
Highly diastereo-/enantioselective Cu-catalyzed propargylic alkylations of propargyl acetates with cyclic enamines†
Cheng
Zhang
ab,
Yun-Ze
Hui
c,
De-Yang
Zhang
b and
Xiang-Ping
Hu
*b
aDepartment of Drug Discovery and Biomedical Sciences, South Carolina College of Pharmacy, Medical University of South Carolina, 280 Calhoun Street MSC140 QF307, Charleston, South Carolina 29425, USA
bDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: xiangping@dicp.ac.cn; xiangping1974@163.com
cFaculty of Engineering and Built Environment, University of Newcastle, Callaghan, NSW 2308, Australia
First published on 28th January 2016
Abstract
Highly diastereo- and enantioselective copper-catalyzed propargylic alkylations of propargylic acetates with morpholine-derived cyclic enamines for the construction of vicinal tertiary stereocenters have been developed. By the employment of a less sterically hindered chiral tridentate P,N,N-ligand, good to excellent diastereo- (up to >98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 dr) and enantioselectivity (up to 99% ee) could be achieved for a wide range of substrates.
:2 dr) and enantioselectivity (up to 99% ee) could be achieved for a wide range of substrates.
Transition-metal-catalyzed propargylic substitutions represent an important class of reactions due to their ability to introduce an electron-rich triple bond that is a versatile entity for further chemical transformations.1 In 2008, van Maarseveen2 and Nishibayashi3 independently reported the first copper-catalyzed asymmetric propargylic amination. Since then, a variety of nitrogen, oxygen, and carbon nucleophiles have proved to be suitable reagents for the copper-catalyzed asymmetric propargylic substitution.4 However, the methods for Cu-catalyzed propargylic substitution that employ prochiral nucleophiles to build two vicinal stereocenters and display high diastereo- and enantioselectivity remain elusive.5
Recently, our group have demonstrated the power of copper catalysis in accessing vicinal tertiary stereocenters with our report on the diastereo- and enantioselective asymmetric propargylic alkylation of acyclic enamines (Scheme 1A).6 However, the use of cyclic enamines was less successful, leading to poor chemoselectivity. The success of this protocol combined with the virtual absence of successful reports describing the application of this transformation to cyclic enamines7 encouraged our further exploration of copper catalysts in the domain of this substrate class. In fact, our previous studies have disclosed that a highly diastereo- and enantioselective [3 + 3] cycloaddition instead of propargylic alkylation preferentially took place for the reaction between propargylic esters and cyclic enamines although this cycloaddition is believed to proceed via the propargylic alkylation as the key step (Scheme 1B).8 It's therefore anticipated that an enantioselective propargylic alkylation of propargylic esters with cyclic enamines could be realized if the last cyclization step can be efficiently inhibited. Herein we report a highly diastereo- and enantioselective copper-catalyzed propargylic alkylation of propargylic acetates with morpholine-derived cyclic enamines by the employment of a less sterically hindered chiral tridentate P,N,N-ligand to build up two vicinal tertiary stereocenters, in which >98:2 dr and up to 99% ee for syn-diastereoisomers have been achieved (Scheme 1C).
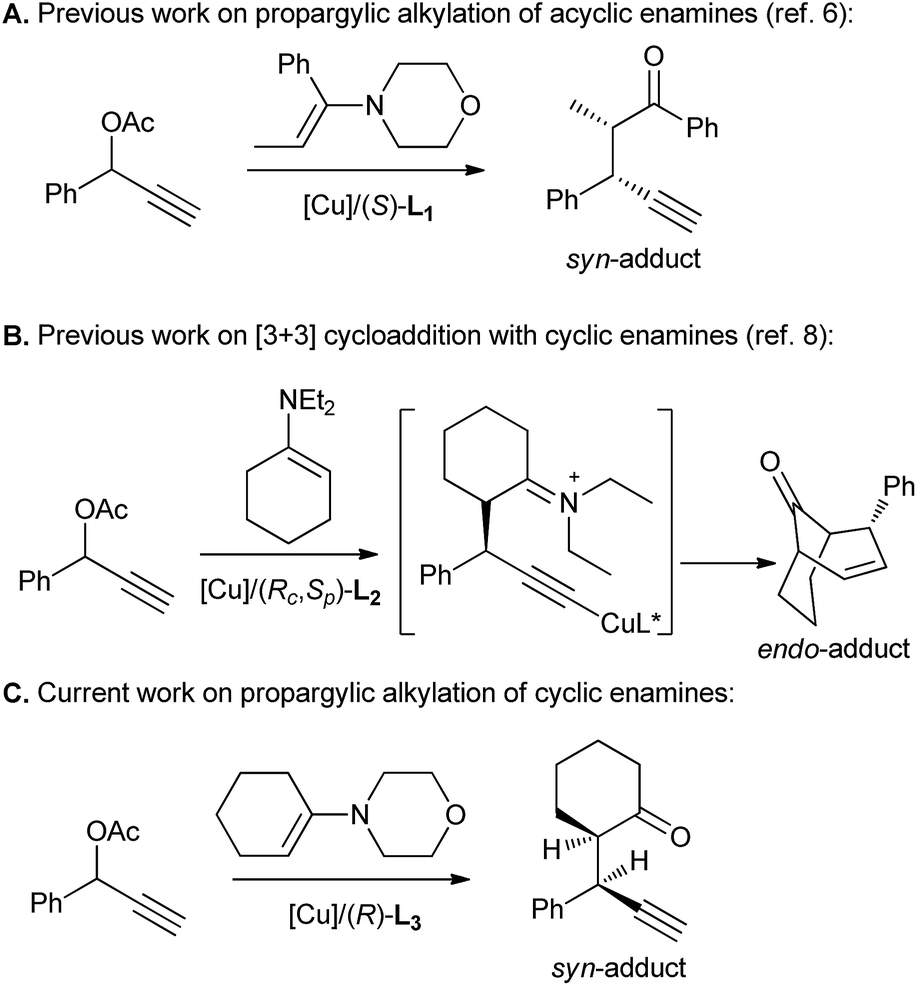 | ||
| Scheme 1 Copper-catalyzed asymmetric reaction between propargylic esters with enamines. | ||
Considering the significant influence of the amino moiety of enamine substrates on the outcomes in the copper-catalyzed [3 + 3] cycloaddition,8 our initial attempt focused on probing the effect of enamines on the reaction. As shown in Table 1, the amino moiety of enamines displayed the crucial role in the chemoselectivity of the reaction. When N,N-diethylamino substituted enamine 2a was used as the reaction partner, only [3 + 3] cycloadduct was obtained and no alkylation product was detected (entry 1). By the introduction of cyclic amino group into enamines, the formation of the alkylation product 3a was observed (entries 2–4). In particular, the use of enamine 2d bearing a morpholine structure as the nucleophile predominantly gave rise to the alkylation product 3a in 80% yield (entry 4).
|
|
|||
|---|---|---|---|
| Entry | Enamine (2) |
3a (syn/anti):4b |
Yield of 3ac (%) |
| a Reaction conditions: Cu(OAc)2·H2O (0.015 mmol), (Rc,Sp)-L2 (0.0165 mmol), 1a (0.3 mmol), 2 (0.36 mmol), iPr2NEt (0.36 mmol) were stirred in 2 mL of MeOH at rt for 12 h. b Determined by 1H NMR. c Isolated yield after column chromatography. | |||
| 1 | 2a | <2 (—):>98 |
— |
| 2 | 2b | 22 (71/29):78 |
16 |
| 3 | 2c | 36 (78/22):64 |
27 |
| 4 | 2d | 94 (91/9):6 |
80 |
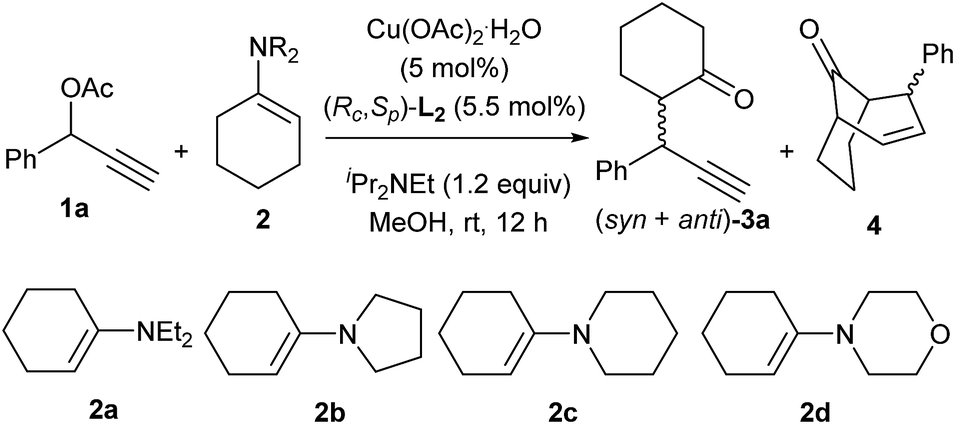
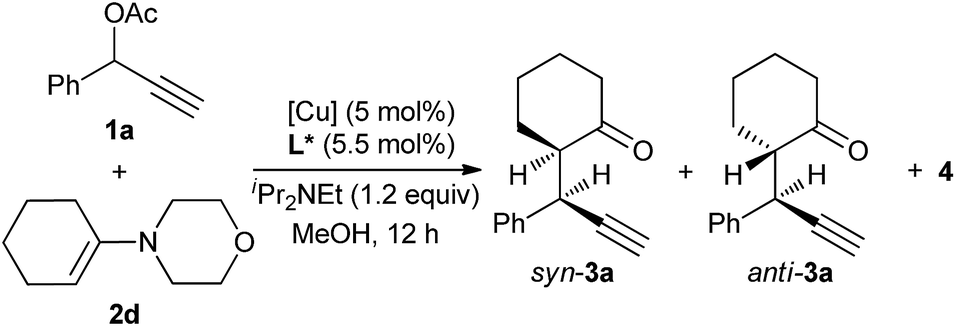
Having established an efficient copper-catalyzed propargylic alkylation of cyclic enamine 2d, we next investigated the effects of different ligands, copper salts, reaction temperature on the efficiency and selectivity of the reaction. 1-Phenyl-2-propynyl acetate 1a and 4-(cyclohex-1-en-1-yl)morpholine 2d were chosen as standard reaction partners, and some selected results of these experiments are summarized in Table 2. Initially, the effect of the ligand was investigated (Fig. 1). As we have disclosed, the use of chiral ferrocenyl P,N,N-ligand L2 led to the predominant formation of the alkylation products 3a with syn-3a as the major diastereoisomer in 91% ee (entry 2). An attempt to improve the reaction outcome by the use of structurally rigid and bulky ketimine P,N,N-ligand L1, which displayed excellent diastereo-/enantioselectivity in the Cu-catalyzed asymmetric propargylic alkylation of acyclic enamines,6 however, was disappointed. Only poor chemoselectivity (3a:4 = 61:39) was obtained although the enantioselectivity for syn-3a was high (entry 1). This should be high reactivity of copper/L1 complex that couldn't efficiently suppress the last cyclization step. We therefore envisioned that ligand having lower reactivity should be more suitable to this reaction. A less sterically hindered P,N,N-ligand L3 was therefore employed in the reaction, and as expected, gave the similar result to that with L2 (entry 3). Excellent chemo- and diasteroselectivity was observed with (S,S)-Ph-pybox L4 as ligand, however, enantioselectivity for syn-3a was low (entry 4). With (S)-SegPhos L5, high chemoselectivity but low diasteteo- and enantioselectivity was achieved (entry 5). Following investigation of copper salts didn't improve the reaction outcome (entries 6–8). Lowering the reaction temperature to 0 °C could significantly improve the chemoselectivity, exclusively giving the alkylation product 3a in good diastereoselectivity, regardless of the use of L2 or L3 (entries 9–10). Especially, ligand L3 led to the alkylation product 3a in 82% yield, and with 94/6 dr and an ee-value of up to 94% for the major syn-diastereoisomer (entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Cu] | L* |
syn-3a:anti-3a:4b |
Yield of 3ac (%) | ee of syn-3ad (%) |
| a Reaction conditions: copper salt (0.015 mmol), L* (0.0165 mmol), 1a (0.3 mmol), 2d (0.36 mmol), iPr2NEt (0.36 mmol) were stirred in 2 mL of MeOH at rt for 12 h unless otherwise specified. b Determined by GC or 1H NMR. c Isolated yield of syn-3a and anti-3a in total. d Determined by HPLC on chiral column. e The reaction was performed at 0 °C for 10 h. f The ee-values of anti-3a in parentheses. | |||||
| 1 | Cu(OAc)2·H2O | L1 | 55:6:39 |
51 | 94 |
| 2 | Cu(OAc)2·H2O | L2 | 86:8:6 |
80 | 91 |
| 3 | Cu(OAc)2·H2O | L3 | 84:8:8 |
76 | 92 |
| 4 | Cu(OAc)2·H2O | L4 | 97:3:0 |
88 | 42 |
| 5 | Cu(OAc)2·H2O | L5 | 55:45:0 |
63 | 52 (51)f |
| 6 | CuI | L3 | 80:20:0 |
74 | 76 (56)f |
| 7 | CuOTf·(C6H6)1/2 | L3 | 89:7:4 |
72 | 92 |
| 8 | Cu(OTf)2 | L3 | 88:7:5 |
72 | 90 |
| 9e | Cu(OAc)2·H2O | L2 | 90:10:0 |
67 | 92 |
| 10e | Cu(OAc)2·H2O | L3 | 94:6:0 |
82 | 94 |
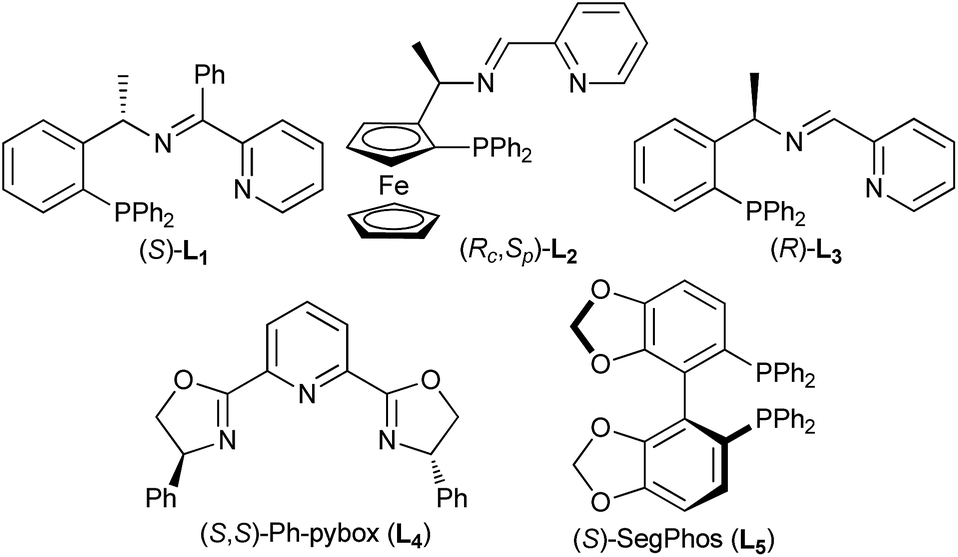 | ||
| Fig. 1 Ligand evaluated in the Cu-catalyzed asymmetric propargylic alkylation. | ||
Having identified the optimal set of reaction conditions, we then investigated the scope of this reaction with respect to propargylic acetates, and the results are shown in Table 3. We found that the reaction proceeded smoothly for all 1-aryl-2-propynyl acetates. In all cases, only alkylation products, predominately syn-diastereoisomers, were obtained and no [3 + 3] cycloadduct was detected. The position of the substituent on the phenyl ring had less influence on the reaction. Thus, all three substrates bearing a Cl-group at the different position on the phenyl ring gave the similarly good results (entries 2–4). The electronic properties of the substituent at the para-position showed little effect on the diastereo-/enantioselectivity. Propargylic acetates bearing either electron-donating or electron-withdrawing substituents on the phenyl ring fared very well in the reaction, delivering the alkylation products in high yield, diastereo-, and enantioselectivity (entries 5–8). 2-Naphthyl substrate 1i also worked well, gave the alkylation product 3i in 88% yield, 94:6 dr and 92% ee for syn-diastereoisomer (entry 9). We were pleased to discover that the use of heteroaryl-substituted propargylic acetates (substrates 1j and 1k) resulted in smooth reactions and delivered the alkylation products 3j and 3k with high yield and diastereoselectivity and with good to excellent enantioselectivity (entries 10–11).
|
|
|||||
|---|---|---|---|---|---|
| Entry | 1 (R) | 3 | Yield of 3b (%) | drc | ee of syn-3d (%) |
| a Reaction conditions: Cu(OAc)2·H2O (0.015 mmol), L3 (0.0165 mmol), 1 (0.3 mmol), 2d (0.36 mmol), iPr2NEt (0.36 mmol) were stirred in 2 mL of MeOH at 0 °C for 10 h. b Isolated yield of syn-3a and anti-3a in total. c Determined by GC or 1H NMR. d Determined by HPLC on chiral column. | |||||
| 1 | 1a: R = Ph | 3a | 82 | 94:6 |
94 |
| 2 | 1b: R = 4-ClC6H4 | 3b | 80 | 93:7 |
95 |
| 3 | 1c: R = 3-ClC6H4 | 3c | 85 | >98:2 |
97 |
| 4 | 1d: R = 2-ClC6H4 | 3d | 88 | >98:2 |
95 |
| 5 | 1e: R = 4-FC6H4 | 3e | 88 | 97:3 |
92 |
| 6 | 1f: R = 4-BrC6H4 | 3f | 90 | 94:6 |
95 |
| 7 | 1g: R = 4-CF3C6H4 | 3g | 84 | 94:6 |
95 |
| 8 | 1h: R = 4-MeC6H4 | 3h | 92 | 92:8 |
91 |
| 9 | 1i: R = 2-naphthyl | 3i | 88 | 94:6 |
92 |
| 10 | 1j: R = 2-furyl | 3j | 90 | 94:6 |
80 |
| 11 | 1k: R = 3-pyridyl | 3k | 86 | 91:9 |
95 |
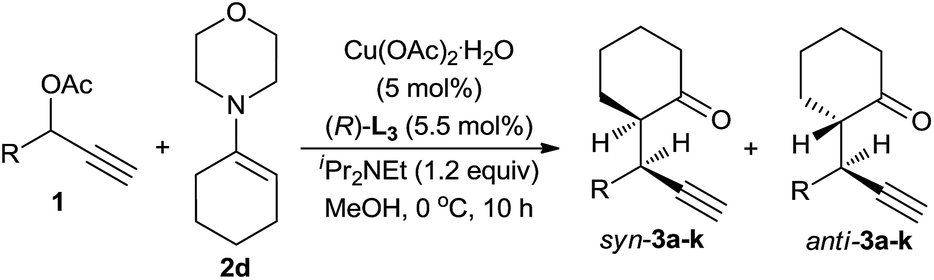
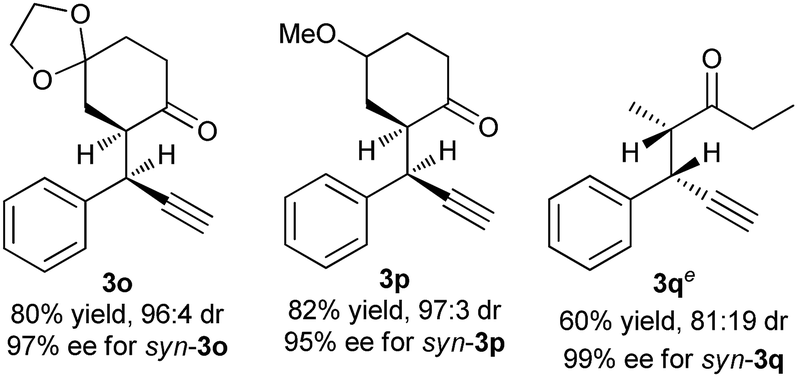
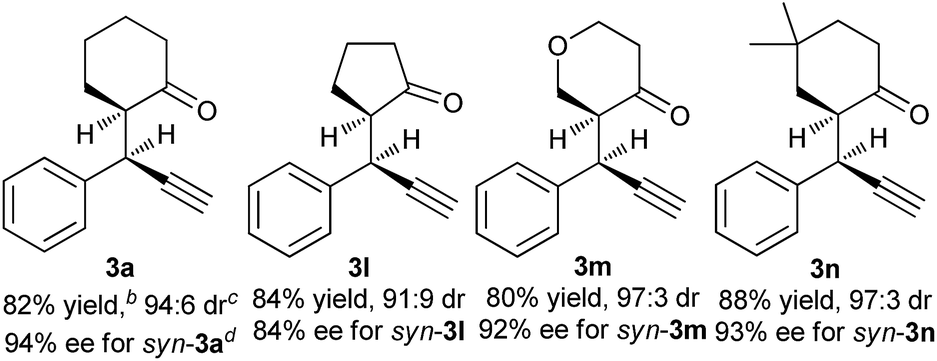
To further evaluate this asymmetric protocol, we next examined the diversity of cyclic enamines permitted in the reaction, and the results are listed in Table 4. In all cases, the reaction proceeded smoothly to afford the alkylation products as the only outcome with syn-diastereoisomers as the major products. In comparison to 4-(cyclohex-1-en-1-yl)morpholine 2d, five-membered cyclic enamines gave the similar yield and diastereoselectivity, but a slightly decreased enantioselectivity. The substituents in cyclohexenyl ring were well tolerated, delivering the corresponding alkylation products in high yield and with excellent diastereo- and enantioselectivity. Importantly, aliphatic acyclic ketone enamine also served well as the reaction partner although the use of (S)-L1 instead of (R)-L3 was required. In this case, (S)-L1 showed better performance than (R)-L3 presumably due to the steric effect, giving the alkylation product 3q in moderate yield and diastereoselectivity but with perfect ee of 99%.
|
|
|---|
| a Reaction conditions: Cu(OAc)2·H2O (0.015 mmol), L3 (0.0165 mmol), 1a (0.3 mmol), 2d–j (0.36 mmol), iPr2NEt (0.36 mmol) were stirred in 2 mL of MeOH at 0 °C for 10 h. b Isolated yield of syn-3a and anti-3a in total. c Determined by GC or 1H NMR. d Determined by HPLC on chiral column. e (S)-L1 was used instead of (R)-L3. |
|
The absolute configuration of the propargylic alkylation product was unambiguously determined by X-ray structure analysis of 3r,9 which was prepared by the reaction of 1-(4-bromophenyl)prop-2-yn-1-yl acetate 1f with 4-(1,4-dioxaspiro[4.5]dec-7-en-8-yl)morpholine in the catalysis of Cu(OAc)2·H2O and (S)-L3. A (S,R)-absolute configuration was assigned as shown in Scheme 2, which is corresponding to syn-diastereoisomer.
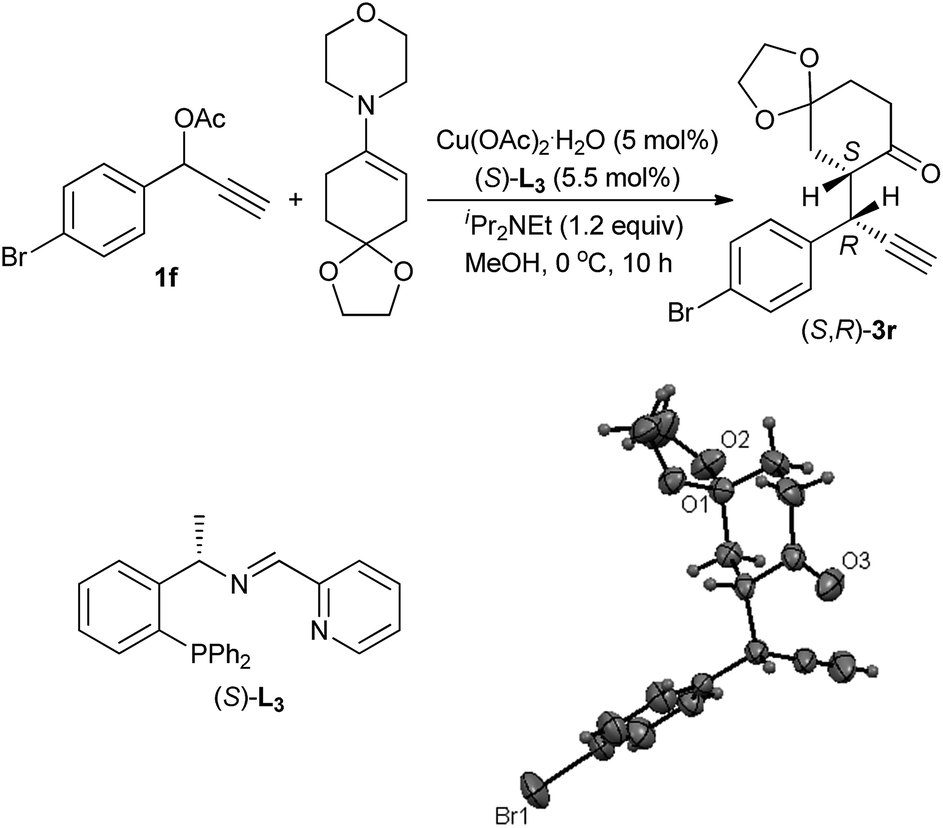 | ||
| Scheme 2 Synthesis of (S,R)-3r and its absolute configuration determined by X-ray analysis. | ||
Based on the experimental results and previous reports,4b,7b a reaction pathway is proposed as shown in Scheme 3. In the first step, a copper complex forms the π-complex A with propargylic acetate 1a. Deprotonation of A with a base then gives a Cu–acetylide complex B, which would explain why a propargylic acetate bearing an internal alkyne moiety such as 1,3-diphenyl-2-propynyl acetate 5 did not react at all. Elimination of an acetoxyl moiety from B affords the copper–allenylidene complex C or its resonance structure, the copper–acetylide complex D bearing a cationic γ-carbon. The nucleophilic attack of β-carbon atom of enamine 2d to the Cγ-atom of the copper–allenylidene complex (C) gives the corresponding copper–acetylide complex (E), which should be the key step for the stereoselection. The intramolecular shift of H atom generates the copper π-alkyne complex F. The starting complex A is then re-generated from F by liberating the propargylic alkylation product G by the ligand exchange with another propargylic acetate 1a.
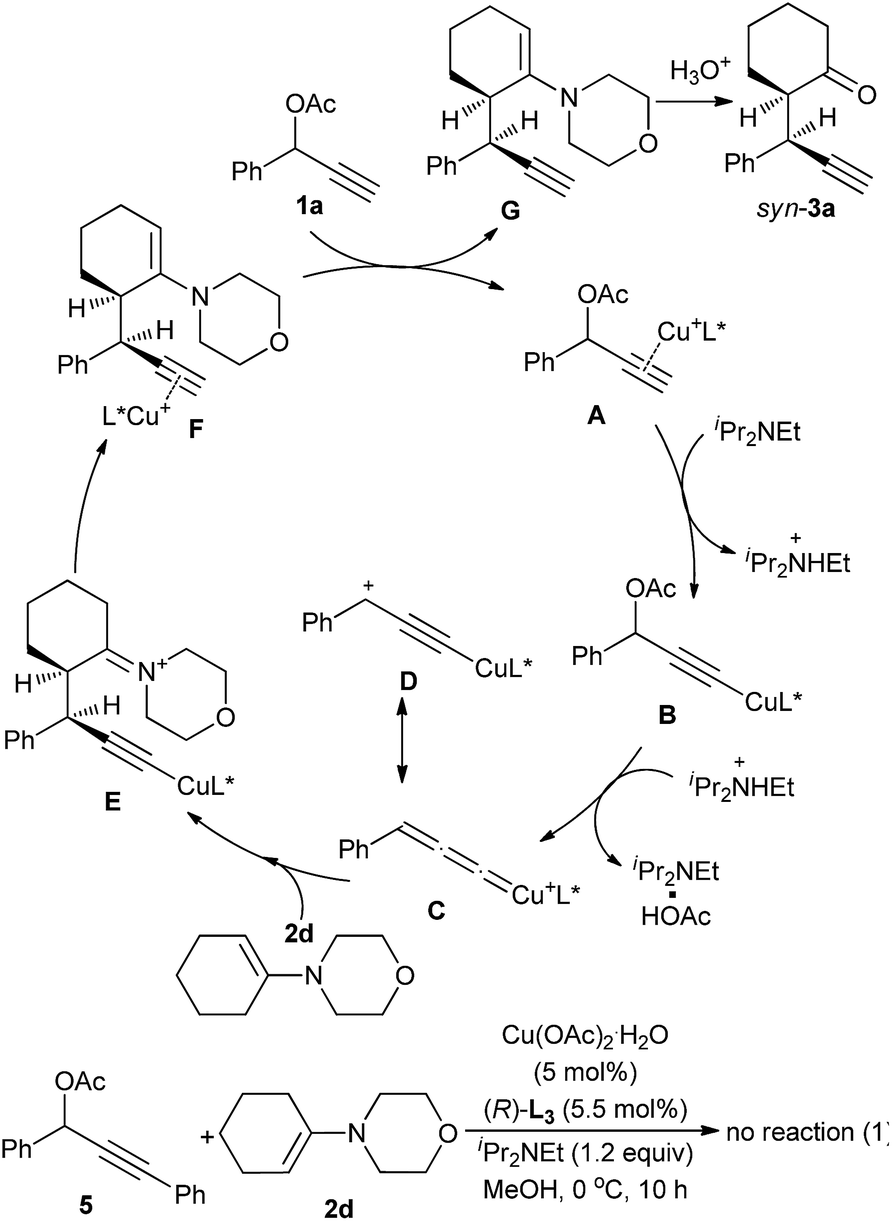 | ||
| Scheme 3 Proposed mechanism. | ||
A transition state of Cu–allenylidene complex with chiral P,N,N-ligand is also proposed to explain the observed stereochemistry as shown in Scheme 4. The edge-to-face aromatic interaction4b and the steric hinderance make the attack of enamine Cβ-nucleophile at the propargylic cation favourably from the Re face viaM-2 mode to generate (R,S)-3a.
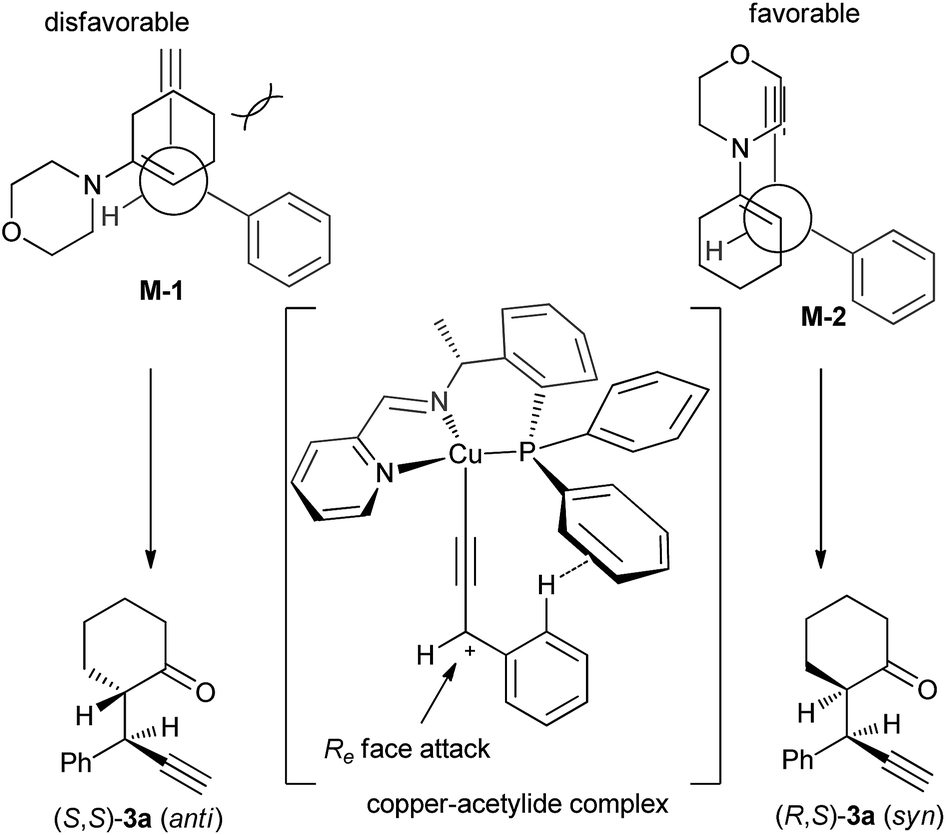 | ||
| Scheme 4 Proposed model for the diastereo-/enantioselectivities. | ||
Conclusions
In conclusion, we have developed a highly diastereo- and enantioselective copper-catalyzed propargylic alkylation of propargylic acetates with morpholine-derived cyclic enamines for the construction of two vicinal tertiary stereocenters. The reaction was preferential to the formation of syn-diastereoisomers of the propargylic alkylation products in the complete chemoselectivity. By the employment of a chiral 1-phenylethylamine-derived tridentate P,N,N-ligand, good to excellent diastereoselectivity (>98:2) and enantioselectivity (up to 99% ee) have been achieved for a wide range of substrates. In comparison with the reaction of acyclic enamines, less sterically hindered P,N,N-ligand showed better performance in the propargylic alkylation of cyclic enamines in term of chemoselectivity, suggesting that its lower reactivity efficiently supressed the last cyclization step. Further development and application of this reaction are underway.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21572226, 21262011) and the Dalian Institute of Chemical Physics.Notes and references
- For reviews, see: (a) Y. Nishibayashi and S. Uemura, Curr. Org. Chem., 2006, 10, 135 CrossRef CAS; (b) N. Ljungdahl and N. Kann, Angew. Chem., Int. Ed., 2009, 48, 642 CrossRef CAS PubMed; (c) Y. Miyake, S. Uemura and Y. Nishibayashi, ChemCatChem, 2009, 1, 342 CrossRef CAS; (d) R. J. Detz, H. Hiemstra and J. H. van Maarseveen, Eur. J. Org. Chem., 2009, 6263 CrossRef CAS; (e) O. Debleds, E. Gayon, E. Vrancken and J.-M. Campagne, Beilstein J. Org. Chem., 2011, 7, 866 CrossRef CAS; (f) C.-H. Ding and X.-L. Hou, Chem. Rev., 2011, 111, 1914 CrossRef CAS; (g) E. B. Bauer, Synthesis, 2012, 44, 1131 CrossRef CAS; (h) Y. Nishibayashi, Synthesis, 2012, 44, 489 CrossRef CAS; (i) D.-Y. Zhang and X.-P. Hu, Tetrahedron Lett., 2015, 56, 283 CrossRef CAS; (j) X.-H. Hu, Z.-T. Liu, L. Shao and X.-P. Hu, Synthesis, 2015, 47, 913 CrossRef CAS.
- R. J. Detz, M. M. E. Delville, H. Hiemstra and J. H. van Maarseveen, Angew. Chem., Int. Ed., 2008, 47, 3777 CrossRef CAS.
- G. Hattori, H. Matsuzawa, Y. Miyake and Y. Nishibayashi, Angew. Chem., Int. Ed., 2008, 47, 3781 CrossRef CAS.
- (a) G. Hattori, A. Yoshida, Y. Miyake and Y. Nishibayashi, J. Org. Chem., 2009, 74, 7603 CrossRef CAS; (b) G. Hattori, K. Sakata, H. Matsuzawa, Y. Tanabe, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2010, 132, 10592 CrossRef CAS; (c) G. Hattori, Y. Miyake and Y. Nishibayashi, ChemCatChem, 2010, 2, 155 CrossRef CAS; (d) R. J. Detz, Z. Abiri, R. le Griel, H. Hiemstra and J. H. van Maarseveen, Chem.–Eur. J., 2011, 17, 5921 CrossRef CAS; (e) A. Yoshida, G. Hattori, Y. Miyake and Y. Nishibayashi, Org. Lett., 2011, 13, 2460 CrossRef CAS; (f) C. Zhang, Y.-H. Wang, X.-H. Hu, Z. Zheng, J. Xu and X.-P. Hu, Adv. Synth. Catal., 2012, 354, 2854 CrossRef CAS; (g) T. Mino, H. Taguchi, M. Hashimoto and M. Sakamoto, Tetrahedron: Asymmetry, 2013, 24, 1520 CrossRef CAS; (h) F.-Z. Han, F.-L. Zhu, Y.-H. Wang, Y. Zou, X.-H. Hu, S. Chen and X.-P. Hu, Org. Lett., 2014, 16, 588 CrossRef CAS; (i) F.-L. Zhu, Y.-H. Wang, D.-Y. Zhang, J. Xu and X.-P. Hu, Angew. Chem., Int. Ed., 2014, 53, 10223 CrossRef CAS; (j) Y. Zou, F.-L. Zhu, Z.-C. Duan, Y.-H. Wang, D.-Y. Zhang, Z. Cao, Z. Zheng and X.-P. Hu, Tetrahedron Lett., 2014, 55, 2033 CrossRef CAS; (k) M. Shibata, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2014, 50, 7874 RSC; (l) F.-L. Zhu, Y.-H. Wang, D.-Y. Zhang, X.-H. Hu, S. Chen, C.-J. Hou, J. Xu and X.-P. Hu, Adv. Synth. Catal., 2014, 356, 3231 CrossRef CAS; (m) B. Wang, C. Liu and H. Guo, RSC Adv., 2014, 4, 53216 RSC; (n) F. Zhu and X. Hu, Chin. J. Catal., 2015, 36, 86 CrossRef CAS; (o) K. Nakajima, M. Shibata and Y. Nishibayashi, J. Am. Chem. Soc., 2015, 137, 2472 CrossRef CAS; (p) D.-Y. Zhang, L. Shao, J. Xu and X.-P. Hu, ACS Catal., 2015, 5, 5026 CrossRef CAS; (q) G. Huang, C. Cheng, L. Ge, B. Guo, L. Zhao and X. Wu, Org. Lett., 2015, 17, 4894 CrossRef CAS.
- For successful examples in the construction of vicinal stereocenters in terms of diastereo- and enantioselectivity, see: (a) L. Zhao, G.-X. Huang, B.-B. Guo, L.-J. Xu, J. Chen, W.-G. Cao, G. Zhao and X.-Y. Wu, Org. Lett., 2014, 16, 5584 CrossRef CAS; (b) W. Shao, H. Li, C. Liu, C.-J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 7684 CrossRef CAS PubMed.
- D.-Y. Zhang, F.-L. Zhu, Y.-H. Wang, X.-H. Hu, S. Chen, C.-J. Hou and X.-P. Hu, Chem. Commun., 2014, 50, 14459 RSC.
- For a singular example of a copper-catalyzed propargylic alkylation of N,N-diethyl-1-cyclohexen-1-amine with 1-phenyl-2-propynyl benzoate in 33% yield and with 10/1 dr and 72% ee, see: (a) P. Fang and X.-L. Hou, Org. Lett., 2009, 11, 4612 CrossRef CAS; for a singular example of a copper-catalyzed decarboxylative propargylic alkylation of 1-phenylprop-2-yn-1-yl 2-oxocyclohexanecarboxylate in 88% yield with 7/1 dr and 86% ee, see: (b) F.-L. Zhu, Y. Zou, D.-Y. Zhang, Y.-H. Wang, X.-H. Hu, S. Chen, J. Xu and X.-P. Hu, Angew. Chem., Int. Ed., 2014, 53, 1410 CrossRef CAS.
- C. Zhang, X.-H. Hu, Y.-H. Wang, Z. Zheng, J. Xu and X.-P. Hu, J. Am. Chem. Soc., 2012, 134, 9585 CrossRef CAS.
- CCDC 1439964 for (S,R)-3r†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data and copies of NMR spectra for all products, and Crystallographic Information Framework (CIF) file of compound (S,R)-3r. CCDC 1439964. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra25627e |
| This journal is © The Royal Society of Chemistry 2016 |