
Nickel-catalyzed alkyl-zincation and carboxylation of diynes†
Tao
Cao
a and
Shengming
Ma
*ab
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn
bDepartment of Chemistry, Fudan University, 220 Handan Lu, Shanghai 200433, P.R. China
First published on 8th October 2016
Abstract
A nickel-catalyzed three-component carbo-carboxylation of aryl-substituted diynes with ZnR2 and CO2 is described. With an ortho-ester functionality in the aryl group as the directing group, the usually observed hydrocarboxylation is completely suppressed with an unexpected C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond isomerization. Based on the X-ray diffraction analysis and mechanistic study, it is believed that the reaction started with the oxidative addition with an aryl-substituted C–C triple bond and transmetalation with dialkyl zinc, followed by syn-carbonickelation with an alkyl-substituted C–C triple bond. Subsequent reductive elimination afforded the corresponding sp2-carbon zinc intermediate. Finally, the dynamic isomerization of a zinc-linked CC bond and the subsequent exclusive reaction of an isomerized sp2-carbon zinc intermediate with CO2 afforded the final carboxylic acids with a high stereoselectivity.
C bond isomerization. Based on the X-ray diffraction analysis and mechanistic study, it is believed that the reaction started with the oxidative addition with an aryl-substituted C–C triple bond and transmetalation with dialkyl zinc, followed by syn-carbonickelation with an alkyl-substituted C–C triple bond. Subsequent reductive elimination afforded the corresponding sp2-carbon zinc intermediate. Finally, the dynamic isomerization of a zinc-linked CC bond and the subsequent exclusive reaction of an isomerized sp2-carbon zinc intermediate with CO2 afforded the final carboxylic acids with a high stereoselectivity.
Introduction
In the past 20 years, CO2 has been broadly utilized as a safe and economic C1 starting material in organic synthesis.1–3 However, although hydrocarboxylation of alkynes has become a powerful method to construct α,β-unsaturated alkenoates,4,5 to the best of our knowledge, the related alkyl-carboxylation with β-H in the alkyl group is still a significant challenge due to the intrinsic, easily occurring β-hydride elimination (Scheme S1, eqn (1)–(4)†).5c,6–8 Recently, we have developed a highly regioselective syn-hydrocarboxylation of 1-aryl-1,6-alkadiynes.9 As expected, when the diyne 1a′ with a para-methoxycarbonyl substituent was treated under the standard conditions, hydrocarboxylation product (Z,E)-A was formed as the major product (Scheme 1, eqn (1)). Surprisingly, when such 1,6-diynes with ortho-methoxycarbonyl group 1 were applied, an interesting unexpected carbo-carboxylation of diynes was observed: the alkyl group from dialkyl zincs was connected to an alkyl-substituted C–C triple bond (a reversed regioselectivity as compared to what was observed in ref. 9) and the stereoselectivity of the aryl-substituted C–C triple bond was also reversed (Scheme 1, eqn (2)). Such a reaction leads to the highly efficient stereoselective construction of two tetra-substituted CC bonds.10
 | ||
| Scheme 1 Hydrocarboxylation vs. alkyl-carboxylation. | ||
Results and discussion
While expanding the reaction scope of ref. 9, we treated diyne 1a bearing an ortho-ester entity with 1 mol% of Ni(cod)2 and 3 equiv. of ZnEt2, which is the most challenging for avoiding β-H elimination,6,7 in DMSO in the presence of CO2 at 60 °C for 3 h. Interestingly the ethyl-carboxylation product (E,Z)-2a was obtained in 73% NMR yield exclusively with a very high selectivity accompanied by 18% of protonolysis product 3a (Table 1, entry 3). The structure and stereochemistry of (E,Z)-2a were established unambiguously by its X-ray single crystal diffraction study,11 which shows clearly that the syn-ethyl metalation reaction occurred highly selectively with the methyl-substituted C–C triple bond while the aryl-substituted C–C triple bond underwent a surprising unusual formal cyclic anti-sp2-carbocarboxylation (Fig. 1).12 | ||
| Fig. 1 ORTEP representation of (E,Z)-2a and the structure of (Z,E)-A′. | ||
|
|
||||
|---|---|---|---|---|
| Entry | T (°C) | Yield of (E,Z)-2a (%) | Yield of (E,Z)-3a (%) | Yield of (Z,Z)-3a (%) |
| a The reaction was carried out with 1a (0.3 mmol), Ni(cod)2 (1 mol%), 3 equiv. of ZnEt2 (1.5 M in toluene), and a balloon of CO2 (about 1 L) in 3 mL of DMSO for 3 h followed by addition of HCl (3 M, 5 mL) to afford the crude products, which were treated with 4 equiv. of TMSCHN2 in 1 mL of MeOH and 4 mL of Et2O at room temperature. b Yields were determined by 1H NMR analysis with CH2Br2 as the internal standard. c The reaction was carried out with 0.5 mmol of 1a in 5 mL of DMSO. d The reaction was carried out with 1 mmol of 1a in 10 mL of DMSO. e Isolated yield. f ZnEt2 (1.5 equiv.) was used. | ||||
| 1 | 40 | 30 | 62 | 4 |
| 2 | 50 | 47 | 46 | 4 |
| 3c | 60 | 73 | 15 | 3 |
| 4 | 70 | 79 | 7 | 3 |
| 5 | 80 | 83 | 7 | 3 |
| 6d | 90 | 86 (79)e | 2 | 1 |
| 7d,f | 90 | 86 (78)e | 6 | 3 |

Encouraged by these results, we turned to further optimize the reaction conditions. We observed a temperature effect: at a lower temperature, protonolysis product 3a was the major product (Table 1, entries 1 and 2) while at a higher temperature, the desired carboxylation product (E,Z)-2a became the major product (Table 1, entries 4–6). When the reaction was conducted at 90 °C, (E,Z)-2a was obtained in 86% NMR yield with only 3% of 3a (Table 1, entry 6). Moreover, the amount of ZnEt2 could be reduced to 1.5 equiv., and the yield of (E,Z)-2a was not influenced (Table 1, entry 7). Finally, 1 mol% of Ni(cod)2 with 1.5 equiv. of ZnEt2 in DMSO at 90 °C has been defined as the standard conditions for further study. Here it should be noted that hydrometalation products (E,E)-2a′, 3a′, and (Z,E)-A′ were not observed.9
With the optimized reaction conditions in mind, we next investigated the reaction scope (Table 2). Besides ZnEt2, ZnMe2 without β-H is, of course, compatible in the reaction (Table 2, entry 3). Furthermore, ZnnBu2 with an elongated alkyl chain reacted smoothly in this reaction, affording cyclizative butyl-carboxylation products (Table 2, entry 4). The reaction may be extended to diynes with additional substituents on the aryl ring (Table 2, entries 5 and 6). The substrates with the linker of NTs (Table 2, entries 7 and 8) and O (Table 2, entry 9 and Scheme 2, eqn (3)) could be tolerated in this reaction to afford tetrahydropyrrole and tetrahydrofuran derivatives. It is important to note that the alkyl substituent of the C–C triple bond (R1) in the substrates could be extended (Table 2, entries 10 and 11) and functionalized with the synthetically active acetoxy group (Table 2, entries 12–14). In addition, except for the methyl ester group, ethyl (Table 2, entry 15), iso-propyl (Table 2, entries 16 and 17) and tert-butyl (Table 2, entries 18 and 19) groups all demonstrated a comparable directing ability. It is easy to conduct the reaction on a one gram scale to afford (E,Z)-2a in 78% yield (Table 2, entry 2).
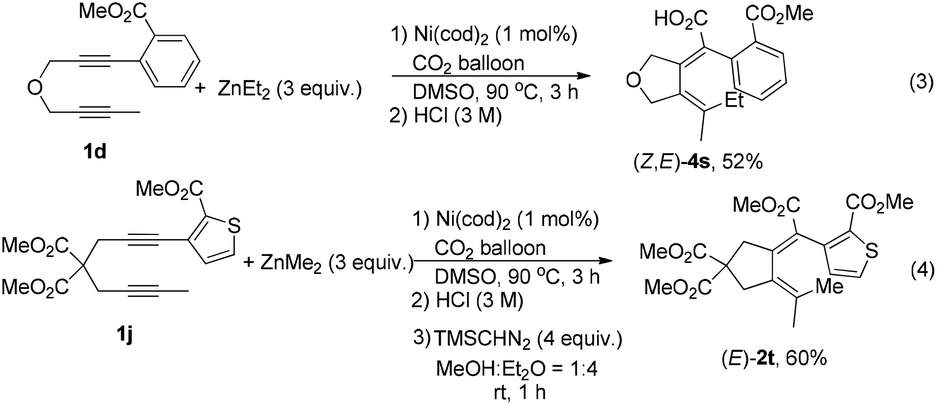 | ||
| Scheme 2 Alkyl-carboxylations of diynes 1d and 1j. | ||
|
|
|||
|---|---|---|---|
| Entry | X; R1; R2; R3 | ZnR42 (equiv.) | Yield (%) |
| a The reaction was carried out with 1 mmol of 1, 1 mol% of Ni(cod)2, 3 equiv. (or 1.5 equiv.) of ZnR42 (1.5 M in toluene for ZnEt2, and 1.0 M in toluene for ZnMe2 and ZnnBu2), and a balloon of CO2 (about 1 L) in 10 mL of DMSO at 90 °C for 3 h followed by addition of HCl (3 M, 10 mL). b Yields of isolated products as the methyl esters 2 after treatment of the crude products with 4 equiv. of TMSCHN2 in 1 mL of MeOH and 4 mL of Et2O at room temperature. c The reaction was carried out with 3 mmol of 1a to afford 1.0403 g of (E,Z)-2a. | |||
| 1 | C(CO2Me)2; Me; H; Me (1a) | ZnEt2 (1.5) | 78 (E,Z)-2a |
| 2c | C(CO2Me)2; Me; H; Me (1a) | ZnEt2 (1.5) | 78 (E,Z)-2a |
| 3 | C(CO2Me)2; Me; H; Me (1a) | ZnMe2 (3) | 61 (E)-2b |
| 4 | C(CO2Me)2; Me; H; Me (1a) | ZnnBu2 (3) | 60 (E,Z)-2c |
| 5 | C(CO2Me)2; Me; 4-MeO; Me (1b) | ZnEt2 (1.5) | 64 (E,Z)-2d |
| 6 | C(CO2Me)2; Me; 4-MeO; Me (1b) | ZnMe2 (3) | 57 (E)-2e |
| 7 | NTs; Me; H; Me (1c) | ZnEt2 (3) | 38 (Z,E)-2f |
| 8 | NTs; Me; H; Me (1c) | ZnMe2 (3) | 40 (Z)-2g |
| 9 | O; Me; H; Me (1d) | ZnMe2 (3) | 57 (Z)-2h |
| 10 | C(CO2Me)2; nPr; H; Me (1e) | ZnEt2 (1.5) | 64 (E,E)-2i |
| 11 | C(CO2Me)2; nPr; H; Me (1e) | ZnMe2 (3) | 66 (E,E)-2j |
| 12 | C(CO2Me)2; AcO(CH2)2; H; Me (1f) | ZnEt2 (3) | 53 (E,E)-2k |
| 13 | C(CO2Me)2; AcO(CH2)2; H; Me (1f) | ZnMe2 (3) | 51 (E,E)-2l |
| 14 | C(CO2Me)2; AcO(CH2)2; H; Me (1f) | ZnnBu2 (3) | 36 (E,E)-2m |
| 15 | C(CO2Me)2; Me; H; Et (1g) | ZnEt2 (1.5) | 67 (E,Z)-2n |
| 16 | C(CO2Me)2; Me; H; iPr (1h) | ZnEt2 (1.5) | 74 (E,Z)-2o |
| 17 | C(CO2Me)2; Me; H; iPr (1h) | ZnnBu2 (3) | 46 (E,Z)-2p |
| 18 | C(CO2Me)2; Me; H; tBu (1i) | ZnEt2 (1.5) | 64 (E,Z)-2q |
| 19 | C(CO2Me)2; Me; H; tBu (1i) | ZnnBu2 (3) | 51 (E,Z)-2r |

In addition, the reaction could be extended to diyne with a heteroaromatic ring (Scheme 2, eqn (4)), indicating that the directing property of the ester group is not affected by the strong coordinating ability of the sulfur atom.
Notably, the tert-butyl group in product (E,Z)-2q could be exclusively removed in the presence of HCO2H, making it possible for further selective derivatization (eqn (5)).
 | (5) |
In order to capture any organometallic intermediates, 1a was treated with 1 mol% of Ni(cod)2 and 1.5 equiv. of ZnEt2 in the absence of CO2 at 40 °C. After 5 min, the reaction was quenched with HCl and D2O, respectively. The starting material 1a was completely consumed, and surprisingly, the protonolysis product (E,Z)-3a and the deuterated product (E,Z)-3a-D were obtained in 84% yield, while the isomerized products (Z,Z)-3a and (Z,Z)-3a-D were obtained in 4% yield, respectively, indicating a very rapid cyclizative alkyl-zincation process to form the corresponding zinc intermediates (Z,Z)-6a and (E,Z)-6a and the non-isomerized (Z,Z)-6a was formed as the major product, which is contradictory to the observed stereoselectivity (Scheme 3, eqn (6)).
 | ||
| Scheme 3 Detection of the key intermediates. | ||
With such an observation, we further monitored the yields of the zinc intermediates (Z,Z)-6a and (E,Z)-6a in the absence of CO2 by measuring the yields of (E,Z)-3a and (Z,Z)-3avs. time, respectively (Fig. 2(a)). Notably, in fact, it was indeed observed that (Z,Z)-6a was formed first, which isomerized to (E,Z)-6a gradually (Fig. 2(a)); for comparison, the reaction was also conducted under a CO2 atmosphere at 40 °C (Fig. 2(b)): After 1 h, in the absence of CO2, 19% of (Z,Z)-3a was formed (Fig. 2(a)) and the ratio of (E,Z)-3a/(Z,Z)-3a was 3.8 (the bottom line in Fig. 2(c)) while in the presence of CO2 3% of (Z,Z)-3a and 77% of (E,Z)-3a were formed accompanied by the exclusive formation of 8% of the carboxylic acid (E,Z)-4a (Fig. 2(b)) and the formation of the stereoisomer carboxylic acid (Z,Z)-4a was not observed. These facts indicated that the final carboxylic acid (E,Z)-4a was generated through the carboxylation of (E,Z)-6a exclusively rather than the carboxylation of (Z,Z)-6a followed by CC bond isomerization (Scheme 3, eqn (7)). These results also suggested that the isomerization of (Z,Z)-6a to form (E,Z)-6a was very slow at 40 °C.
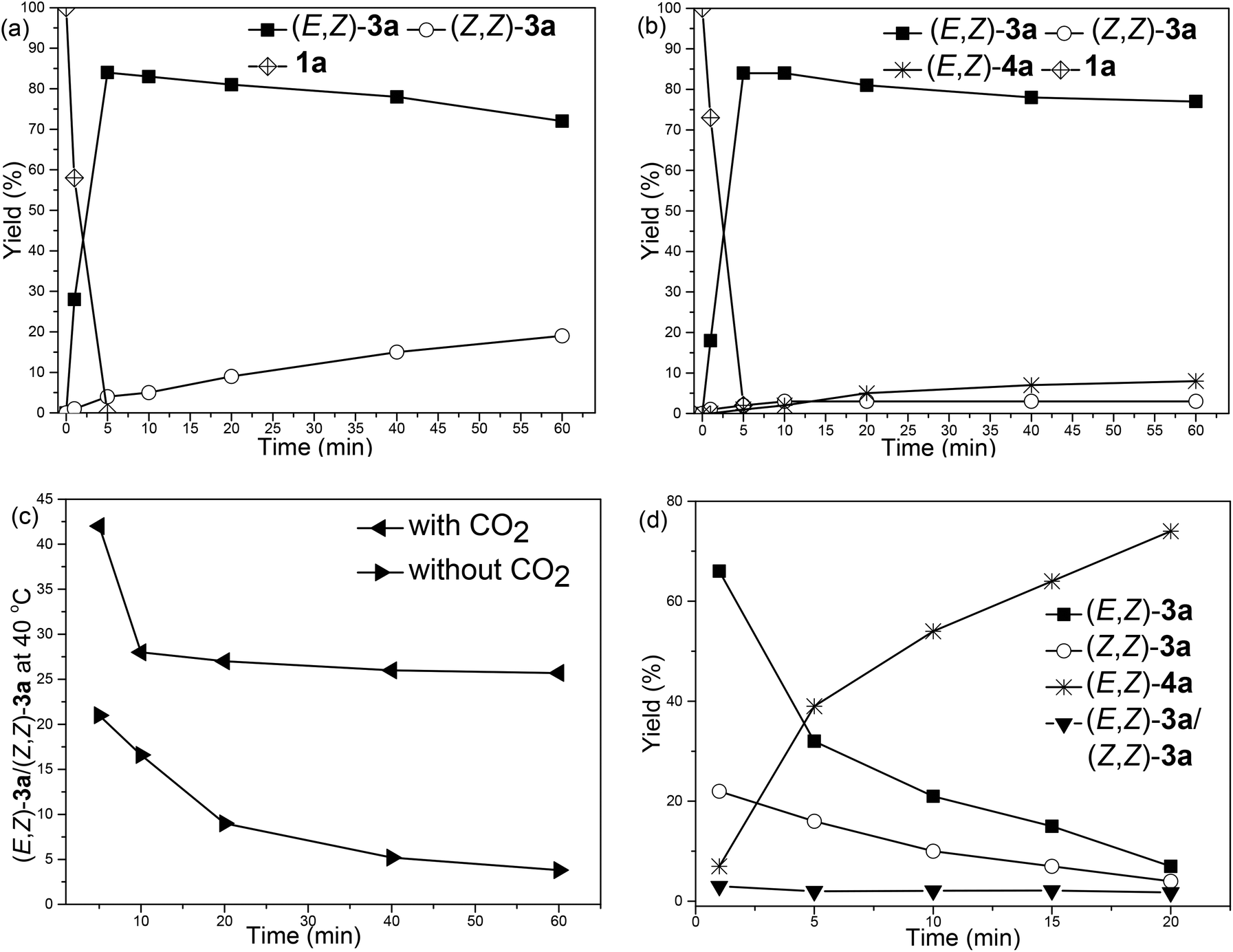 | ||
| Fig. 2 Yields of (E,Z)-3a, (Z,Z)-3a and (E,Z)-4avs. time. (a) Reaction at 40 °C without CO2. (b) Reaction at 40 °C in the presence of CO2. (c) Ratio of (E,Z)-3a to (Z,Z)-3a at 40 °C. (d) Reaction at 90 °C in the presence of CO2. | ||
In addition, when the reaction was conducted at 90 °C in the presence of CO2 (Fig. 2(d)), the disappearance of 1a was extremely fast (<1 min), and the ratio of (E,Z)-3a to (Z,Z)-3a became constant (about 2.0) after 5 min, indicating that the isomerization of (Z,Z)-6a to (E,Z)-6a was much faster at 90 °C.
Based on the observed stereoselectivity and control experiments, we proposed a possible mechanism for the ester group-directed alkylative carboxylation of diynes (Scheme 4): at first, the substrate 1a was reacted with Ni(cod)2 to regioselectively generate nickellacyclopropene Int 1 with the aid of the ester group coordinating to Ni(II), which switched the regioselectivity of the two C–C triple bonds. The nickellacyclopropene Int 1 would transmetalate with ZnEt2, forming the zinc-nickelation product Int 2. Due to the coordination of the ester group, Ni(II) in Int 2 lacked the vacant coordination site, which is required for β-H elimination, resulting in the insertion of the remaining triple bond into the Ni–Et bond to generate Int 3 without β-H elimination. Reductive elimination of Int 3 formed the vinylic zinc intermediate (Z,Z)-6a. However, (Z,Z)-6a could not react with CO2 most probably due to the steric hindrance and underwent isomerization to form (E,Z)-6a, which underwent carboxylation to form the final zinc carboxylate.2d,e,13,14
 | ||
| Scheme 4 A proposed mechanism. | ||
Conclusions
In conclusion, we have developed an example of a rather challenging CO2-based alkyl-carboxylation without β-H elimination. Secondly, the reaction enjoys excellent chemo and/or regioselectivity with the aryl-substituted C–C triple bond reacting first followed by the transmetalation/insertion with the alkyl-substituted C–C triple bond and reductive elimination. Last but not least the reaction also demonstrated excellent stereoselective-syn-insertion and complete CC bond isomerization, which was caused by the higher reactivity of the (E,Z)-6a-type of intermediate towards CO2 probably due to the steric effect. Owing to the easily prepared starting materials, high efficiency and selectivity, and the unique mechanism, this reaction may begin a new era of selectivity-controlled carbo-carboxylation. Related research including exploring the scope further and the reactivity of the in situ generated zinc is ongoing in this laboratory.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21232006) and the National Basic Research Program (2015CB856600) is greatly appreciated. We thank Tonghao Zhu in this group for reproducing the results of entries 4, 8, and 13 in Table 2.Notes and references
- For reviews of CO2 utilization, see: (a) J. Louie, Curr. Org. Chem., 2005, 9, 605 CrossRef CAS; (b) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed; (c) M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC; (d) A. Correa and R. Martin, Angew. Chem., Int. Ed., 2009, 48, 6201 CrossRef CAS PubMed; (e) S. N. Riduan and Y. Zhang, Dalton Trans., 2010, 39, 3347 RSC; (f) C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254 CrossRef CAS PubMed; (g) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kuhn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed; (h) K. Huang, C. Sun and Z. Shi, Chem. Soc. Rev., 2011, 40, 2435 RSC; (i) Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956 RSC; (j) X. Cai and B. Xie, Synthesis, 2013, 3305 CrossRef CAS; (k) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933, DOI:10.1038/ncomms6933; (l) D. Yu, S. P. Teong and Y. Zhang, Coord. Chem. Rev., 2015, 293–294, 279 CrossRef CAS.
- For selected examples on catalytic carboxylation of organometallics, see: (a) M. Shi and K. M. Nicholas, J. Am. Chem. Soc., 1997, 119, 5057 CrossRef CAS; (b) K. Ukai, M. Aoki, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2006, 128, 8706 CrossRef CAS PubMed; (c) T. Ohishi, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 5792 CrossRef CAS PubMed; (d) C. S. Yeung and V. M. Dong, J. Am. Chem. Soc., 2008, 130, 7826 CrossRef CAS PubMed; (e) H. Ochiai, M. Jang, K. Hirano, H. Yorimitsu and K. Oshima, Org. Lett., 2008, 10, 2681 CrossRef CAS PubMed; (f) B. Miao and S. Ma, Chem. Commun., 2014, 50, 3285 RSC; (g) B. Miao and S. Ma, Org. Chem. Front., 2015, 2, 65 RSC; (h) B. Miao and S. Ma, Chem. – Eur. J., 2015, 21, 17224 CrossRef CAS PubMed.
- For selected examples on catalytic carboxylation of electrophiles, see: (a) A. Correa and R. Martin, J. Am. Chem. Soc., 2009, 131, 15974 CrossRef CAS PubMed; (b) T. Fujihara, K. Nogi, T. Xu, J. Terao and Y. Tsuji, J. Am. Chem. Soc., 2012, 134, 9106 CrossRef CAS PubMed; (c) T. León, A. Correa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1221 CrossRef PubMed; (d) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062 CrossRef CAS PubMed.
- For examples on stoichiometric nickel-mediated hydrocarboxylation of alkynes, see: (a) G. Burkhart and H. Hoberg, Angew. Chem., Int. Ed. Engl., 1982, 21, 76 CrossRef; (b) S. Saito, S. Nakagawa, T. Koizumi, K. Hirayama and Y. Yamamoto, J. Org. Chem., 1999, 64, 3975 CrossRef CAS; (c) M. Aoki, M. Kaneko, S. Izumi, K. Ukai and N. Iwasawa, Chem. Commun., 2004, 2568 RSC.
- For examples on catalytic hydrocarboxylation of alkynes, see: (a) E. Duñach, S. Dérien and J. Périchon, J. Organomet. Chem., 1989, 364, C33 CrossRef; (b) S. Dérien, E. Duñach and J. Périchon, J. Am. Chem. Soc., 1991, 113, 8447 CrossRef; (c) S. Li, W. Yuan and S. Ma, Angew. Chem., Int. Ed., 2011, 50, 2578 CrossRef CAS PubMed; (d) S. Li and S. Ma, Chem. – Asian J., 2012, 7, 2411 CrossRef CAS PubMed; (e) T. Fujihara, T. Xu, K. Semba, J. Terao and Y. Tsuji, Angew. Chem., Int. Ed., 2011, 50, 523 CrossRef CAS PubMed; (f) X. Wang, M. Nakajima and R. Martin, J. Am. Chem. Soc., 2015, 137, 8924 CrossRef CAS PubMed.
- For pioneering stoichiometric nickel-mediated alkyl-carboxylation of 4-benzyloxy-1-butyne with 2 equiv. of DBU, see: M. Takimoto, K. Shimizu and M. Mori, Org. Lett., 2001, 3, 3345 CrossRef CAS PubMed , in which the reaction with n-BuZnI is fine while the reaction with Et2Zn the hydrocarboxylation product is predominant (Scheme S1, eqn (1)†).
- In 2005 and 2006 Mori also reported the alkyl-carboxylation of 1-trimethylsilyl-4-phenylbutyne or 1-(p-methoxyphenyl)-4-phenylbutyne under the catalysis of 20 mol% of Ni(cod)2, which is again limited to dibutyl zinc with an issue of regioselectivity (Scheme S1, eqn (2)†) or a serious competition with the hydrocarboxylation reaction (Scheme S1, eqn (3)), respectively, with the help of 10 equiv. of DBU. In these catalytic systems, 10 equiv. of DBU were required to coordinate with dialkyl zinc to in situ form the dialkyl zinc reagents-R2Zn(DBU)2. See: (a) K. Shimizu, M. Takimoto, Y. Sato and M. Mori, Org. Lett., 2005, 7, 195 CrossRef CAS PubMed; (b) K. Shimizu, M. Takimoto, Y. Sato and M. Mori, Synlett, 2006, 3182 CAS.
- For a recent example on Cu-catalyzed alkylative carboxylation of ynamides with dialkyl zinc (Scheme S1, eqn (4)), see: M. Takimoto, S. S. Gholap and Z. Hou, Chem. – Eur. J., 2015, 21, 15218 CrossRef CAS PubMed.
- T. Cao and S. Ma, Org. Lett., 2016, 18, 1510 CrossRef CAS PubMed.
- For reviews on stereoselective construction of tetrasubstituted CC bonds, see:
(a) A. B. Flynn and W. W. Ogilvie, Chem. Rev., 2007, 107, 4698 CrossRef CAS PubMed;
(b) M. Mori, Eur. J. Org. Chem., 2007, 4981 CrossRef CAS;
(c) E.-i. Negishi, Z. Huang, G. Wang, S. Mohan, C. Wang and H. Hattori, Acc. Chem. Res., 2008, 41, 1474 CrossRef CAS PubMed;
(d) M. Shindo and K. Matsumoto, Top. Curr. Chem., 2012, 327, 1 CrossRef CAS PubMed;
(e) S.-M. Paek, Molecules, 2012, 17, 3348 CrossRef CAS PubMed.
- Crystal data for (E,Z)-2a: C24H28O8, MW = 444.46, monoclinic, space group P21/c, final R indices [I > 2σ(I)], R1 = 0.0570, wR2 = 0.1473; R indices (all data), R1 = 0.0716, wR2 = 0.1612; a = 16.1503(18) Å, b = 8.3426(9) Å, c = 18.1574(19) Å, α = 90.00°, β = 108.184(2)°, γ = 90.00°, V = 2324.3(4) Å3, T = 293(2) K, Z = 4, reflections collected/unique 13
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 362/4564 (Rint = 0.0637), number of observations [>2σ(I)] 3494, parameters: 296. Supplementary crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, CCDC 1443804.
362/4564 (Rint = 0.0637), number of observations [>2σ(I)] 3494, parameters: 296. Supplementary crystallographic data have been deposited at the Cambridge Crystallographic Data Centre, CCDC 1443804. - For reviews on carbozincation of alkynes, see: (a) J. F. Normant and A. Alexakis, Synthesis, 1981, 841 CrossRef CAS; (b) K. Murakami and H. Yorimitsu, Beilstein J. Org. Chem., 2013, 9, 278 CrossRef CAS PubMed. For selected examples on anti-carbozincation of alkynes, see: (c) J. Maury, L. Feray and M. P. Bertrand, Org. Lett., 2011, 13, 1884 CrossRef CAS PubMed; (d) C. W. Cheung, F. E. Zhurkin and X. Hu, J. Am. Chem. Soc., 2015, 137, 4932 CrossRef CAS PubMed.
- M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 636 RSC.
- C. M. Williams, J. B. Johnson and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14936 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, spectroscopic data, and the 1H, 13C NMR spectra of all the products. CCDC 1443804. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00484a |
| This journal is © the Partner Organisations 2016 |