
In situ synthesis and photocatalytic performance of WO3/ZnWO4 composite powders
Abstract
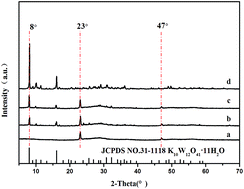
The WO3/ZnWO4 composite powders were synthesized through an in situ reaction process with tunnel structure K10W12O41·11H2O filiform crystallites used as a precursor. At first, Zn2+ ions was intercalated into the K10W12O41·11H2O crystal by exchanging K+ ions, then these Zn2+-exchanged samples were transformed into WO3/ZnWO4 composite powders during heat-treatment. The formation reaction and structure of these samples were characterized by X-ray diffraction (XRD), field-emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM) and energy dispersive X-ray spectrometer (EDS). The results showed that the WO3/ZnWO4 composite powders consisted of WO3 nanoparticles and ZnWO4 nanorods. Photocatalytic experiments exhibited an excellent photocatalytic performance for the degradation of methylene blue (MB) and the degradation efficiency was about 95% after 70 min under simulated sunlight.
 Please wait while we load your content...
Please wait while we load your content...

