DOI:
10.1039/C6RA02258H
(Paper)
RSC Adv., 2016,
6, 28737-28744
Chemoenzymatic modification of silk fibroin with poly(2,6-dimethyl-1,5-phenylene ether) using horseradish peroxidase†
Received
25th January 2016
, Accepted 11th March 2016
First published on 14th March 2016
Abstract
Chemical modification of silk materials is a powerful method for tailoring the desired physical properties for possible application in various fields. In this work, we modified silk fibroin with poly(2,6-dimethyl-1,5-phenylene ether) (PPE) in order to imbue the silks with hydrophobicity so as to resist the absorption of humidity. This modification was achieved by chemoenzymatic polymerization of 2,6-dimethylphenol (DMP) using horseradish peroxidase (HRP) as a catalyst in the presence of silk fibroin obtained from Bombyx mori. The PPE chain content in the modified silk was tuned by varying the feed concentration of DMP. Wide-angle X-ray scattering measurements revealed that β-sheet crystalline structures were formed in the PPE-modified silk, even after the introduction of bulky PPE chains. The PPE-modified silk showed glass transitions derived from the PPE domains, which enabled the formation of self-standing films upon thermal processing. Films of the PPE-modified silk exhibited higher static contact angles of water droplets compared to the native silk films, indicating that the film surface of silk fibroin became more hydrophobic due to the introduction of PPE. These improved physical properties were achieved without sacrificing the inherent secondary structure of silk fibroin, namely, the β-sheet structure that is largely responsible for the mechanical properties of silk materials.
Introduction
Silk fibroin is a naturally occurring protein obtained from silkworms that is widely utilized as a high-quality fabric material in the textile industry. The major component of silk fibroin is a heavy-chain, long and robust polypeptide containing 5263 amino acid residues with a molecular weight of 391 kDa.1 The heavy chain has a glycine-rich sequence that is composed primarily of GAGAGX hexapeptide repetitive units (X = S, Y, or A),2 which form pleated β-sheet crystalline domains in the fibrillar structure.3–5 The antiparallel β-sheet structure assembled by the repetitive GAGAGX motif imparts high tensile strength to silk fibroin.6,7 Because of silk fibroin's excellent properties, such as its versatile optical properties, biocompatibility, and high mechanical strength, intensive efforts have been made to develop novel applications of silk materials in various fields in addition to in the textile industry.
Chemical modification of silk fibroin is desirable methods to modulate its properties for different applications. Various approaches have been used to modify silk fibroin with functional groups, and the resulting materials have been used for bio-applications, including tissue engineering and drug delivery.8–10 In addition to the repetitive motif that consists mainly of glycine and alanine, silk fibroin possesses various amino acid residues, such as lysine, serine, and tyrosine, and thus, chemical modifications can be achieved by functionalizing reactive amine and hydroxy groups. Numerous studies have exploited reactive amino acid residues, such as serine and/or lysine, to introduce functional groups on silk backbones by utilizing coupling methods with reagents such as cyanuric chloride derivatives,11–14 carbodiimides,15–17 alkoxysilanes,18 and epoxides.19,20 Another such study demonstrated that tyrosine residues could be modified by electrophilic aromatic substitution.21,22 This technique was able to selectively introduce various unnatural functional groups at the aromatic rings of tyrosine residues through diazotization. Introducing hydrophilic or hydrophobic groups into silk hydrogels as cell culture scaffolds has been shown to enable fine control over their affinity for living cells.22
Chemoenzymatic reactions using enzymes as catalysts have been widely applied in the development of various biomaterials and constitute as a powerful method for the synthesis and modification of proteins and polypeptides.23–25 Enzyme specificity and selectivity enable sophisticated modifications of proteins at certain sites. Tyrosinase, an oxidase, catalyzes the oxidation of tyrosine residues in the presence of oxygen and has been effectively exploited for the site-specific modification of polypeptide materials.26–28 We demonstrated that the tyrosine residues of poly(tyrosine-r-lysine) could be converted to 3,4-dihydroxyphenylalanine (DOPA) residues through selective oxidation using tyrosinase to develop novel adhesive materials inspired by a natural protein found in mussels.28 The conversion of tyrosine into DOPA residues was also achieved using natural silk fibroin.26,27 Horseradish peroxidase (HRP) catalyzes the oxidation of phenolic substrates into radical species in the presence of hydrogen peroxide. Phenolic radicals are related to physiological functions involving the cross-linking of biopolymers in plant cell walls through dityrosine linkages and the biosynthesis of lignin through the polymerization of phenolic monomers in natural plants.29 Because of its relatively wide substrate specificity, HRP has been utilized in various in vitro synthetic chemistry reactions. HRP-catalyzed oxidative coupling of phenols has been shown to produce polymeric materials in both organic and aqueous media.30–33 It has been demonstrated that poly(2,6-dimethylphenylene ether) (PPE), a widely used engineering plastic, could be synthesized by the oxidative coupling polymerization of 2,6-dimethylphenol (DMP) using HRP.34,35
Silk materials are candidates for structural materials because of their excellent mechanical properties. However, their polar amide structures allow for the absorption of atmospheric moisture by the structures, which alters their higher-order structure36 and deteriorates the material's properties, such as thermal stability and mechanical strength.37,38 Therefore, imparting hydrophobicity to silk materials, particularly at the surface, is important for their applications as structural materials. To this end, we focused on the chemoenzymatic modification of silk fibroin using HRP because the tyrosine residues in the primary structure could be effectively and selectively utilized. If silk fibroin and a phenolic compound coexist in the presence of HRP, both the tyrosine residues in the silk and the phenolic compound will be simultaneously oxidized to their corresponding radical species, which could couple to generate phenol-modified residues. In this article, the successful grafting of silk fibroin with PPE was demonstrated by means of the chemoenzymatic polymerization of DMP using HRP in the presence of silk fibroin. Grafting polymer chains from the silk has been reported only for soft and hydrophilic polymers, such as poly(methyl methacrylate)39 and poly(ethylene oxide),40 this is the first report describing the modification of silk materials with a rigid and hydrophobic polymer. The film of the resulting PPE-modified silk fibroin showed a higher water droplet contact angle than that of the native film, revealing that the introduction of PPE effectively imbued the silk fibroin with hydrophobicity.
Experimental methods
Materials
HRP (EC no. 1.11.1.7) was purchased from Wako Pure Chemical Industries (Osaka, Japan) and used as received. The activity of HRP was approximately 100 units per mg. Silk fibroin was obtained from the silk cocoons of Bombyx mori, and its aqueous solution (1.0 wt%) was prepared according to a previously reported protocol.41 The other reagents were used as received without purification unless otherwise noted.
Chemoenzymatic modification of silk fibroin with PPE in two-phase reactions
To a glass tube equipped with a stirring bar and a silicon stopper were added HRP (3 mg), a carbonate buffer solution (1.5 mL, 1 M, pH = 10), and the silk fibroin solution (1.5 mL, 1.0 wt%). The final concentrations of HRP and silk fibroin in the aqueous phase were 1 mg mL−1 and 0.5 wt%, respectively. To this aqueous solution, we added a solution of DMP in toluene (3 mL) with various concentrations ranging from 0.02 to 0.1 M. The resulting two-phase mixture was vigorously stirred at 800 rpm using an EYELA ChemiStation PPS-5511 (Tokyo Rikakikai Co. Ltd., Tokyo, Japan) at 40 °C in air. Then, hydrogen peroxide (30% aqueous solution, in equivalent molar amounts to DMP ranging from 6.7 to 33 μL in total) was added in 10 portions over 15 min intervals. The reaction was carried out for 2.5 h after the first addition of hydrogen peroxide. Subsequently, the resulting mixture was centrifuged at 9000 rpm for 15 min at 4 °C to remove both the aqueous and organic supernatants. The precipitate was washed with water, methanol, and toluene to remove unreacted silk fibroin and the homopolymer of DMP, and the sample was lyophilized to obtain PPE-modified silk fibroin as a yellow-to-orange solid.
Static contact angle measurements of the PPE-modified silk films
A film of the PPE-modified silk fibroin was prepared by a thermal press method using an MP-SNL mini test press-10 (Toyo Seiki Seisaku-sho, Ltd., Tokyo, Japan). The silk samples were placed in a frame with a Teflon sheet with an area of 5 mm2 and a width of 50 μm that was covered with Teflon sheets on both sides. The sample was then subjected to thermal pressing at 2.5 MPa and 220 °C for 30 min. Contact angle measurements on the PPE-modified silk films were performed with a DMe-201 contact angle meter (Kyowa Interface Science Co. Ltd., Saitama, Japan). A water droplet (∼1 μL) was placed on the film surface, and the static contact angle of the droplet was recorded. The measurement was replicated three times for each film sample. Statistical differences in contact angle were determined by an unpaired t-test with a two-tailed distribution and differences were considered statistically significant at p < 0.05 and p < 0.01. The data in the contact angle measurements are expressed as means ± standard deviation.
Measurements
Infrared (IR) spectra of the bulk samples were recorded on an IRPrestige-21 Fourier transform infrared spectrophotometer (Shimadzu Corporation, Kyoto, Japan) with a MIRacle A single reflection ATR unit using a Ge prism. 1H nuclear magnetic resonance (NMR) spectra were recorded on a Varian NMR System 500 (Varian Medical Systems, Palo Alto, CA) at 25 °C and a frequency of 500 MHz. Trifluoroacetic acid-d (TFA-d) was used as a solvent and tetramethylsilane as an internal standard. Synchrotron wide-angle X-ray diffraction (WAXD) measurements of the PPE-modified silk powder samples were performed by a BL45XU beamline at SPring-8, Harima, Japan, using an X-ray energy of 12.4 keV (wavelength: 0.1 nm). Differential scanning calorimetry (DSC) measurements were carried out using a TGA/DSC 2 Star system (Mettler Toledo, Greifensee, Switzerland). After the samples were dried at 60 °C for 24 h, 3 mg portions were encapsulated in aluminum pans and heated under a nitrogen atmosphere from 30 to 500 °C at a heating rate of 20 °C min−1.
Results and discussion
Chemoenzymatic modification using HRP
For the chemoenzymatic modification of silk fibroin, silkworm cocoons of Bombyx mori were washed with a sodium bicarbonate solution to remove sericin, dissolved in a LiBr solution, and dialyzed to obtain an aqueous solution of silk fibroin (1.0 wt%) according a reported procedure.41 The silk fibroin contained 5.2% tyrosine residues,1 which are a suitable substrate for HRP oxidation. Small phenolic compounds, including DMP, are also oxidized by HRP, usually resulting in a polymerized product.42 Therefore, we assumed that the simultaneous oxidation of silk fibroin and DMP using HRP could allow for the graft polymerization of DMP from the tyrosine residues in silk fibroin. The polymerization of DMP catalyzed with HRP in the presence of silk fibroin was performed as shown in Fig. 1. The polymerization reaction was initially carried out in 1 M sodium hydroxide, which dissolved both DMP and silk fibroin. The reaction proceeded at 40 °C for 2.5 h with periodic additions of hydrogen peroxide (H2O2) as an oxidant. However, no changes were observed in the reaction mixture, and only a small amount of unreacted silk fibroin was recovered after dialyzing the aqueous solution. This low yield was due to the hydrolysis of silk fibroin under highly basic conditions (pH > 13). Next, a two-phase reaction medium consisting of toluene and carbonate buffer (pH = 10) was used for the polymerization reaction. DMP was most likely to dissolve in toluene, and DMP oxidation could effectively occur at the toluene/aqueous buffer interface.43 The concentration of silk in the aqueous phase was constant at 0.5 wt%, whereas the concentration of DMP in toluene was varied from 0.02 to 0.1 M. The two-phase mixture was vigorously stirred at 40 °C for 2.5 h with periodic additions of H2O2. As the reaction proceeded, a yellow-to-orange powder precipitated at the interface of the two-phase mixture (Fig. 1b). Finally, the modified silk product was obtained after centrifugation followed by washing with toluene, methanol, and water to remove DMP, unreacted silk fibroin, and the PPE homopolymer (Fig. 1c). The results of the chemoenzymatic modification are summarized in Table 1. The yield increased as the initial concentration of DMP increased. The color of the product also changed from white to orange as the amount of DMP increased, as shown in Fig. 1c. The product was soluble in TFA but completely insoluble in water (buffer solutions) and toluene, which are good solvents for silk fibroin and PPE homopolymer, respectively. Thus, the silk fibroin modification using HRP and DMP induced a substantial change in the silk material's solubility.
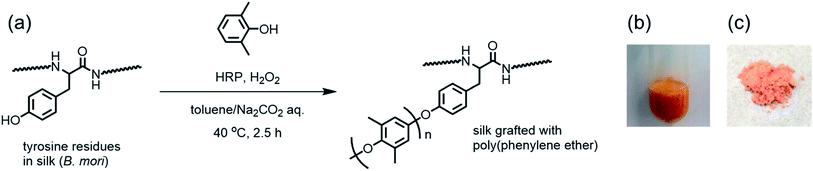 |
| | Fig. 1 (a) Scheme of the chemoenzymatic modification of silk fibroin with PPE using HRP and photographic images of (b) the two-phase reaction after 2.5 h and (c) the product obtained by centrifugation. | |
Table 1 Results of the chemoenzymatic modification of silk fibroin using HRP at various DMP feed concentrationsa
| Run |
[DMP]b (M) |
[H2O2]c (M) |
Yieldd (mg) |
Ratio of amide/ethere |
Color |
|
The two-phase reaction was carried out using HRP (3 mg) and silk fibroin (15 mg) in a carbonate buffer (3 mL, pH = 10) and DMP in toluene (3 mL) at 40 °C for 2.5 h with periodic additions of H2O2 every 15 min.
Feed concentration.
Final concentration after addition of all reagents.
Precipitate collected by centrifugation after washing with water, methanol, and toluene.
Determined by IR.
|
| 1 |
0 |
0 |
5.6 |
— |
White |
| 2 |
0.02 |
0.02 |
17.2 |
1/0.94 |
Yellow |
| 3 |
0.04 |
0.04 |
23.5 |
1/1.24 |
Orange |
| 4 |
0.06 |
0.06 |
24.7 |
1/1.44 |
Orange |
| 5 |
0.08 |
0.08 |
28.4 |
1/1.74 |
Orange |
| 6 |
0.1 |
0.1 |
30.3 |
1/1.72 |
Orange |
Characterization of the chemical structures
The chemical structure of the product was characterized by IR and 1H NMR spectroscopy. The IR spectra of PPE-modified silk fibroin are shown in Fig. 2. For native silk fibroin, characteristic peaks were observed at 1658, 1526, and 1234 cm−1, which were derived from the amide I (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), II (C–N–H bending), and III (C–N stretching) modes, respectively.21 Additionally, a broad peak assignable to the N–H stretching of amide bonds appeared between 3700 and 3100 cm−1. In contrast, PPE showed two strong peaks that were assignable to the aromatic CC and C–O stretching modes at 1593 and 1192 cm−1, respectively. These two characteristic peaks were derived from the PPE chain and were clearly observed in the spectra of all the silk samples subjected to chemoenzymatic modifications, in addition to the peaks of the silk backbone. Thus, silk fibroin was successfully modified with PPE chains via HRP-catalyzed modification. The amide/ether ratio was determined from the IR spectra by calculating the absorbance ratio between the peaks at 1658 and 1192 cm−1 as an index of the silk backbone and PPE chains, respectively (Table 1). As the feed concentration of DMP increased from 0.02 to 0.1 M, the PPE chain contents in the PPE-modified silk increased. The PPE-modified silk generated with DMP feed concentrations of 0.08 and 0.1 M showed similar amide/ether ratios. The amount of DMP involved in the silk modification was probably saturated at a feed concentration of approximately 0.08 M.
O stretching), II (C–N–H bending), and III (C–N stretching) modes, respectively.21 Additionally, a broad peak assignable to the N–H stretching of amide bonds appeared between 3700 and 3100 cm−1. In contrast, PPE showed two strong peaks that were assignable to the aromatic CC and C–O stretching modes at 1593 and 1192 cm−1, respectively. These two characteristic peaks were derived from the PPE chain and were clearly observed in the spectra of all the silk samples subjected to chemoenzymatic modifications, in addition to the peaks of the silk backbone. Thus, silk fibroin was successfully modified with PPE chains via HRP-catalyzed modification. The amide/ether ratio was determined from the IR spectra by calculating the absorbance ratio between the peaks at 1658 and 1192 cm−1 as an index of the silk backbone and PPE chains, respectively (Table 1). As the feed concentration of DMP increased from 0.02 to 0.1 M, the PPE chain contents in the PPE-modified silk increased. The PPE-modified silk generated with DMP feed concentrations of 0.08 and 0.1 M showed similar amide/ether ratios. The amount of DMP involved in the silk modification was probably saturated at a feed concentration of approximately 0.08 M.
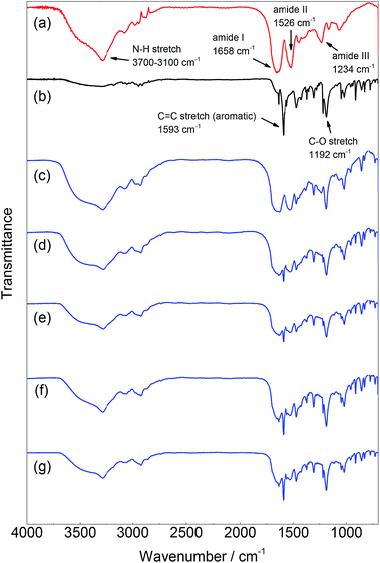 |
| | Fig. 2 IR spectra of (a) silk fibroin, (b) PPE, and PPE-modified silk with different feed concentrations of DMP: (c) 0.02 M, (d) 0.04 M, (e) 0.06 M, (f) 0.08 M, and (g) 0.1 M. | |
The structure of the PPE-modified silk was also confirmed by an 1H NMR spectrum obtained in TFA-d, as shown in Fig. 3. The native silk fibroin showed multiple signals assignable to aliphatic and α-methine protons mainly dominated by alanine and glycine residues at approximately 1.5 and 4.5 ppm, respectively. The two signals derived from the aromatic protons of the tyrosine residues in the silk appeared at 7.16 and 6.95 ppm. After the silk was modified with the PPE chains, no significant changes were observed in the signals of the silk backbone except that the signal intensities of the tyrosine residues decreased. However, new signals appeared at 7.2 and 2.3 ppm, which could be attributed to the aromatic and methyl protons, respectively, of the PPE chains. Because the PPE homopolymer was removed by washing the sample with toluene, this finding strongly supports the successful modification of silk fibroin with PPE chains at the tyrosine residues. The signal at 2.3 ppm was accompanied by a small signal at 2.2 ppm, which probably resulted from an undesired overoxidized PPE moiety. This overoxidized product may have caused the color of the PPE-modified silk to change from white to orange.
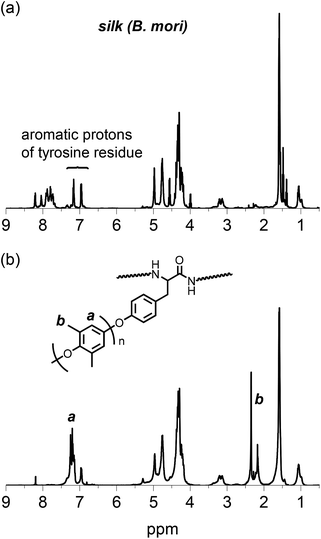 |
| | Fig. 3
1H NMR spectra of (a) silk fibroin and (b) PPE-modified silk (feed concentration of DMP: 0.06 M) in TFA-d. | |
A plausible mechanism
Scheme 1 illustrates the mechanism of the HRP-catalyzed generation of PPE-modified silk in the presence of H2O2. HRP's catalytic site is activated by H2O2 and subsequently oxidizes DMP and the tyrosine residues in silk to generate phenoxyl radical species.29 These radicals are immediately consumed in a radical coupling reaction. C–O coupling generally dominates the coupling reaction to yield a phenoxyphenol moiety, and the coupling between DMP and tyrosine radicals results in the chain elongation of PPE from silk fibroin (path A in Scheme 1). In contrast, C–C coupling between the radicals at the para- or ortho-positions in the resonance structures occurs to some extent, producing the branching structures shown in path B in Scheme 1.44,45 The resulting structure of the C–C coupling reaction can be further oxidized into an undesired keto-dimer by HRP, which probably causes product discoloration. Because there is no significant selectivity among the radical species, the C–O/C–C coupling reactions between DMP radicals also occur, resulting in PPE homopolymers, which can be completely removed by washing with a selective solvent, such as toluene or chloroform.
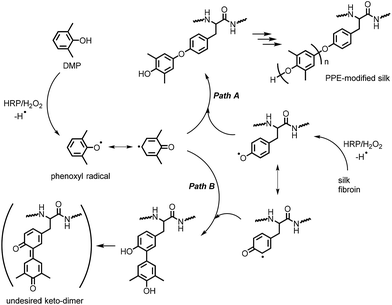 |
| | Scheme 1 Plausible mechanism of the HRP-catalyzed modification of silk with PPE. | |
Structural characterization by WAXD measurement
Silk materials' excellent mechanical properties are attributed to the higher-order structures assembled by the secondary structures, which predominantly consist of β-sheets. Therefore, the existence of structures comparable to those of native silk is important for the application of PPE-modified silk materials as structural materials. Because tyrosine residues typically exist at the ends of the repetitive units (GAGAGY) that form the β-sheet structures in silk, modifications of tyrosine residues' side groups would change or collapse the structures and conformations. The structure of PPE-modified silk was investigated by WAXD measurements and compared with that of silk fibroin. The WAXD profiles of the representative PPE-modified silk (DMP feed conc.: 0.06 M) and the silk fibroin fiber obtained by removing sericin from the silk cocoon are shown in Fig. 4. The characteristic peaks derived from the silk II structure were detected at d spacings of 4.3 and 3.6 Å in the profile of the silk fibroin fiber; these peaks were assignable to the reflections of (020) and (211) in the antiparallel β-sheet crystal domains, respectively.4,46 In contrast, the PPE-modified silk's profile was similar to that of silk fibroin, with slightly long d spacings of 4.5, 3.8, 2.4, and 2.2 Å (Fig. 4b). Therefore, the modification of silk with PPE at the tyrosine residues did not hamper the formation of antiparallel β-sheet structures. However, the bulky PPE chains branched from tyrosine residues probably caused a slight expansion in the d spacings of the β-sheet crystal domains. The PPE-modified silks with lower (0.02 M) and higher (0.08 M) DMP feed concentrations showed similar WAXD profiles, revealing the existence of β-sheet structures (Fig. S1 in ESI†). No significant change in the peaks of β-sheet was observed among the PPE modified silks in the tested range of the PPE content.
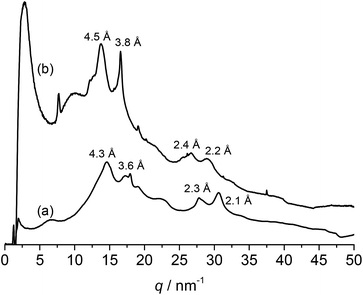 |
| | Fig. 4 WAXD profiles of (a) silk fibroin fiber and (b) PPE-modified silk (feed conc. of DMP: 0.06 M). | |
Thermal analysis
The thermal properties of the PPE-modified silks were investigated by DSC measurements after drying the samples at 60 °C. The DSC profile of the PPE-modified silk in the first heating scan is shown in Fig. 5. Decomposition of the PPE-modified silk occurred at approximately 250 °C. The decomposition behavior almost corresponded to that of silk fibroin.37 In addition, a baseline shift corresponding to the glass transition temperature (Tg) of the PPE chains appeared at around 200 °C,47 indicating that the amorphous domains of the PPE branches segregated from the polypeptide main chains. Polypeptide materials, which form β-sheet structures, including natural structural proteins, generally show no melting and/or glass transitions below their decomposition temperatures, resulting in poor thermal processability.48 Therefore, imparting Tg to silk materials without sacrificing their distinctive structures is advantageous for various applications in terms of enlarging processing windows. Indeed, self-standing rigid films were obtained from the PPE-modified silks using a thermal press at 220 °C, as shown in Fig. 6a. However, film preparation via a casting method from a TFA solution failed because of the material's poor solubility.
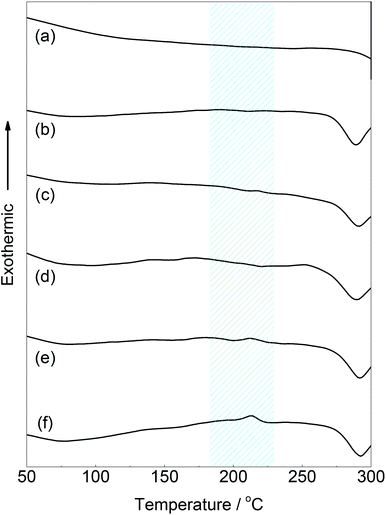 |
| | Fig. 5 DSC traces of (a) the native silk fibroin and PPE-modified silks with different feed concentrations of DMP (b: 0.02 M, c: 0.04 M, d: 0.06 M, e: 0.08 M, and f: 0.10 M) in the first heating scan at 20 °C min−1. | |
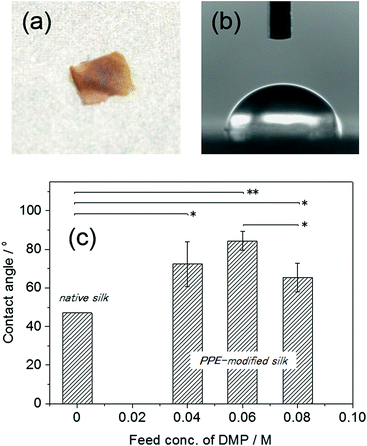 |
| | Fig. 6 (a) The PPE-modified silk film (DMP feed conc.: 0.06 M) prepared by thermal press at 2.5 MPa and 200 °C for 30 min, (b and c) the contact angles of water droplets on the PPE-modified silk films produced with different DMP feed concentrations. The contact angle of the native silk was obtained from ref. 12 (p value: *p < 0.05, **p < 0.01). | |
Static contact angle measurements on the PPE-modified films
To evaluate the hydrophobicity of the PPE-modified silk, the static contact angles of water droplets on the films were measured (Fig. 6b and c). The contact angles of the PPE-modified silk films ranged from 65 ± 7.4° to 84 ± 4.8°, and the highest value (84 ± 4.8°) was obtained for the film with a DMP feed concentration of 0.06 M. Unfortunately, some of the films (DMP feed concentrations of 0.02 and 0.1 M) were so brittle that the contact angles could not be measured. The Tg of the PPE-modified silk was not clearly observed for the DMP feed concentration of 0.02 M, which caused the less processability by the lack of mobility. On the other hand, the PPE formed large domains at the highest PPE content (DMP feed concentration of 0.1 M), resulting in the brittle film probably because of the phase separation between PPE and silk. No significant relationship between the contact angle and the PPE chain contents was observed, although the angle was slightly decreased at a DMP feed concentration of 0.08 M. As a control, we also attempted to measure the contact angle on a native silk film prepared by casting the aqueous silk fibroin solution. However, the silk film swelled upon the addition of the water droplet and completely dissolved, even after immersion in methanol and/or drying in a vacuum oven. Thus, the native silk film exhibited a highly hydrophilic surface, which easily absorbed the humidity. The static contact angle on the silk fibroin film was previously determined to be 47°,12 and all the PPE-modified films had higher contact angles than the reference value. This result indicated that the chemoenzymatic modification of silk with PPE chains effectively provided the silk-based film with a hydrophobic surface. Unlike the native silk film, the PPE-modified silk films were stable when in contact with a water droplet over a long period (>1 h) and did not exhibit any swelling or dissolution.
Conclusions
The chemoenzymatic polymerization of DMP by the HRP/H2O2 system in the presence of silk fibroin was carried out in a two-phase reaction medium of a toluene/carbonate buffer. Characterization of the product by IR and 1H NMR spectroscopy revealed that the resulting silk material was successfully modified with PPE chains. As the DMP feed concentration increased, PPE-modified silk materials with higher PPE chain contents were obtained. The PPE-modified silks formed β-sheet crystalline domains similar to those of native silk fibers, indicating that the introduction of PPE grafting chains did not impede the formation of secondary structures in the silk domains. In addition, the modification with PPE imbued the silk materials with glass transitions, enabling the PPE-modified silks to convert into self-standing films upon thermal processing. The films showed higher contact angles of up to 84° than the native silk film because of the highly hydrophobic surface induced by the PPE domains. This simple enzymatic system-based modification technique can be applied not only to silk fibroin but also to other types of structural proteins, enabling these natural materials to be utilized for possible applications requiring strong mechanical properties. The modified silks are expected to be applicable to reinforcing agents for the engineering plastics or surface coatings for the polymeric structural materials. Some of the films prepared from the PPE-modified silks were brittle because of the phase separation of the PPE domains. Therefore, a further study on the optimization for the degree of polymerization and the graft density of the PPE chains is ongoing in order to control the physical properties as well as processability of the modified silks.
Acknowledgements
The authors acknowledge Dr Hiroyasu Masunaga and Dr Takaaki Hikima for conducting measurements on the synchrotron WAXD at SPring-8, Harima, Japan. This work was financially supported by Impulsing Paradigm Change through the Disrupt Technologies Program (ImPACT).
Notes and references
- B. Lotz and F. Colonna Cesari, Biochimie, 1979, 61, 205–214 CrossRef CAS PubMed.
- C.-Z. Zhou, F. Confalonieri, M. Jacquet, R. Perasso, Z.-G. Li and J. Janin, Protein Struct. Funct. Genet., 2001, 44, 119–122 CrossRef CAS PubMed.
- H. Saito, R. Tabeta, T. Asakura, Y. Iwanaga, A. Shoji, T. Ozaki and I. Ando, Macromolecules, 1984, 17, 1405–1412 CrossRef CAS.
- A. Martel, M. Burghammer, R. J. Davies, E. Di Cola, C. Vendrely and C. Riekel, J. Am. Chem. Soc., 2008, 130, 17070–17074 CrossRef CAS PubMed.
- Z. Gong, L. Huang, Y. Yang, X. Chen and Z. Shao, Chem. Commun., 2009, 48, 7506–7508 RSC.
- C. Zhao, J. Yao, H. Masuda, R. Kishore and T. Asakura, Biopolymers, 2003, 69, 253–259 CrossRef CAS PubMed.
- Z. Zhu, T. Imada and T. Asakura, Mater. Chem. Phys., 2009, 117, 430–433 CrossRef CAS.
- G. H. Altman, F. Diaz, C. Jakuba, T. Calabro, R. L. Horan, J. Chen, H. Lu, J. Richmond and D. L. Kaplan, Biomaterials, 2003, 24, 401–416 CrossRef CAS PubMed.
- A. R. Murphy and D. L. Kaplan, J. Mater. Chem., 2009, 19, 6443–6450 RSC.
- K. Numata and D. L. Kaplan, Adv. Drug Delivery Rev., 2010, 62, 1497–1508 CrossRef CAS PubMed.
- Y. Gotoh, M. Tsukada, S.-i. Aiba and N. Minoura, Int. J. Biol. Macromol., 1996, 18, 19–26 CrossRef CAS PubMed.
- Y. Gotoh, M. Tsukada, N. Minoura and Y. Imai, Biomaterials, 1997, 18, 267–271 CrossRef CAS PubMed.
- C. Acharya, B. Hinz and S. C. Kundu, Biomaterials, 2008, 29, 4665–4675 CrossRef CAS PubMed.
- F. Galeotti, A. Andicsova, F. Bertini and C. Botta, J. Mater. Sci., 2013, 48, 7004–7010 CrossRef CAS.
- S. Sofia, M. B. McCarthy, G. Gronowicz and D. L. Kaplan, J. Biomed. Mater. Res., 2001, 54, 139–148 CrossRef CAS PubMed.
- K. Cai, K. Yao, S. Lin, Z. Yang, X. Li, H. Xie, T. Qing and L. Gao, Biomaterials, 2002, 23, 1153–1160 CrossRef CAS PubMed.
- C. P. Vepari and D. L. Kaplan, Biotechnol. Bioeng., 2006, 93, 1130–1137 CrossRef CAS PubMed.
- A. Sagnella, M. Zambianchi, M. Durso, T. Posati, A. Del Rio, A. Donnadio, A. Mazzanti, A. Pistone, G. Ruani, R. Zamboni, V. Benfenati and M. Melucci, RSC Adv., 2015, 5, 63401–63406 RSC.
- H. Shiozaki, M. Tsukada, Y. Gotoh, N. Kasai and G. Freddi, J. Appl. Polym. Sci., 1994, 52, 1037–1045 CrossRef CAS.
- Z. Cai, G. Jiang and Y. Qiu, J. Appl. Polym. Sci., 2004, 91, 3579–3586 CrossRef CAS.
- A. R. Murphy, P. S. John and D. L. Kaplan, Biomaterials, 2008, 29, 2829–2838 CrossRef CAS PubMed.
- P. N. Atterberry, T. J. Roark, S. Y. Severt, M. L. Schiller, J. M. Antos and A. R. Murphy, Biomacromolecules, 2015, 16, 1582–1589 CrossRef CAS PubMed.
- D. Rabuka, Curr. Opin. Chem. Biol., 2010, 14, 790–796 CrossRef CAS PubMed.
- K. Yazawa and K. Numata, Molecules, 2014, 19, 13755 CrossRef PubMed.
- K. Numata, Polym. J., 2015, 47, 537–545 CrossRef CAS.
- S. Sampaio, P. Taddei, P. Monti, J. Buchert and G. Freddi, J. Biotechnol., 2005, 116, 21–33 CrossRef CAS PubMed.
- G. Freddi, A. Anghileri, S. Sampaio, J. Buchert, P. Monti and P. Taddei, J. Biotechnol., 2006, 125, 281–294 CrossRef CAS PubMed.
- K. Numata and P. J. Baker, Biomacromolecules, 2014, 15, 3206–3212 CrossRef CAS PubMed.
- N. C. Veitch, Phytochemistry, 2004, 65, 249–259 CrossRef CAS PubMed.
- J. S. Dordick, M. A. Marletta and A. M. Klibanov, Biotechnol. Bioeng., 1987, 30, 31–36 CrossRef CAS PubMed.
- Y.-P. Xu, G.-L. Huang and Y.-T. Yu, Biotechnol. Bioeng., 1995, 47, 117–119 CrossRef CAS PubMed.
- H. Uyama, N. Maruichi, H. Tonami and S. Kobayashi, Biomacromolecules, 2002, 3, 187–193 CrossRef CAS PubMed.
- K. Won, Y. H. Kim, E. S. An, Y. S. Lee and B. K. Song, Biomacromolecules, 2004, 5, 1–4 CrossRef CAS PubMed.
- R. Ikeda, J. Sugihara, H. Uyama and S. Kobayashi, Macromolecules, 1996, 29, 8702–8705 CrossRef CAS.
- S. Nanayakkara, Z. Zhao, A. F. Patti, L. He and K. Saito, ACS Sustainable Chem. Eng., 2014, 2, 1947–1950 CrossRef CAS.
- J. Ming, F. Pan and B. Zuo, Int. J. Biol. Macromol., 2015, 75, 398–401 CrossRef CAS PubMed.
- N. Agarwal, D. A. Hoagland and R. J. Farris, J. Appl. Polym. Sci., 1997, 63, 401–410 CrossRef CAS.
- C. Fu, D. Porter and Z. Shao, Macromolecules, 2009, 42, 7877–7880 CrossRef CAS.
- M.-R. Buga, C. Zaharia, M. Bălan, C. Bressy, F. Ziarelli and A. Margaillan, Mater. Sci. Eng., C, 2015, 51, 233–241 CrossRef CAS PubMed.
- I. Drachuk, R. Calabrese, S. Harbaugh, N. Kelley-Loughnane, D. L. Kaplan, M. Stone and V. V. Tsukruk, ACS Nano, 2015, 9, 1219–1235 CrossRef CAS PubMed.
- D. N. Rockwood, R. C. Preda, T. Yucel, X. Wang, M. L. Lovett and D. L. Kaplan, Nat. Protoc., 2011, 6, 1612–1631 CrossRef CAS PubMed.
- J. A. Nicell, K. W. Saadi and I. D. Buchanan, Bioresour. Technol., 1995, 54, 5–16 CrossRef CAS.
- J. Nunoshige, H. Akahoshi, Y. Shibasaki and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5278–5282 CrossRef CAS.
- H. Tonami, H. Uyama, S. Kobayashi, H. Higashimura and T. Oguchi, J. Macromol. Sci., Part A: Pure Appl.Chem., 1999, 36, 719–730 CrossRef.
- K. Saito, S. Pant and M. T. W. Hearn, J. Appl. Polym. Sci., 2011, 122, 2174–2180 CrossRef CAS.
- A. Martel, M. Burghammer, R. J. Davies and C. Riekel, Biomacromolecules, 2007, 8, 3548–3556 CrossRef CAS PubMed.
- T. Fukuhara, Y. Shibasaki, S. Ando and M. Ueda, Polymer, 2004, 45, 843–847 CrossRef CAS.
- J. Hwang and T. J. Deming, Biomacromolecules, 2001, 2, 17–21 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra02258h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence





