimes of B4B in solutions and in solid states; UV-vis and fluorescence emission spectra, as well as the photos of B2B, B3B, B24B, B25B, B345B during aggregation process; crystal structures of B3B, B24B and B25B; XRD patterns of B4B in different solid states. CCDC 1446478–1446483. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra03722d |
|
| This journal is © The Royal Society of Chemistry 2016 |









