
An insight into the protospacer adjacent motif of Streptococcus pyogenes Cas9 with artificially stimulated RNA-guided-Cas9 DNA cleavage flexibility†
Abstract
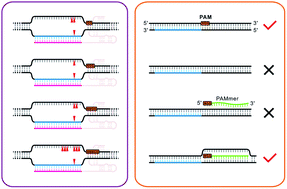
The CRISPR (clustered regularly interspaced short palindromic repeats)-associated (Cas) protein, Cas9, is a RNA-guided endonuclease that uses RNA–DNA base pairing to recognize and cleave double-stranded DNA (dsDNA) with a protospacer adjacent motif (PAM). It is widely accepted that the most commonly used Streptococcus pyogenes Cas9 (SpyCas9) protein recognizes a canonical 5′-NGG-3′ sequence in the PAM. In this study, we discovered another critical characteristic required for SpyCas9 cleavage i.e. the interspace between the protospacer and NGG. The results generated from DNA cleavage assays showed that both interspace length and the presence of a GG dinucleotide (particularly the upstream guanosine) are critical components in permitting SpyCas9-mediated cleavage. Interestingly, the interspace length significantly affects the selection of SpyCas9 cleavage sites on the non-complementary strand. Additionally, the complementary strand cleavage site is determined by the location of the single-molecular guide RNA (sgRNA). This indicates that PAM and sgRNA play different roles in determining the SpyCas9 specific cleavage site. Importantly, we also revealed for the first time that in vitro annealing of dsDNA with exogenous PAM-presenting oligonucleotides (PAMmers) stimulated SpyCas9 cleavage of target dsDNA without PAM. This study pertaining to PAM and SpyCas9 is expected to improve our understanding of SpyCas9 with an associated impact on related bioengineering capabilities.
 Please wait while we load your content...
Please wait while we load your content...

