
Palladium-catalyzed N-arylsulfonamide formation from arylsulfonyl hydrazides and nitroarenes†
Feng
Zhao
a,
Bin
Li
a,
Huawen
Huang
a and
Guo-Jun
Deng
*ab
aKey Laboratory of Environmentally Friendly Chemistry and Application of Ministry of Education, College of Chemistry, Xiangtan University, Xiangtan 411105, China. E-mail: gjdeng@xtu.edu.cn; Fax: +86-0731-5829-2251; Tel: +86-0731-5829-8601
bKey Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100080, China
First published on 26th January 2016
Abstract
A palladium-catalyzed construction for N-arylsulfonamide from nitroarenes and arylsulfonyl hydrazides is developed. In this protocol, abundant and stable nitroarenes serve as the nitrogen sources by in situ reduction reaction of hydrogen released from arylsulfonyl hydrazides. No external oxidants or reductants are needed for this kind of transformation.
It is well-known that N-arylsulfonamides are a class of crucial building blocks that are ubiquitous in many biologically active compounds and pharmaceutical intermediates, and these structures have important roles in organic synthesis and biological and medicinal study.1 Therefore, the exploration of efficient and green methods for the synthesis of sulfonamides under mild conditions is an attractive area for organic and pharmaceutical chemists. During the past years, various efforts have been made for the construction of sulfonamides.2 The conventional route mainly relies on the reaction of sulfonyl chlorides3 or sodium sulfinates4 with amino compounds due to the simplicity and practicability. The transition-metal-catalyzed couplings of sulfonamides with aryl halides,5 aryl boronic acids,6 cyclohexanones,7 alcohols8 and hydrocarbons9 provide alternative methods to synthesize N-arylsulfonamides. However, there are still many shortcomings existing in the current research, such as harsh reaction conditions, unstable starting materials, poor functional group tolerance, requirements of additional ligand and/or base. Some of approaches suffer from the utility of amino groups as nitrogen sources, which are potentially toxic, volatile and thus leading to limitations and tedious procedures. To overcome these drawbacks, it is highly desirable to develop a method for the synthesis of N-arylsulfonamides from cheap and stable nitrogen sources.
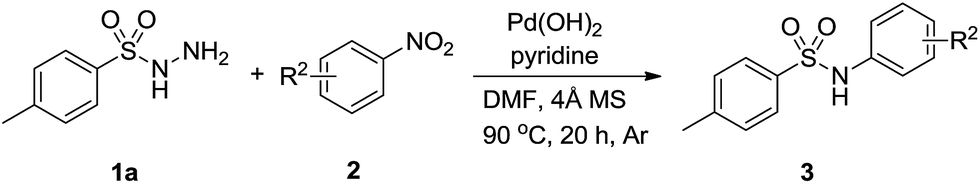
As we known, aromatic nitro compounds can undergo reductive reactions for the preparation of the corresponding arylamines. In recent years, there have been significant attractions to transition-metal-catalyzed C–N bond formations by utilizing nitroarenes as starting materials. The nitroarenes were reduced in situ via borrowing hydrogen strategy (hydrogen transfer)10,11 or by adding external reducing agent.12 We envisioned that it might be rational using nitroarenes instead of aromatic amines as abundant and stable nitrogen sources for the selective construction of S–N bond. Previously, our group demonstrated a series of protocols by harnessing alcohols and nitroarenes to form second amines,13 tertiary amines,14 amides15 and N-containing heterocycles.16 We also reported a palladium-catalyzed one-pot synthesis of diarylamines from nitroarenes and cyclohexanones.17 In these methods, nitroarenes successfully acted as the hydrogen acceptors and were reduced to amines in situ through hydrogen transfer methodology, and no external reductants were needed to the reactions. As part of our continuing efforts in using nitroarenes as the coupling partners to construct C–N and N-hetero bonds, herein, we report a palladium-catalyzed formation of N-arylsulfonamides from nitroarenes and arylsulfonyl hydrazides using the hydrogen transfer strategy (Scheme 1).
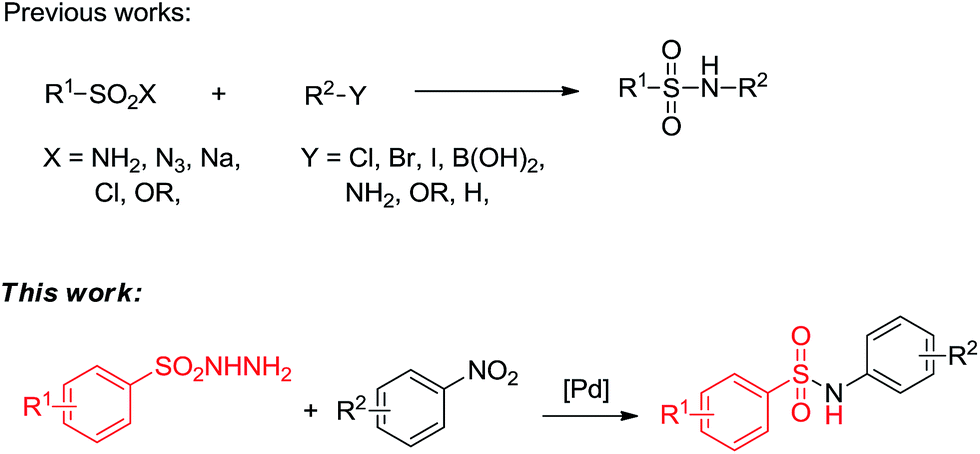 | ||
| Scheme 1 Different pathways for the synthesis of sulphonamides. | ||
We begin our research by investigating the reaction of p-toluenesulfonyl hydrazides (1a) with nitrobenzene (2a) in DMF by using Pd(OAc)2 as catalyst and the desired product was obtained in 19% yield as detected by GC and NMR methods (Table 1, entry 1). Afterwards, a variety of palladium catalysts were examined for this reaction. Similar results were achieved when employing PdCl2, PdBr2, Pd(COD)Cl2 as the catalyst (Table 1, entries 2–4). Pd(acac)2 and Pd(TFA)2 improved the reaction yield to 36% and 35%, respectively (entries 5 and 6). Among the catalysts screened, Pd(OH)2 showed the most effective and afford 3a in 42% yield (entry 7). Solvents also played an important role and the effect of solvents on this transformation was also examined. DMA, NMP and pyridine gave a similar result (entries 8–10). A lower yield was obtained when DMSO was employed as the solvent (entry 11). Unfortunately, when the reaction was conducted in weak polar solvent such as dioxane and toluene, only a trace amount of desired product was acquired under the similar reaction conditions (entries 12 and 13). In order to improve the reaction yield, we attempted to investigate some additives. Encouraged by the result of entry 10, we used 1 equiv. of pyridine as the additive in DMF. To our delight, the yield of 3a was increased to 58% (entry 14). We tested some external reductants such as hydrazine hydrate and isopropanol to enhance the reaction yield, however both of them were not efficient (entries 15 and 16). We proposed that the role of pyridine was acted as a base so we tried organic base triethylamine and inorganic base potassium carbonate, but the results were also unsatisfactory (entries 16–17). Notably, a desired yield could be obtained when the reaction was performed under an argon atmosphere and molecular sieve was added to the reaction system (entries 18–20).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Solvent | Yieldb (%) |
| a Conditions: 1a (0.4 mmol), 2a (0.2 mmol), catalyst (5 mol%), additive (0.2 mmol), solvent (0.8 mL), 90 °C, 20 h under air unless otherwise noted. b GC yield based on 2a. c 100 mg 4 Å molecular sieves was added. d Under argon. | ||||
| 1 | Pd(OAc)2 | DMF | 19 | |
| 2 | PdCl2 | DMF | 25 | |
| 3 | PdBr2 | DMF | 24 | |
| 4 | Pd(COD)Cl2 | DMF | 42 | |
| 5 | Pd(acac)2 | DMF | 36 | |
| 6 | Pd(TFA)2 | DMF | 35 | |
| 7 | Pd(OH)2 | DMF | 42 | |
| 8 | Pd(OH)2 | DMF | 36 | |
| 9 | Pd(OH)2 | NMP | 34 | |
| 10 | Pd(OH)2 | Pyridine | 37 | |
| 11 | Pd(OH)2 | DMSO | 23 | |
| 12 | Pd(OH)2 | Toluene | Trace | |
| 13 | Pd(OH)2 | Dioxane | Trace | |
| 14 | Pd(OH)2 | Pyridine | DMF | 58 |
| 15 | Pd(OH)2 | N2H4·H2O | DMF | Trace |
| 16 | Pd(OH)2 | i-PrOH | DMF | 19 |
| 17 | Pd(OH)2 | Et3N | DMF | 21 |
| 18 | Pd(OH)2 | K2CO3 | DMF | Trace |
| 19c | Pd(OH)2 | Pyridine | DMF | 66 |
| 20d | Pd(OH)2 | Pyridine | DMF | 72 |
| 21c,d | Pd(OH)2 | Pyridine | DMF | 84 |
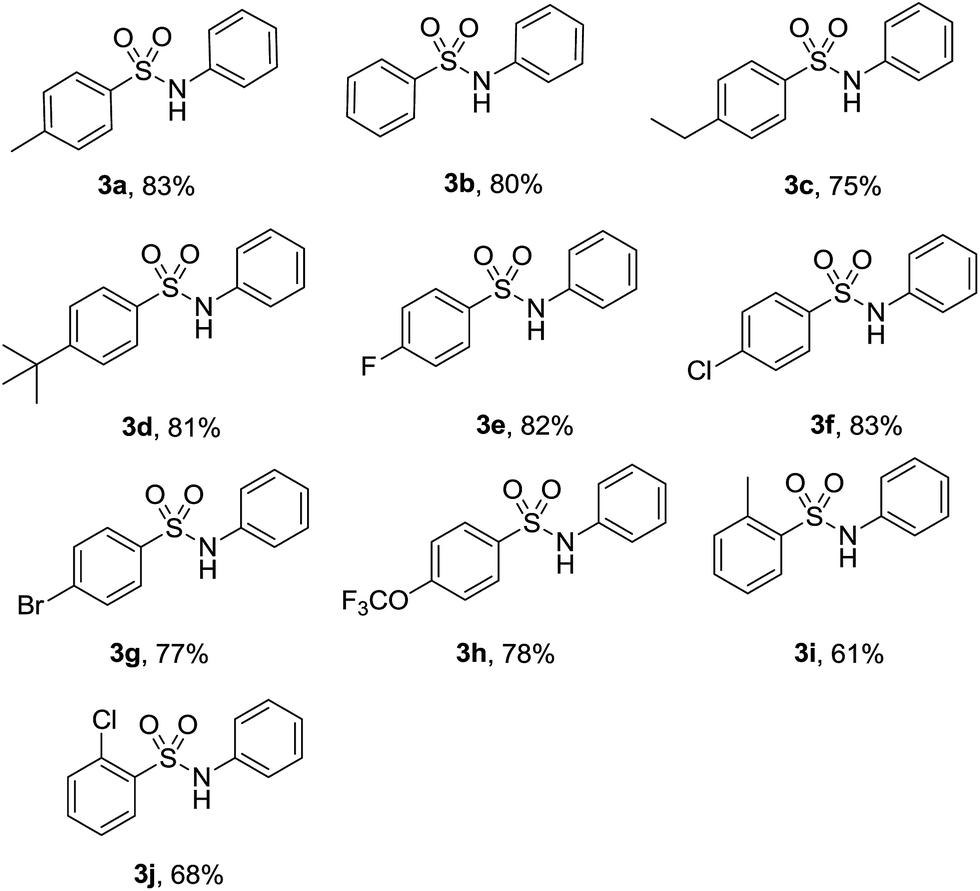
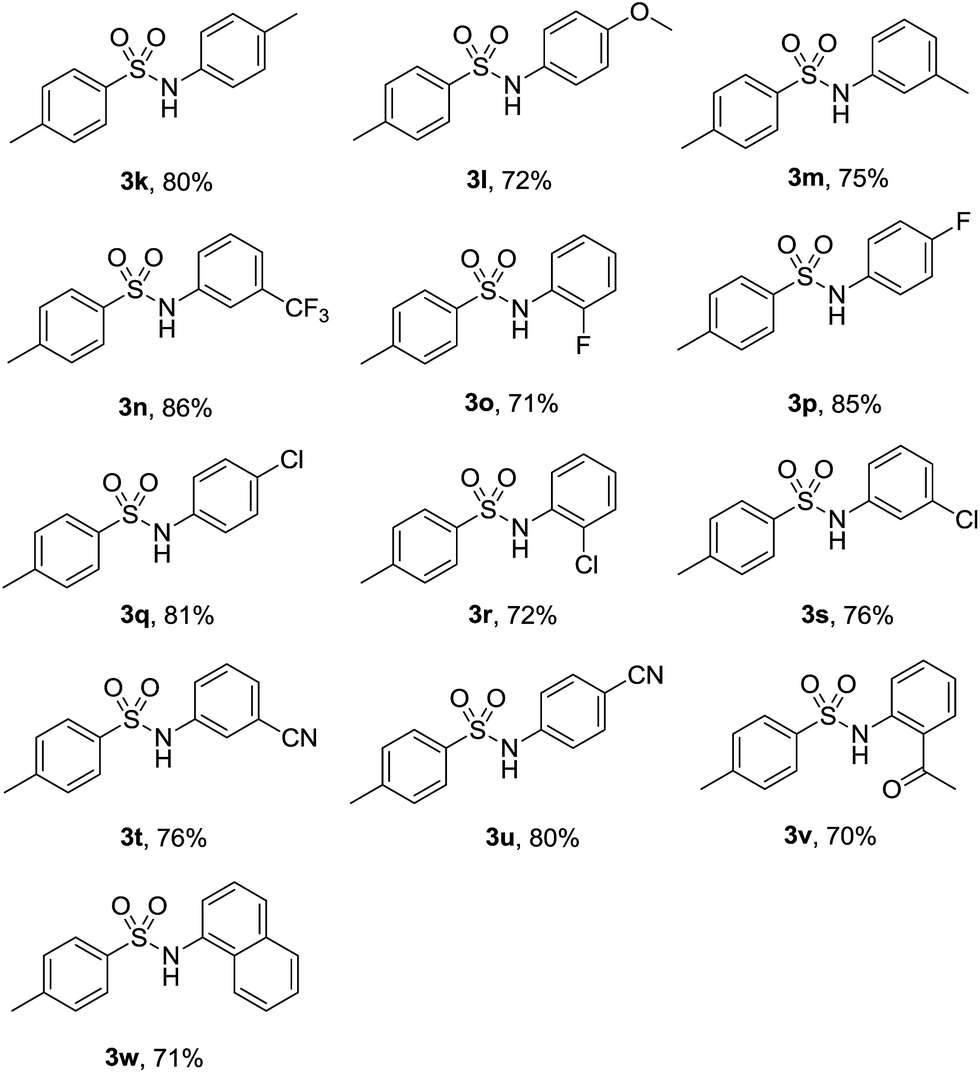
With the optimized reaction conditions in hand, we explored the scope of this transformation. As presented in Table 2, a variety of arylsulfonyl hydrazides were successfully reacted with nitrobenzene (2a) to produce corresponding N-arylsulfonamides in moderate to good yields. Generally, alkyl substituents at the para position of sulfonyl hydrazide slightly affected the reaction yield (3a–3d). Functional groups such as halogens and trifluoromethoxy were well tolerated under the optimal reaction conditions (3e–3h). It should be noted that cleavage of C-halogen bond was not observed during the reaction process. When ortho-substituted arylsulfonyl hydrazides reacted with nitrobenzene, relatively lower yields were obtained probably due to the steric effect of the substituents (3i-3j).
|
|
|---|
| a Conditions: 1 (0.4 mmol), 2a (0.2 mmol), Pd(OH)2 (5 mol%), pyridine (0.2 mmol), DMF (0.8 mL), 4 Å MS (100 mg), 90 °C, 20 h, under argon; isolated yield based on 2a. |
|
Subsequently, a number of substituted nitroarenes were employed to react with p-toluenesulfonyl hydrazide and the results are shown in Table 3. Methyl and methoxy nitrobenzenes reacted smoothly with 1a to give the corresponding products in reasonable yields (3k–3m). Halogen substituents such as trifluoromethyl, fluoro and chloro were well tolerated under the optimized reaction conditions, and the target products were obtained in good yields (3n–3s). Other substrates bearing electron-withdrawing group such as cyano remained effective and gave the corresponding arylsulfonamides in good yields (3t–3u). Notably, when 1-(2-nitrophenyl)ethanone was subjected to this reaction system, the desired product could be achieved in 70% yield (3v). Finally, a more steric bulky substrate such as 1-nitronaphthelene also successfully afforded the product in 71% yield (3w).
|
|
|---|
| a Conditions: 1a (0.4 mmol), 2 (0.2 mmol), Pd(OH)2 (5 mol%), pyridine (0.2 mmol), DMF (0.8 mL), 4 Å MS (100 mg), 90 °C, 20 h, under argon; isolated yield based on 2. |
|
For the mechanism of this transformation, we proposed a catalytic process that includes the following steps (Scheme 2). The reaction of arylsulfonyl hydrazide with a Pd catalyst results in a Pd complex A and sulfonyl diazene 3, which is formed through successive hydrogen transferring and β-hydride elimination. In the presence of a Pd catalyst, liberation of N2 and H+ from 3 leads to the formation of arylsulfonylpalladium B.18 On the other hand, nitrobenzene is reduced to nitrosobenzene with the help of complex A.17,19 Then, the reaction of complex B with nitrosobenzene gives N-arylsulfonyl-N-phenylhydroxyamine 6. At last, hydrogenation of 6 affords the desired product and regenerates the Pd catalyst.
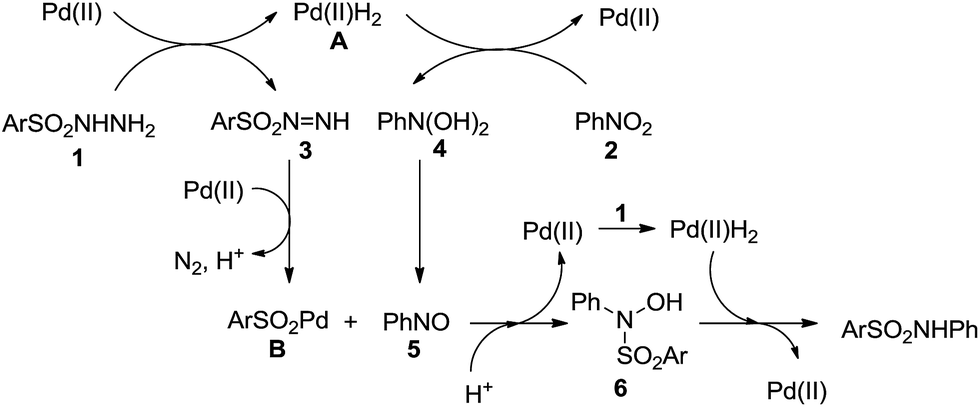 | ||
| Scheme 2 Proposed reaction mechanism. | ||
To gain a mechanistic insight, deuterium labelling experiments were conducted and the results were summarized in Scheme 3. When TsNDND2 (ref. 20) (1a–D) reacted with 2a under the standard reaction conditions, N-deuterated product was obtained (Scheme 3, eqn (1)). When 1a reacted with 2a in the presence of D2O, no deuterated product 3a was observed (Scheme 3, eqn (2)). Treatment of 3a with D2O led to 3a recovered and no deuterated 3a was obtained (Scheme 3, eqn (3)). These results indicated that the hydrogen transfer process very likely proceeded according to the proposed mechanism.
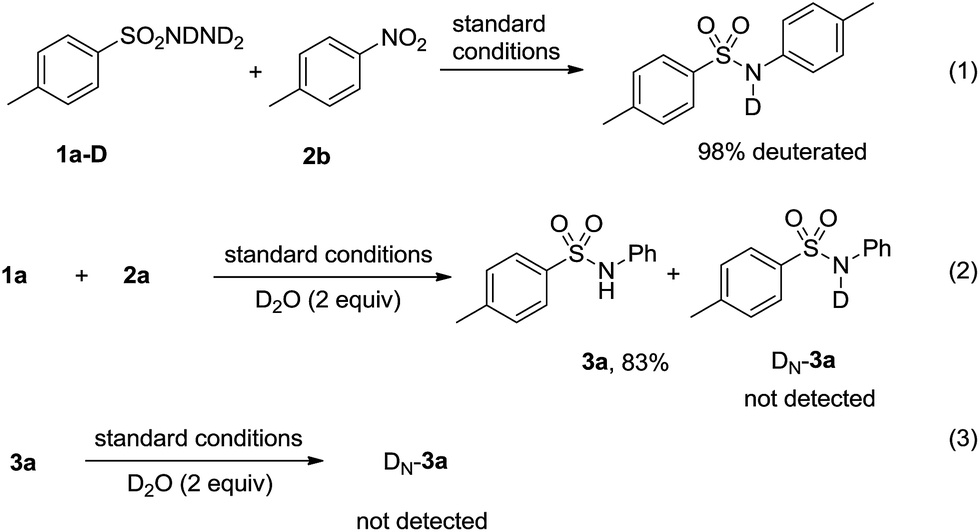 | ||
| Scheme 3 Deuterium labelling experiments. | ||
Conclusions
In summary, we have developed a novel approach for the synthesis of N-arylsulfonamides from nitroarenes and arylsulfonyl hydrazides under palladium-catalysis conditions. Nitroarenes were reduced in situ by hydrogen released from arylsulfonyl hydrazides and no external reducing agent is necessary. The reaction showed good substrate scope and various functional groups such as halogens, acetyl, trifluoromethyl and cyano were well tolerated under the optimized reaction conditions. This method affords an efficient approach using readily available and stable nitroarenes for facile construction of N-arylsulfonamides under mild reaction conditions with good selectivity. Further investigations of the scope and mechanism for this transformation are undergoing in our laboratory.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21172185, 21372187, 21572194), the Hunan Provincial Innovative Foundation for Postgraduate (CX2014B258), and the Research Fund for the Doctoral Program of Higher Education of China, Ministry of Education of China (20124301110005).Notes and references
- (a) D. A. Smith and R. M. Jones, Curr. Opin. Drug Discovery Dev., 2008, 11, 72 CAS; (b) A. Scozzafava, T. Owa, A. Mastrolorezo and C. T. Supuran, Curr. Med. Chem., 2003, 10, 925 CrossRef CAS; (c) J. D. Wilden, J. Chem. Res., 2010, 34, 541 CrossRef CAS; (d) J. B. Buckingham, Dictionary of Natural Products, Chapman and Hall, London, 1994, vol. 1 Search PubMed.
- For selected reviews, see: (a) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC; (b) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS; (c) S. Caddick, J. D. Wilden and D. B. Judd, J. Am. Chem. Soc., 2004, 126, 1024 CrossRef CAS; (d) J. Wilden, L. Geldeard, C. Lee, D. Judd and S. Caddick, Chem. Commun., 2007, 10, 1074 RSC; (e) S. Lavy, J. Miller, M. Pazicky, A. Rodrigues, C. Jaekel, D. Serra, N. Vinokurov, M. Limbach and F. Rominger, Adv. Synth. Catal., 2010, 352, 2993 CrossRef CAS; (f) S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080 CrossRef CAS; (g) K. Bahrami, M. Khodaei and M. Soheilizad, J. Org. Chem., 2009, 74, 9287 CrossRef CAS.
- (a) K. K. Andersen, in Comprehensive Organic Chemistry, ed. D. N. Jones, Pergamon, Oxford, 1979, vol. 3, pp. 331–340, For recent selected examples, see: Search PubMed; (b) J. R. DeBergh, N. Niljianskul and S. L. Buchwald, J. Am. Chem. Soc., 2013, 135, 10638 CrossRef CAS; (c) J. Baffoe, M. Hoe and B. Touré, Org. Lett., 2010, 12, 1532 CrossRef CAS; (d) X. Deng and N. Mani, Green Chem., 2006, 8, 835 RSC; (e) R. Sridhar, B. Srinivas, V. Kumar, M. Narender and K. Rao, Adv. Synth. Catal., 2007, 349, 1873 CrossRef CAS; (f) A. Kamal, J. Reddy, E. Bharathi and D. Dastagiri, Tetrahedron Lett., 2008, 49, 348 CrossRef CAS.
- (a) X. Tang, L. Huang, C. Qi, X. Wu, W. Wu and H. Jiang, Chem. Commun., 2013, 49, 6102 RSC; (b) C. Buathongjan, D. Beukeaw and S. Yotphan, Eur. J. Org. Chem., 2015, 1575 CrossRef CAS; (c) X. Pan, J. Gao, J. Liu, J. Lai, H. Jiang and G. Yuan, Green Chem., 2015, 17, 1400 RSC; (d) K. Yang, M. Ke, Y. Lin and Q. Song, Green Chem., 2015, 17, 1395 RSC.
- (a) J. Yin and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 6043 CrossRef CAS; (b) G. Burton, P. Cao, G. Li and R. Rivero, Org. Lett., 2003, 5, 4373 CrossRef CAS; (c) B. R. Rosen, J. C. Ruble, T. J. Beauchamp and A. Navarro, Org. Lett., 2011, 13, 2564 CrossRef CAS; (d) W. Deng, L. Liu, C. Zhang, M. Liu and Q.-X. Guo, Tetrahedron Lett., 2005, 46, 7295 CrossRef CAS.
- (a) P. Y. S. Lam, G. Vincent, C. G. Clark, S. Deudon and P. K. Jadhav, Tetrahedron Lett., 2001, 42, 3415 CrossRef CAS; (b) C. Pan, J. Cheng, H. Wu, J. Ding and M. Liu, Synth. Commun., 2009, 39, 2082 CrossRef CAS; (c) S. Y. Moon, J. Nam, K. Rathwell and W. S. Kim, Org. Lett., 2014, 16, 338 CrossRef CAS.
- X. Cao, Y. Bai, Y. Xie and G. J. Deng, J. Mol. Catal. A: Chem., 2014, 383–384, 94 CrossRef CAS.
- For selected examples, see: (a) P. Qu, C. Sun, J. Ma and F. Li, Adv. Synth. Catal., 2014, 356, 447 CrossRef CAS; (b) A. J. A. Watson, A. C. Maxwell and J. M. J. Williams, J. Org. Chem., 2011, 76, 2328 CrossRef CAS; (c) H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2007, 46, 409 CrossRef CAS; (d) S. Shekhar, T. B. Dunn, B. J. Kotecki, D. K. Montavon and S. C. Cullen, J. Org. Chem., 2011, 76, 4552 CrossRef CAS.
- (a) B. Kalita, A. A. Lamar and K. M. Nicholas, Chem. Commun., 2008, 44, 4291 RSC; (b) B. Xiao, T. J. Gong, J. Xu, Z. J. Liu and L. Liu, J. Am. Chem. Soc., 2011, 133, 1466 CrossRef CAS PubMed; (c) H. Zhao, Y. Shang and W. Su, Org. Lett., 2013, 15, 5106 CrossRef CAS; (d) Q. Z. Zheng, Y. F. Liang, C. Qin and N. Jiao, Chem. Commun., 2013, 49, 5654 RSC.
- For selected reviews on hydrogen transfer reaction, see: (a) T. Nixon, M. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 5, 753 RSC; (b) G. Guillena, D. J. Ramon and M. Yus, Chem. Rev., 2010, 110, 1611 CrossRef CAS; (c) A. Watson and J. M. J. Williams, Science, 2010, 329, 635 CrossRef CAS.
- For selected examples, see: (a) J. Oakdale, D. Solano, J. Fettinger, M. Haddadin and M. J. Kurth, Org. Lett., 2009, 11, 2760 CrossRef CAS; (b) X. J. Cui, Y. Zhang, F. Shi and Y. Q. Deng, Chem.–Eur. J., 2011, 17, 2587 CrossRef CAS; (c) A. Zanardi, J. A. Mata and E. Peris, Chem.–Eur. J., 2010, 16, 10502 CrossRef CAS; (d) R. Cano, D. J. Ramon and M. Yus, J. Org. Chem., 2011, 76, 5574 CrossRef; (e) C. H. Tang, L. He, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, Chem.–Eur. J., 2011, 17, 7172 CrossRef CAS; (f) T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, J. Am. Chem. Soc., 2013, 135, 118 CrossRef CAS; (g) T. B. Nguyen, L. Ermolenko and A. Al-Mourabit, Org. Lett., 2013, 15, 4218 CrossRef CAS; (h) T. B. Nguyen, P. l. Retailleau and A. Al-Mourabit, Org. Lett., 2013, 15, 5238 CrossRef CAS; (i) T. B. Nguyen, J. L. Bescont, L. Ermolenko and A. Al-Mourabit, Org. Lett., 2013, 15, 6218 CrossRef CAS; (j) X. J. Cui, Y. Q. Deng and F. Shi, ACS Catal., 2013, 3, 808 CrossRef CAS; (k) Q. L. Peng, Y. Zhang, F. Shi and Y. Q. Deng, Chem. Commun., 2011, 47, 6476 RSC.
- For an excellent example on olefin hydroamination with nitroarenes using PhSiH3 as the reductant, see: J. Gui, C. Pan, Y. Jing, T. Qin, J. Lo, B. Lee, S. Spergel, M. Mertzman, W. Pitts, T. Cruz, M. Schmidt, N. Darvatkar, S. Natarajan and P. Baran, Science, 2015, 348, 886 CrossRef CAS.
- Y. Liu, W. Chen, C. Feng and G. J. Deng, Chem.–Asian J., 2011, 6, 1142 CrossRef CAS.
- C. Feng, Y. Liu, S. Peng, Q. Shuai, G. J. Deng and C. J. Li, Org. Lett., 2010, 12, 4888 CrossRef CAS.
- F. Xiao, Y. Liu, C. Tang and G. J. Deng, Org. Lett., 2012, 14, 984 CrossRef CAS.
- (a) H. Wang, H. Chen, Y. Chen and G. J. Deng, Org. Biomol. Chem., 2014, 12, 7792 RSC; (b) H. Wang, X. Cao, F. Xiao and G. J. Deng, Org. Lett., 2013, 15, 4900 CrossRef CAS; (c) M. Wu, X. Hu, J. Liu, Y. Liao and G. J. Deng, Org. Lett., 2012, 14, 2722 CrossRef CAS.
- Y. Xie, S. Liu, Y. Liu, Y. Wen and G. J. Deng, Org. Lett., 2012, 14, 1692 CrossRef CAS PubMed.
- (a) F. Yang, X. Ma and S. K. Tian, Chem.–Eur. J., 2012, 18, 1582 CrossRef CAS; (b) F. Yang and S. K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929 CrossRef CAS; (c) B. Liu, J. Li, F. Song and J. S. You, Chem.–Eur. J., 2012, 18, 10830 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332 CrossRef CAS.
- For synthesis of TsNDND2: J. Wang, G. Burdzinski, T. L. Gustafson and M. S. Platz, J. Am. Chem. Soc., 2007, 129, 2597 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra26588f |
| This journal is © The Royal Society of Chemistry 2016 |