
Investigation into the adsorption of partially hydrolyzed polyacrylamide onto in situ formed magnesium hydroxide particles†
Abstract
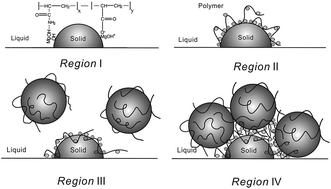
In situ formed magnesium hydroxide (Mg(OH)2) particles were investigated through the direct wet precipitation method in the presence of two partially hydrolyzed polyacrylamides (HPAM) of different molecular weights. The influence of the reaction parameters, such as MgCl2 concentration, pH, and reaction temperature, on the interactions between in situ formed Mg(OH)2 particles and HPAM molecules was investigated. The Mg(OH)2 particles obtained in the absence and presence of polymers were characterized in terms of their morphology, particle size, and crystal habit by high-resolution transmission electron microscopy, X-ray diffraction, Fourier transform infrared spectroscopy, dynamic light scattering, and zeta potential measurements. It was found that different pH values of the reaction solution led to different predominant species distribution for MgCl2. The adsorbed polymer led to the formation of four distinct adsorption regions as the concentration of polymer increased. The adsorption of partially hydrolyzed polyacrylamide on the oppositely charged Mg(OH)2 particles was explained by electrostatic attraction, hydrogen bonding, and bridging.
 Please wait while we load your content...
Please wait while we load your content...

