DOI:
10.1039/C6QO00319B
(Research Article)
Org. Chem. Front., 2016,
3, 1392-1415
A new approach toward the synthesis of 2,4-bis(fluoroalkyl)-substituted quinoline derivatives using fluoroalkyl amino reagent chemistry†
Received
30th June 2016
, Accepted 27th July 2016
First published on 29th July 2016
Abstract
The present work describes the unprecedented use of Fluoroalkyl Amino Reagents (FARs) to afford 2,4-bis(fluoroalkyl)-substituted quinoline derivatives in two steps. In contrast to the Combes reaction, this approach allows for the synthesis of numerous quinoline derivatives bearing two identical or different fluoroalkyl substituents in the 2 and 4 positions, under mild reaction conditions, in good yields and with a very good regioselectivity. This reaction is easily scalable and suitable for an industrial process.
Introduction
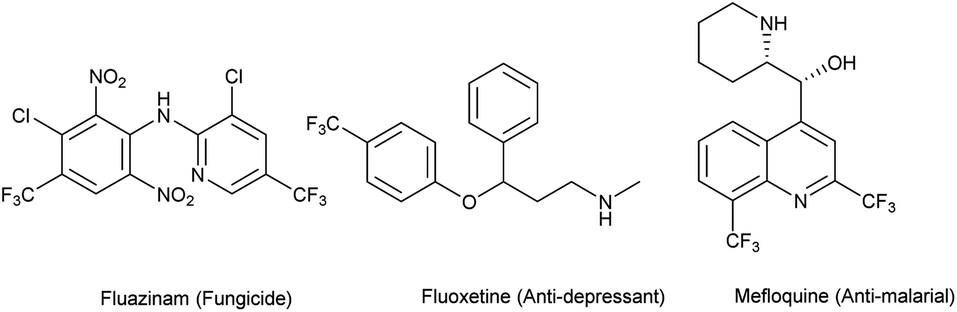
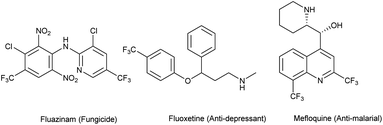
Since the last decade, fluorine has started to become an alternative solution to many issues in agrochemistry, medicinal chemistry and materials science. Indeed, its specific properties such as its high electronegativity, the strength of the C–F bond due to its low polarizability, and its size comparable to that of a hydrogen atom gives it a special appeal. The introduction of fluorine into a lead molecule may considerably improve its physicochemical properties. For example, the metabolic stability of C–F bonds toward oxidative degradation is well-known, and the acidity or basicity of adjacent functional groups can be modified. Likewise, the lipophilicity of molecules can be enhanced by introducing fluorine atoms in order to facilitate transport through membranes.1–18 Therefore, interest in fluorine chemistry has encountered a swift rise, and developing new strategies for the introduction of emergent fluorinated groups remains a challenge to overcome. A recent survey has shown that 20% of pharmaceuticals and 40% of agrochemicals (e.g. fluazinam (fungicide), fluoxetine (anti-depressant)) are F-containing compounds (Fig. 1). Among the 155 F-containing compounds listed as agrochemicals, 43% bear a trifluoromethyl group, 32% a single fluorine atom and around 11% are difluoromethyl derivatives.19,20 On the other hand, the quinoline motif is present in many natural products and drugs already commercialized (e.g. mefloquine, used as an antimalarial drug). For example, they are used in the treatment of cancer, inflammatory diseases, bacterial or viral infections, etc. Quinoline-derived molecules are known for their various biological activities which make them important pharmaceutical and agrochemical precursors.21–27 The combination of an aza-heterocycle and various fluorinated substituents may enhance the properties of previously known compounds and make them highly attractive as agrochemical ingredients.15,28–31
 |
| | Fig. 1 Bioactive compounds containing fluorinated groups. | |
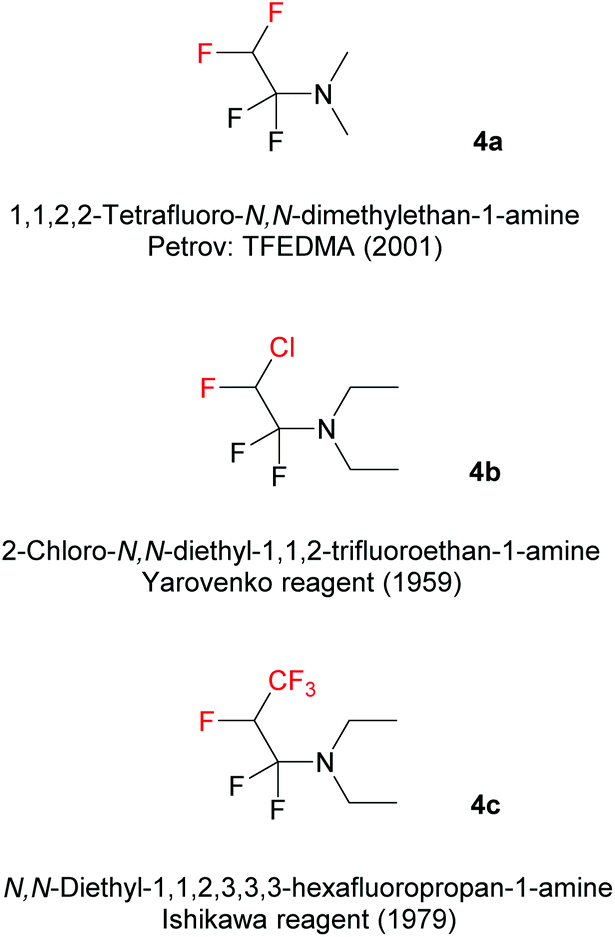
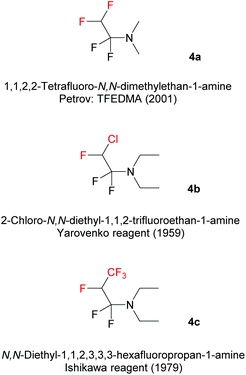
In this paper, we report on a new method to synthesize unprecedented 2,4-bis(fluoroalkyl)-substituted quinoline derivatives, based on the reaction of Fluoroalkyl Amino Reagents (FARs) (Scheme 1) with N-aryl imines 1. FARs represent a powerful tool for the introduction of various fluorinated substituents. In the literature, three different FARs were used: 1,1,2,2-tetrafluoro-N,N-dimethylethan-1-amine (4a; TFEDMA), 2-chloro-N,N-diethyl-1,1,2-trifluoroethan-1-amine (4b; Yarovenko reagent), and N,N-diethyl-1,1,2,3,3,3-hexafluoropropan-1-amine (4c; Ishikawa reagent). FARs are prepared from commercially available fluoroolefins and dimethyl- or diethylamine, and were initially used as fluorinating agents in order to convert hydroxyl or carbonyl groups into fluoroakyl substituents under mild conditions.32–36
 |
| | Scheme 1 Commercially available Fluoroalkyl Amino Reagents (FARs). | |
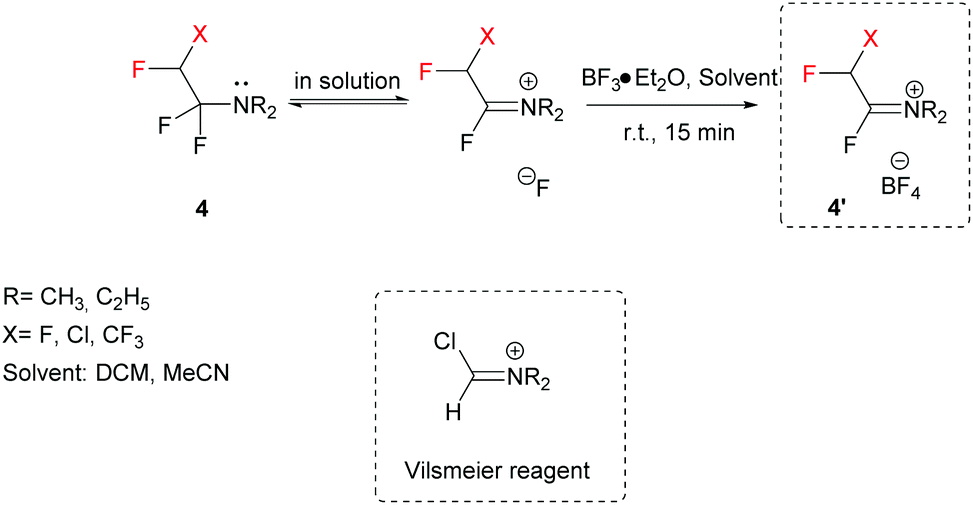
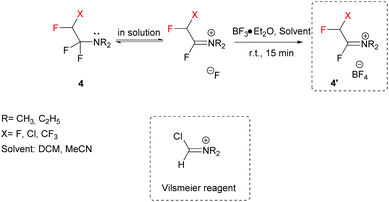
A key feature of FARs is their activation by Lewis acids (e.g. BF3·Et2O), preferentially in an aprotic solvent, to provide highly reactive Vilsmeier-like iminium salts (Scheme 2).37–39 After activation, the electrophilic character of FARs is enhanced, and the iminium salts can undergo nucleophilic attack by various compounds.
 |
| | Scheme 2 Activation of FARs by Lewis acids converting them into their corresponding iminium salts. | |
Of the three existent FARs, TFEDMA is the most stable at ambient temperature, the purer after activation and it is also the most reactive one, due to the presence of the dimethyl moiety that makes it less hindered and thus more prone to nucleophilic attack.
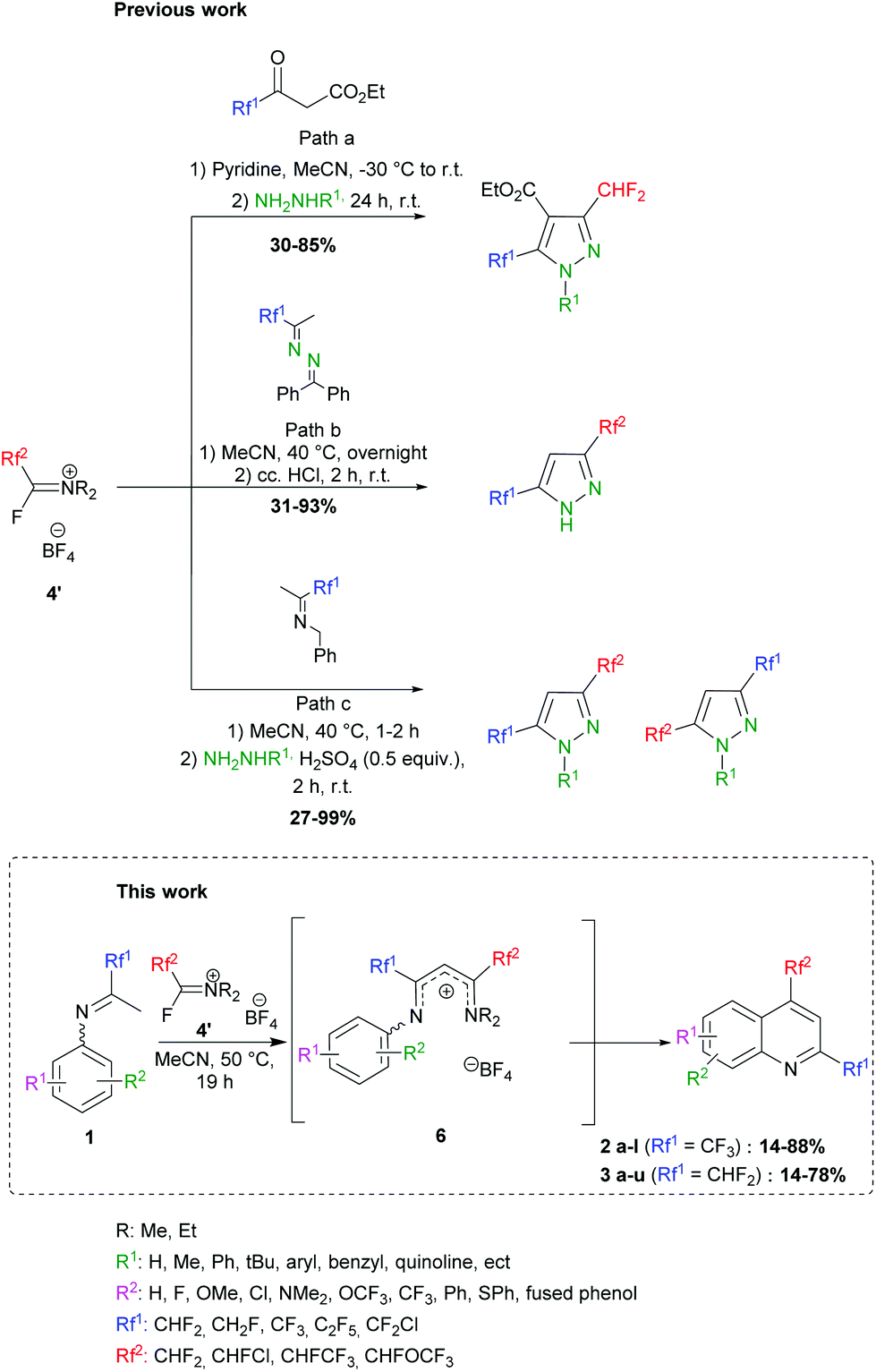
In 1979, Ishikawa employed the Yarovenko reagent (4b) in cyclization reactions with 2-amino-anilines, -phenols and -thiophenols to provide various benzazoles.40 More recently, we further demonstrated the ability of FARs to behave as α-fluoroacyl equivalents for the introduction of fluorinated substituents (CHF2, CHFCl and CHFCF3) in the preparation of a new family of compounds for agrochemistry.41–46 We were able to synthesize fluorinated heterocycles, especially 3,5-bis(fluoroalkyl) pyrazoles following 3 strategies (Scheme 3). In path a, pyrazole carboxylates were formed by reaction of FARs with fluorinated acetoacetates, followed by cyclization with hydrazines.42,43 The limited availability of fluorinated acetoacetates encouraged us to turn towards other strategies. NH-pyrazoles could also be obtained from the condensation of benzophenone-derived fluorinated azines with activated FARs, followed by cyclization under acidic conditions (path b). However, a major drawback of this reaction was the residual benzophenone, which proved difficult to remove even after several purifications.44,45 This method was then improved in terms of yield, reaction time and waste by the use of fluorinated N-benzyl imines (path c).46,47
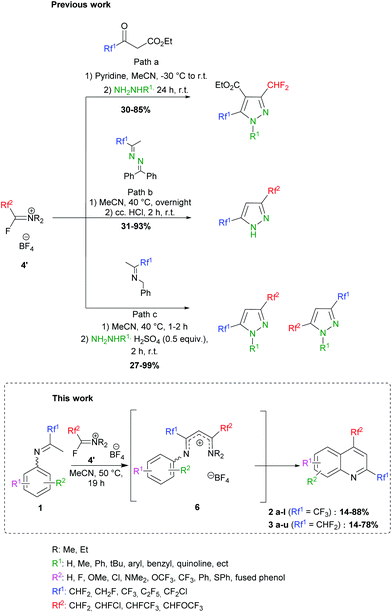 |
| | Scheme 3 Previous work: formation of fluorinated pyrazoles involving activated FARs: (a) from fluorinated acetoacetates, (b) from fluorinated azines and (c) from fluorinated imines. This work: formation of fluorinated quinoline derivatives involving activated FARs. | |
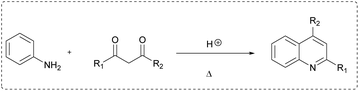
To further exploit the reactivity of FARs, we decided to use N-aryl fluoroketimines. The reactions afforded bis-fluorinated quinolines 2 and 3 (Scheme 3). Such fluorinated quinolines are commonly prepared by first constructing the quinoline core (via the Skraup, Döbner-von Miller, Pfitzinger, Conrad–Limpach, Friedlander or Combes syntheses, to cite a few),21,48,49 mostly under harsh conditions, and sometimes with low yields. Then, fluorine can be introduced by means of fluorinating agents, the Balz–Schiemann reaction, the Halex process or HF-mediated process. Nevertheless, such fluorinations often require multi-step reactions and particular equipment.31,50–59 Among the existing syntheses of fluorinated quinolines, none combines a short synthetic pathway, mild reaction conditions, the possibility to introduce different fluorinated substituents in the same molecule and industrially suitable conditions. In particular, the synthesis of quinoline derivatives bearing two fluorinated groups in both positions 2 and 4 is scarcely described in the literature; only a few bis(trifluoromethylated)quinolines can be found.57,58,60,61 Some examples reported on the use of anilines and fluorinated β-diketones, which are difficultly accessible. The products were formed in low yields and their isolation proved to be tricky because of their volatility. Moreover, the cyclization step is usually performed under harsh conditions in polyphosphoric acid, which produces P-containing waste in large amounts, and the use of unsymmetrical β-diketones leads to mixtures of regioisomers.62–64 In the present work, we describe the access to quinolines substituted in positions 2 and 4 with different fluoroalkyl groups, under mild conditions and with complete regioselectivity.
Results and discussion
Synthesis of quinoline derivatives
Substituted anilines were condensed onto di- or trifluoroacetone 5 at room temperature in anhydrous dichloromethane (DCM) in the presence of a desiccant (e.g. 4 Å MS, MgSO4) following the publication of Perrone et al. (Scheme 4).65 The desired fluorinated imines 1 were obtained in moderate to excellent yields (Tables 1 and 2). Ketimines obtained from 1,1-difluoroacetone gave better results than those from trifluoroacetone. This can be explained by the weaker destabilization of the carbocation – due to the less intense electron-withdrawing effect of the CHF2 moiety compared to CF3 – during the hemiaminal dehydration step. In some cases, the purity of the ketimine was low, either because of incomplete conversion—even after introduction of an additional ketone or prolonged reaction time—or side reactions, e.g. self-condensation of the ketone. This can be avoided by performing the reaction at 0 °C; however, under these conditions, the condensation between anilines and the ketones is either very slow or totally ineffective. Ketimines cannot be purified due to their sensitivity to moisture or silica. Nonetheless, the presence of side products in 1 did not seem to affect the next step. The stability of the ketimines is acceptable and they can be stored under argon for a few weeks without hydrolysis or degradation.
 |
| | Scheme 4 Synthesis of fluorinated ketimines. | |
Table 1 Trifluoromethylated ketimines
| Entry |
Substrate |
Yield (%) |
Entry |
Substrate |
Yield (%) |
| 1 |
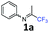
|
87 |
7 |
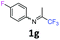
|
93 |
| 2 |
|
90 |
8 |
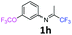
|
82 |
| 3 |
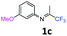
|
92 |
9 |
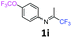
|
93 |
| 4 |
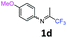
|
93 |
10 |
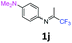
|
80 |
| 5 |
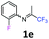
|
70 |
11 |
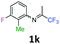
|
57 |
| 6 |
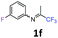
|
62 |
12 |
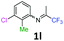
|
50 |
Table 2 Difluoromethylated ketimines
| Entry |
Substrate |
Yield (%) |
Entry |
Substrate |
Yield (%) |
|
Mixture (SM, impurities).
|
| 1 |
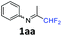
|
96 |
12 |
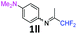
|
94 |
| 2 |
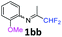
|
98 |
13 |
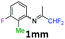
|
90 |
| 3 |
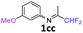
|
98 |
14 |
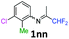
|
91 |
| 4 |
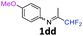
|
98 |
15 |
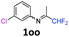
|
|
| 5 |
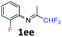
|
78 |
16 |
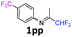
|
29 |
| 6 |
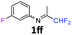
|
91 |
17 |
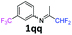
|
93 |
| 7 |
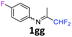
|
83 |
18 |
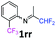
|
40 |
| 8 |
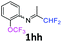
|
75 |
19 |
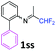
|
|
| 9 |
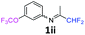
|
93 |
20 |
|
|
| 10 |
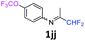
|
91 |
21 |
|
91 |
| 11 |
|
|
|
|
|
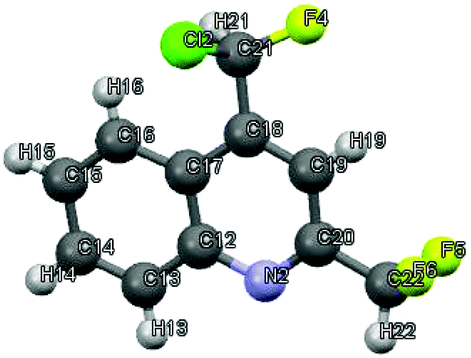
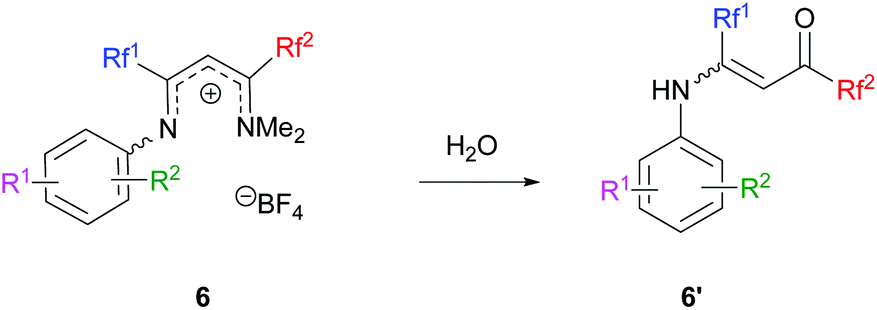
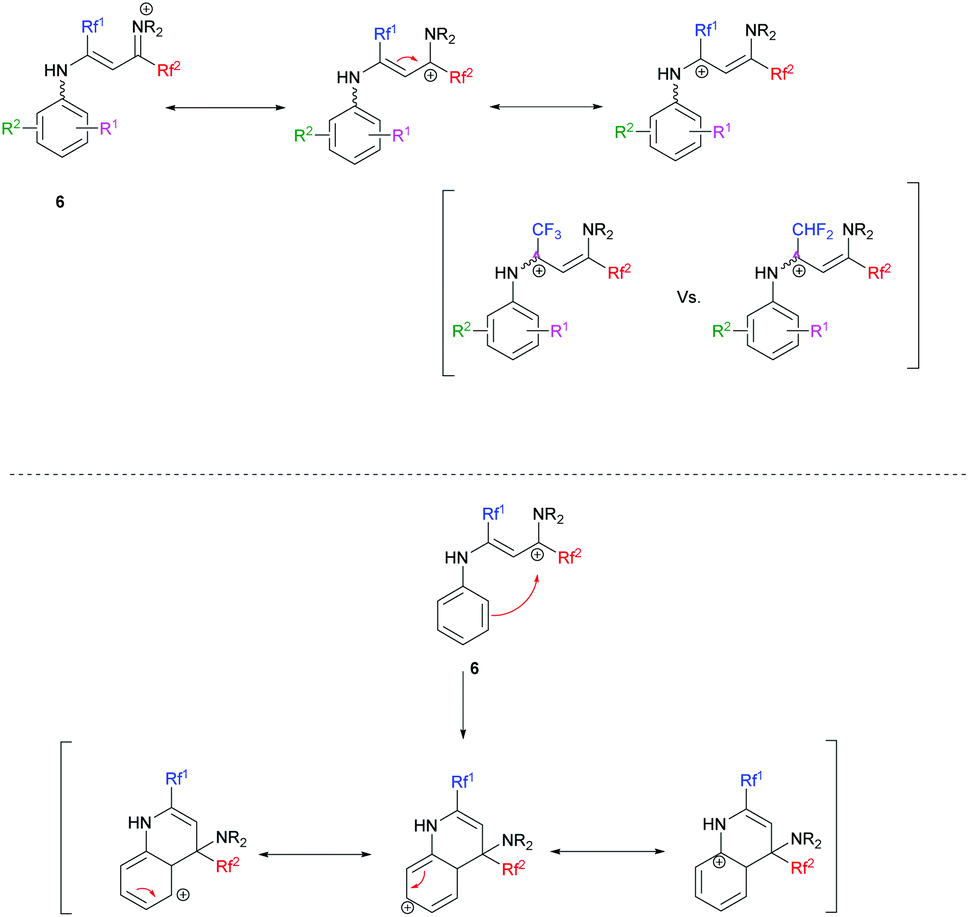
Next, ketimines 1 were reacted with activated FARs. In practice, FARs were treated beforehand with BF3·Et2O in MeCN for 15 min and directly mixed with fluorinated imines. Upon nucleophilic attack of the enamine tautomer of 1 onto the iminium salt, the resulting vinamidinium intermediate 6 undergoes electrophilic aromatic substitution yielding quinolines 2 and 3, following a Combes-like rearrangement (Schemes 3, 5 and Fig. 2).66
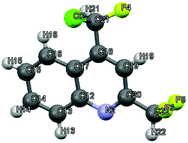 |
| | Fig. 2 Crystal structure of 4-[chloro(fluoro)methyl]-2-(difluoromethyl)quinoline 3aii.67 | |
 |
| | Scheme 5 Combes reaction. | |
In certain cases, the transformation was not complete and hydrolysis of the reaction mixture provided vinamides 6′, thus confirming the mechanistic pathway (Scheme 6).
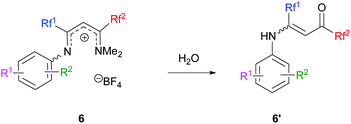 |
| | Scheme 6 Vinamidinium hydrolysis. | |
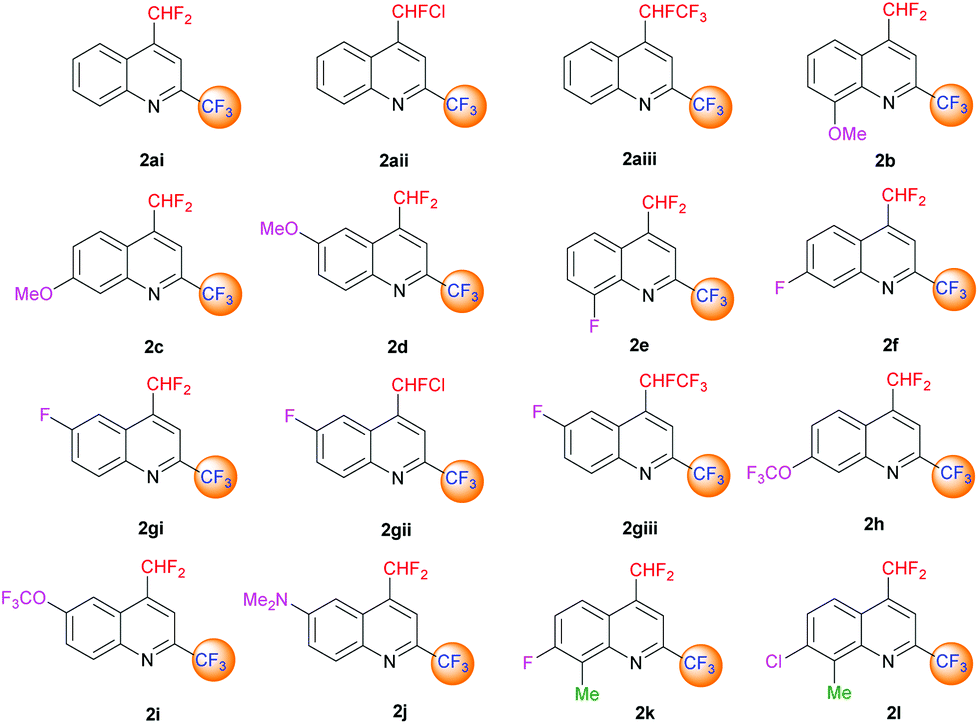
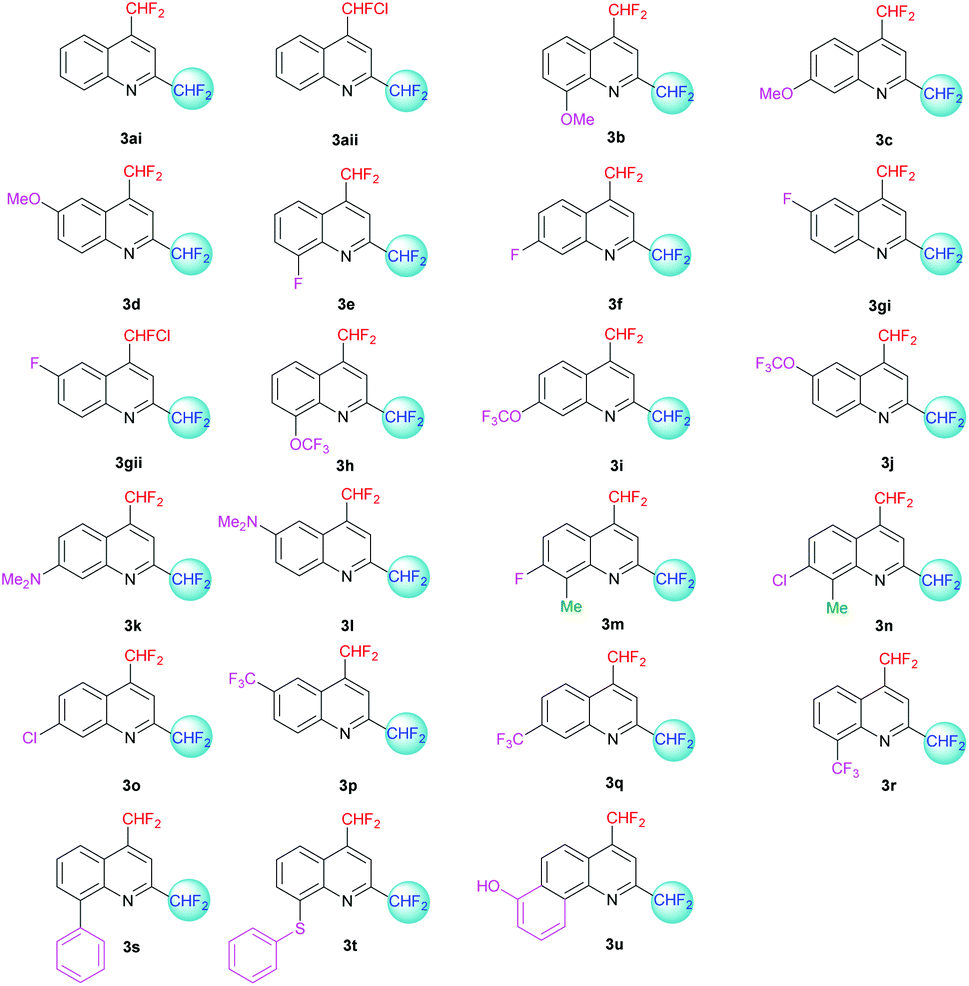
The desired quinolines were obtained in moderate to good yields. All results are summarized below (Tables 3 and 4, Fig. 3 and 4).
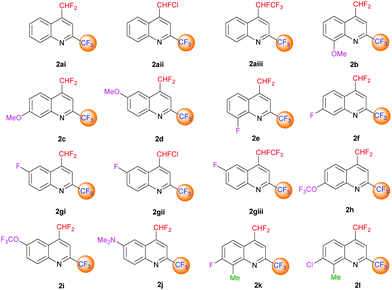 |
| | Fig. 3 4-(Difluoromethyl)-2-(trifluoromethyl)-substituted quinoline derivatives 2ai–l. | |
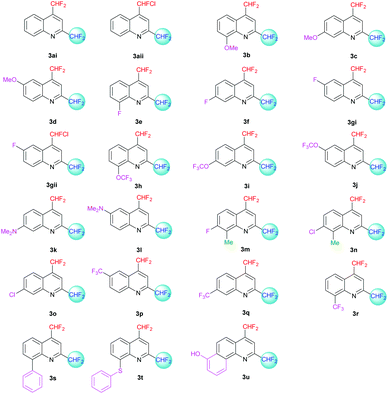 |
| | Fig. 4 2,4-Bis(difluoromethyl)-substituted quinoline derivatives 3ai–u. | |
Table 3 Synthesis of quinolines from 1,1,1-trifluoroacetone-derived imines 1a–l
| Entry |
Substrate |
FAR |
Product |
Yielda (%) |
Isolated yield unless indicated otherwise.
19F NMR yield by using fluorobenzene as internal standard.
1H NMR ratio (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6′), not isolated. :6′), not isolated.
|
| 1 |
1a
|
4a
|
2ai
|
62 |
| 2 |
4b
|
2aii
|
57 |
| 3 |
4c
|
2aiii
|
14 |
| 4 |
1b
|
4a
|
2b
|
88 |
| 5 |
1c
|
2c
|
64 |
| 6 |
1d
|
2d
|
85 |
| 7 |
1e
|
2e
|
70 |
| 8 |
1f
|
2f
|
62 |
| 9 |
1g
|
4a
|
2gi
|
67 |
| 10 |
4b
|
2gii
|
35b |
| 11 |
4c
|
2giii
|
1:3c |
| 12 |
1h
|
4a
|
2h
|
36 |
| 13 |
1i
|
2i
|
49 |
| 14 |
1j
|
2j
|
37 |
| 15 |
1k
|
2k
|
38 |
| 16 |
1l
|
2l
|
38 |
Table 4 Synthesis of quinolines from 1,1-difluoroacetone-derived imines 1aa–uu
| Entry |
Substrate |
FAR |
Product |
Yielda (%) |
|
Isolated yield unless indicated otherwise.
1H NMR ratio (3:6′), not isolated.
|
| 1 |
1aa
|
4a
|
3ai
|
77 |
| 2 |
4b
|
3aii
|
78 |
| 3 |
1bb
|
4a
|
3b
|
72 |
| 4 |
1cc
|
3c
|
78 |
| 5 |
1dd
|
3d
|
71 |
| 6 |
1ee
|
3e
|
39 (3:1)b |
| 7 |
1ff
|
3f
|
70 |
| 8 |
1gg
|
3gi
|
39 |
| 9 |
4b
|
3gii
|
Traces |
| 10 |
1hh
|
4a
|
3h
|
39 |
| 11 |
1ii
|
3i
|
71 |
| 12 |
1jj
|
3j
|
58 |
| 13 |
1kk
|
3k
|
15 |
| 14 |
1ll
|
3l
|
14 |
| 15 |
1mm
|
3m
|
76 |
| 16 |
1nn
|
3n
|
75 |
| 17 |
1oo
|
3o
|
45 |
| 18 |
1pp
|
3p
|
23 |
| 19 |
1qq
|
3q
|
28 |
| 20 |
1rr
|
3r
|
Traces |
| 21 |
1ss
|
3s
|
28 |
| 22 |
1tt
|
3t
|
52 |
| 23 |
1uu
|
3u
|
51 |
In general, quinoline derivatives obtained from either the Yarovenko 4b or Ishikawa 4c reagents (Table 3: entries 2, 3, 10 and 11; Table 4: entries 2 and 9) were formed in lower yields than those obtained from the reaction with TFEDMA 4a. This confirms the higher reactivity of the latter reagent.
The synthesis of quinolines derived from CF3 substituted anilines (Table 4: entries 18–20) was performed, however the yields were not satisfying due to the high volatility of these compounds.
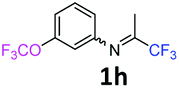
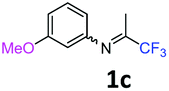
Starting from imines bearing a 3-substituent on the arene (Table 3: entries 5, 8 and 12; Table 4: entries 4, 7, 11, 13 and 19), only one regioisomer was observed after purification, namely 7-substituted quinolines. Indeed, the cyclisation of vinamidinium intermediate 6 occurs at the less hindered position, i.e. para to the 3-substituent of the imine.
Overall, the relative yields of quinolines in Tables 3 and 4 can be rationalized in part by an interplay of destabilizing and stabilizing effects operating on the vinamidinium intermediate on the one hand, and on the Wheland intermediate obtained during cyclisation on the other hand (Scheme 7).
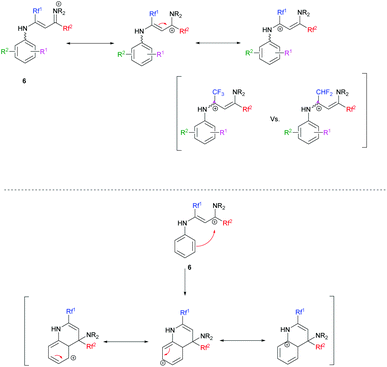 |
| | Scheme 7 Stabilization/destabilization of intermediate 6 during the cyclization process. | |
In the case where Rf1 is a CF3 group, the withdrawing effect of CF3 destabilizes the vinamidinium intermediate 6. The addition of mesomeric donor substituents on 6 might stabilize it. We indeed obtained similar or better results than when starting from the corresponding N-phenyl substrate (Table 3, entries 1, 4–9). A notable exception is the dimethylamino-substituted substrate (entry 14). When Rf1 is a CF3 group, the best results in terms of yields were obtained when mesomeric donor substituents are in positions 2 and 4 (Table 3, entries 4 and 6 vs. entry 5, entries 7 and 9 vs. entry 8). The introduction of electron-withdrawing substituents destabilizes the vinamidinium even further and the cyclization is less efficient (Table 3, entries 1, 12 and 13).
When Rf1 is a CHF2, the vinamidinium intermediate 6 is less destabilized. Cyclization would therefore be facilitated by mesomeric donor substituents (Table 4, entries 1, 3–5, 7 vs. 10, 12, 18–20). As expected quinoline derivatives which possess mesomeric donor substituents in position 7 gave the best results. Indeed the carbocation is stabilized during the cyclization process (Table 4, entry 4 vs. entries 3 and 5, entry 7 vs. entries 6 and 8). However, once again the dimethylamino group fails to obey this rule (entry 13), and a 3-OCF3 substituent leads to a surprisingly high yield of 71% (entry 11).
Clearly, other subtle effects have yet to be invoked to account for the observed reactivities. In general, higher yields are obtained for 7-substituted quinolines compared to 6- or 8-substituted ones with Rf1 = CHF2 (Table 4, entry 4 vs. entries 3 and 5; entry 7 vs. entries 6 and 8), while the opposite applies when Rf1 is a CF3 (Table 3, entry 5 vs. entries 4 and 6; entry 8 vs. entries 7 and 9).
Introduction of new functionalities in position 8 by oxidation
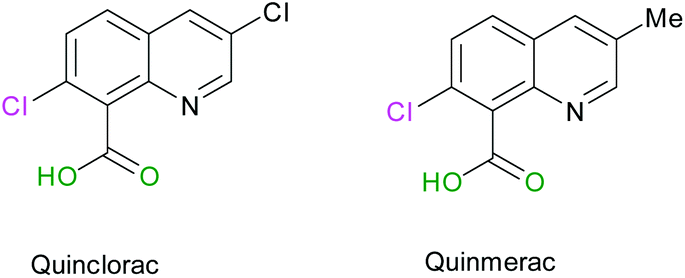
Commercially available quinolines named Quinmerac and Quinclorac, developed by BASF, have shown some activities as selective herbicides and are used on cereals to control weeds growth.68,69 The particularity of these molecules is that they bear a carboxylic acid function in position 8 (Scheme 8).
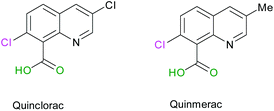 |
| | Scheme 8 Molecular structures of Quinclorac and Quinmerac. | |
Thus, we were interested in the introduction of a carboxylic acid function in the same position on our fluorinated quinoline derivatives. Bis-substituted quinolines 3n and 3m were chosen as precursors for the synthesis of bis(fluoroalkyl) analogues of Quinmerac and Quinclorac in order to test their potential activity as phytosanitary ingredients.
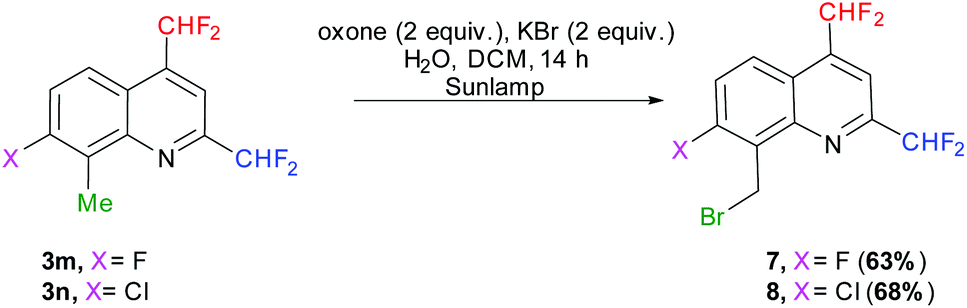
We first attempted to oxidize the methyl group of quinolines 3m–n under photochemical conditions, following the procedure of Togo et al., which should afford the desired carboxylic acids in the presence of oxone and potassium bromide in a mixture of water and dichloromethane.70 Nevertheless, the reaction did not allow access to the desired compounds but to the 8-(bromomethyl)-2,4-bis(difluoromethyl)-7-halo-quinolines 7 (X = F) and 8 (X = Cl) (Scheme 9). Alternately, the oxidation conditions of Sudalai et al. (aqueous sulfuric acid, sodium periodate and lithium bromide in the absence of a solvent)71 provided the same 8-(bromomethyl)quinoline derivatives 7 and 8 in 50 and 55% yields, respectively. Although it requires a slightly longer reaction time, the photochemical procedure was preferred as it gave better results and is also easier to carry out.
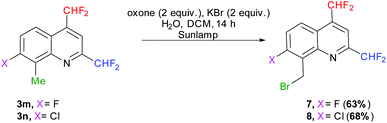 |
| | Scheme 9 Methyl group oxidation by photochemistry. | |
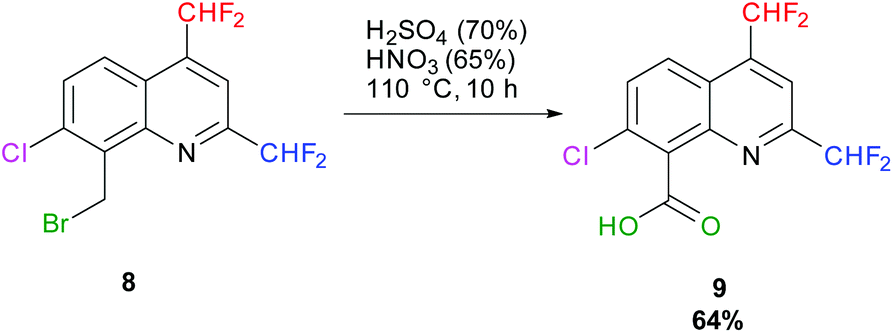
In 1984, H. Hagen and coworkers described the oxidation of a bromomethyl quinoline derivative into its corresponding carboxylic acid (Quinmerac) thanks to a mixture of concentrated acids at 110 °C.69 Following this procedure, 7-chloro-2,4-bis(difluoromethyl)quinoline-8-carboxylic acid 9 was obtained in 64% yield (Scheme 10).
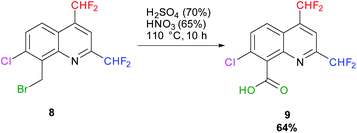 |
| | Scheme 10 Oxidation of 8-(bromomethyl)-7-chloro-2,4-bis(difluoromethyl)quinoline 8 into its corresponding carboxylic acid 9. | |
The structure of the desired compound was confirmed by X-ray diffraction crystallography, which allowed us to observe the presence of a reciprocal intermolecular hydrogen bonding between the hydroxyl group of one molecule and the nitrogen of the other (Fig. 5).
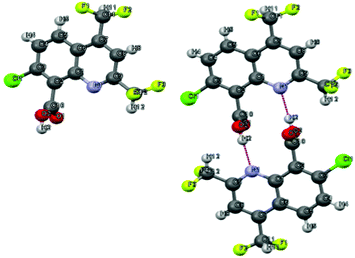 |
| | Fig. 5 Single crystal X-ray diffraction structure of 9.67 | |
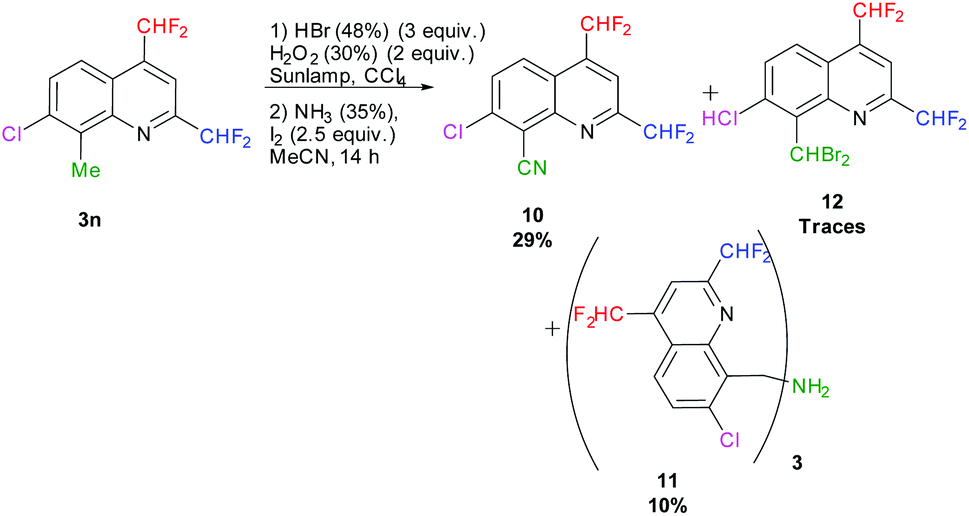
In parallel, we attempted the transformation of the methyl in position 8 into a nitrile according to Togo et al., employing aq. HBr and aq. H2O2 in CCl4 followed by the addition of aq. NH3 and I2 in MeCN.72 After 14 hours of reaction under sunlamp irradiation at 60 °C, 29% of the desired 7-chloro-2,4-bis(difluoromethyl)quinoline-8-carbonitrile 10 was obtained. After purification, traces of 8-(dibromomethyl)-7-chloro-2,4-bis(difluoromethyl)quinoline 12 and 10% of the tris(quinoline-8-ylmethyl)amine 11 were isolated (Scheme 11).
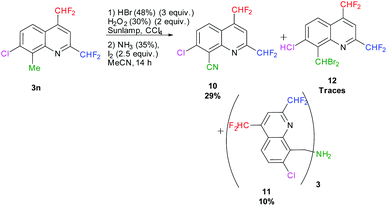 |
| | Scheme 11 One-pot transformation of 8-methylquinoline 3n into the corresponding quinoline-8-carbonitrile 10. | |
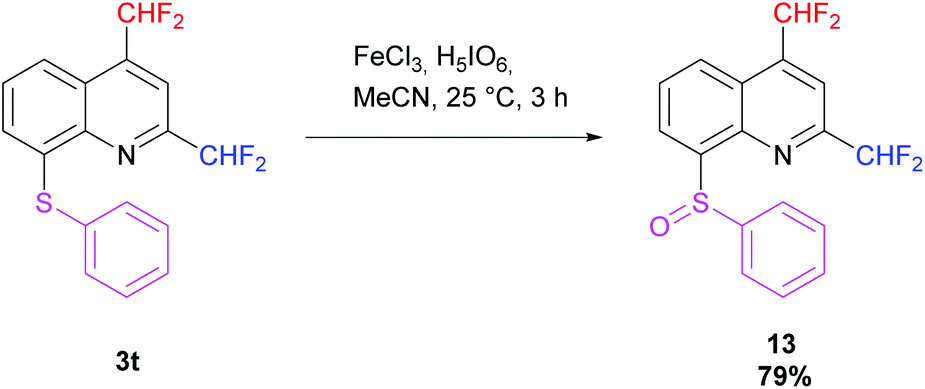
Then, oxidation of the 2,4-bis(difluoromethyl)-8-(phenylsulfanyl)quinoline 3t was performed following the procedure of Kim et al. using iron(III) chloride and periodic acid.73 The desired sulfoxide 13 was obtained in 79% yield in a racemic form. The diastereotopic fluorine atoms observed in 19F NMR confirm the presence of a stereocenter on the molecule (Scheme 12).
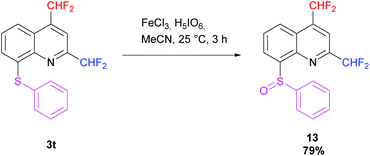 |
| | Scheme 12 Oxidation of 3t into sulfoxide 13. | |
Several other oxidation protocols were attempted in order to functionalize the position 8 of quinolines 7 and 8 but all failed. The low electron-density of these bis(fluoroalkyl)quinolines render them difficult to oxidize, as attested by the need of really harsh conditions, even at high temperature, to afford the desired compounds in moderate yields only.
Comparison between the use of FARs and carboxylic anhydrides or oxalate derivatives in the synthesis of quinolines
Furthermore, we studied the reaction of ketimine 1aa with carboxylic anhydrides and acyl chloride. Indeed, various anhydrides or acyl chlorides are commercially available and could enlarge the scope of the approach. The purpose was to study the possibility to access to new quinoline derivatives by using this abundant starting material.
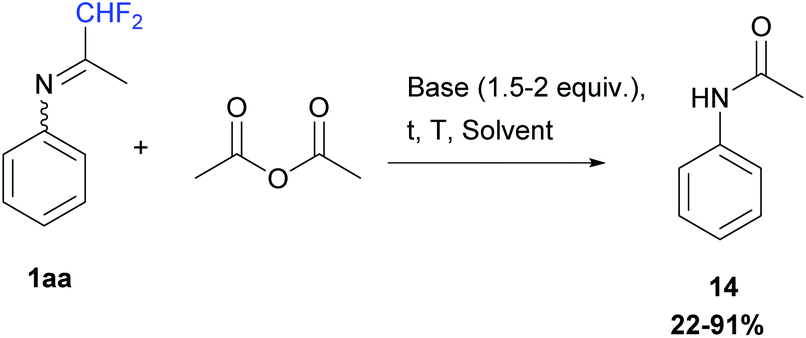
Our first attempt used acetic anhydride (1 equiv.) in either dichloromethane or acetonitrile, from −5 to 50 °C, in the presence of bases such as pyridine or DIPEA (1.5 to 2 equiv.) or without. At the end of the reaction, N-phenylacetamide 14 was always obtained after N-acylation and hydrolysis of the ketimine 1aa in 22 to 91% yields (Scheme 13).
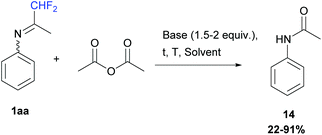 |
| | Scheme 13 Reaction between ketimine 1aa and acetic anhydride. | |
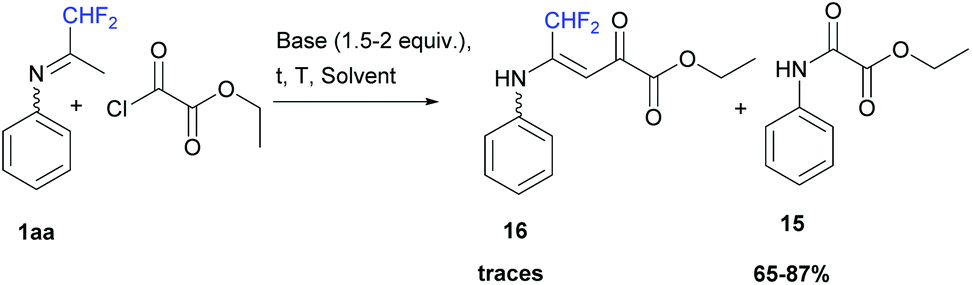
When ketimine 1aa was reacted with ethyl oxalyl monochloride (1 equiv.) under the same series of conditions as those for acetic anhydride, the corresponding ethyl-2-oxo-2-(phenylamino)acetate 15 was obtained in 65 to 87% yields (Scheme 14). However, when 1aa was dissolved in acetonitrile in the presence of pyridine (1.5 equiv.) at room temperature, a small amount of vinamide 16 was obtained as a minor fraction in combination with unidentified impurities.
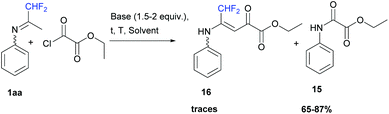 |
| | Scheme 14 Reaction between ketimine 1aa and ethyl oxalyl monochloride. | |
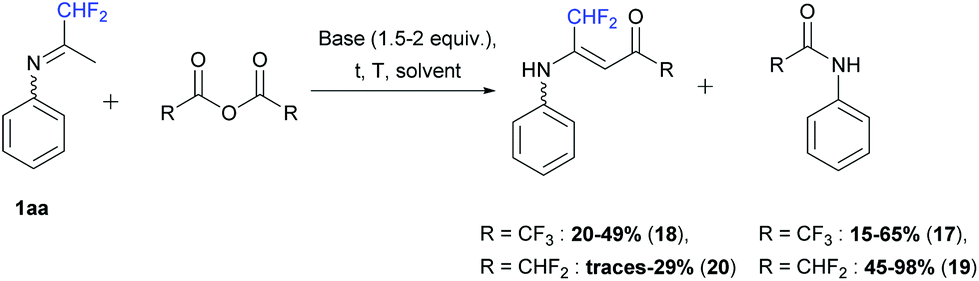
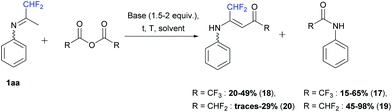
In the second part, we decided to study the reactivity of ketimine 1aa in the presence of fluorinated acetic anhydrides (1 equiv.). First, when trifluoroacetic anhydride was used, the corresponding 2,2,2,-trifluoro-N-phenylacetamide 17 was obtained. In dichloromethane in the presence of pyridine (1.5 equiv.) at −5 to 50 °C and in acetonitrile in the presence of DIPEA (2 equiv.) at room temperature, only compound 17 was formed in 65 and 35% yields, respectively. However, in acetonitrile in the presence of pyridine (1.5 equiv.) or without a base, at room temperature or at 50 °C, the desired vinamide 18 was obtained in 20 to 49% yields (Scheme 15).
 |
| | Scheme 15 Reaction between ketimine 1aa and fluorinated anhydrides. | |
Next, difluoroacetic anhydride was reacted with ketimine 1aa under the same reaction conditions (Scheme 15). In dichloromethane, in the presence of pyridine (1.5 equiv.) at −5 to 50 °C, 45% yield of 2,2-difluoro-N-phenylacetamide 19 was obtained. Vinamide 20 was observed in a small amount in combination with degradation products and isolated in 29% yield after purification by column chromatography. DIPEA, used instead of pyridine also led to the formation of amide 19 as a major product in 47% yield and only traces of vinamide 20 were isolated.
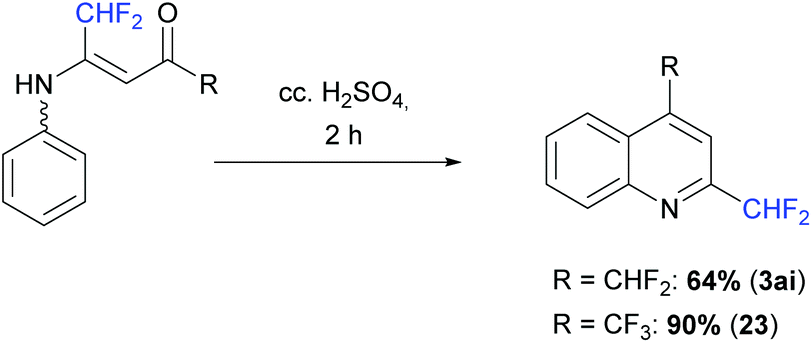
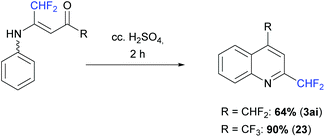
The vinamides 18 and 20 were finally involved in a cyclization reaction in the presence of concentrated H2SO4 (10 equiv.) at 50 °C for 2 h. The corresponding quinoline derivatives 3ai and 23 were obtained in 64 and 90% yield, respectively (Scheme 16).
 |
| | Scheme 16 Cyclization reaction of vinamide. | |
These results confirm the superiority of FARs in the synthesis of quinoline derivatives in one step with overall good yields. Although the use of anhydrides or acyl chlorides was a synthetic asset, they have not allowed access to the desired quinoline derivatives in one step, as they first afforded the intermediate vinamides which had to be converted into the quinolines under harsh conditions.
Conclusions
In summary, this work describes a new and efficient approach to access quinolines bearing two different or identical fluoroalkyl groups in positions 2 and 4. FARs proved to be highly useful tools in the synthesis of these fluorinated heterocycles and afforded them in moderate to high yield (14–88%) under mild conditions.
Experimental section
Materials and methods
All reactions were performed in flame-dried glassware using Schlenk techniques. Liquids and solutions were transferred with syringes. Air- and moisture-sensitive materials were stored and handled under an atmosphere of argon. Solvents were purified and dried following standard procedures: dichloromethane (DCM) and tetrahydrofuran (THF) were distilled from CaH2 or sodium + benzophenone prior to use. Desiccants (4 Å molecular sieves (4 Å MS) or magnesium sulphate (MgSO4)) were previously activated in an oven. Technical grade solvents for extraction and chromatography (cyclohexane, dichloromethane, n-pentane, ether, toluene, and ethyl acetate) were used without purification. Starting materials, if commercial, were purchased from standard suppliers (Sigma-Aldrich, Acros, Alfa Aesar and Apollo Scientific) and used as such, provided that adequate checks (NMR) had confirmed the claimed purity. Analytical thin-layer chromatography (TLC) was carried out on 0.25 mm Merck silica-gel (60-F254). Flash column chromatography was performed on silica gel 60 (40–63 μm, 230–400 mesh, ASTM) by Merck using the indicated solvents. 1H, 13C, and 19F-NMR spectra were recorded in CDCl3 on Bruker AV 400 instruments (1H: 400 MHz, 19F: 376 MHz, 13C: 101 MHz). Chemical shifts are reported in parts per million (ppm) and are referenced to the residual solvent resonance as the internal standard (chloroform (δ [1H] = 7.26 and accordingly δ [13C] = 77.16 ppm). Data are reported as follows: chemical shift, multiplicity (br s = broad singlet, s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, td = triplet of doublets, dd = doublet of doublets), coupling constant (Hz) and integration. The spectra were processed with the program MestReNova (version 6.0.2–5475). Melting points (MPs) were determined for crystalline compounds with a Büchi Melting Point Apparatus M-560 and are not corrected. IR spectra were measured with a Perkin Elmer Spectrum UATR two (diamond detection). HRMS analysis (measurement accuracy ≤15 ppm) and EA were performed by the analytical facility at the University of Strasbourg. Crystal X-ray diffraction analysis was carried out by the Radiocrystallography Service of the University of Strasbourg.
General procedure for the synthesis of ketimine derivatives 1
Under an argon atmosphere, an excess of cold perfluoroacetone (2 equiv.) was added to aniline derivatives (1 equiv.) in anhydrous DCM (2 mL per 1 mmol) in the presence of a desiccant (e.g. (4 Å MS) or MgSO4). Then the reaction mixture was stirred for the indicated time at room temperature. The desiccant was then filtered off on Celite and washed with ether. The filtrate was concentrated under reduced pressure to provide the desired product. Ketimines cannot be purified due to their sensitivity to moisture or silica, so the compound was used without any further purification.
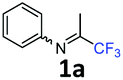
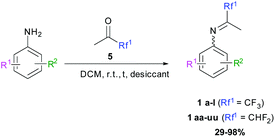
N-(1,1,1-Trifluoropropan-2-ylidene)aniline 1a.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 1.92 mL, 21.4 mmol) and aniline (1 equiv., 0.98 mL, 10.7 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 3 h at room temperature. N-(1,1,1-Trifluoropropan-2-ylidene)aniline 1a was provided as a brown oil (1.74 g, 87%, estimated). 1H NMR δH = 7.38 (2 H, t, 3JH–H = 7.9 Hz, C(3,5)H), 7.18 (1 H, t, 3JH–H = 7.5 Hz, C(4)H), 6.82–6.74 (2 H, m, C(2,6)H), 2.02 (3 H, s, Me) ppm. 19F NMR δF = −74.68 (3 F, s, CF3) ppm. 13C NMR δC = 157.41 (q, 2JC–F = 33.9 Hz, CCF3), 147.72 (s, C-1), 129.33 (2 C, s, C-3,5), 125.29 (s, C-4), 119.87 (q, 1JC–F = 278.4 Hz, CF3) 118.87 (2 C, s, C-2,6), 14.50 (s, Me) ppm.
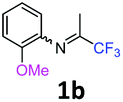
2-Methoxy-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1b.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 1.46 mL, 16.2 mmol) and 2-methoxyaniline (1 equiv., 0.92 mL, 8.12 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 21 h at room temperature. 2-Methoxy-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1b was provided as a brown oil (1.59 g, 90%). 1H NMR δH = 7.16–7.12 (1 H, m, C(4)H), 7.00–6.90 (2 H, m, C(5,6)H), 6.78–6.75 (1 H, m, C(3)H), 3.78 (3 H, s, OMe), 1.95 (3 H, s, Me) ppm. 19F NMR δF = −74.33 to −74.42 (3 F, m, CF3) ppm. 13C NMR δC = 158.86 (q, 2JC–F = 33.7 Hz, CCF3), 148.19 (s, C-2), 136.43 (s, C-1), 126.15 (s, C-4), 120.90 (s, C-5), 120.14 (s, C-3), 119.77 (q, 1JC–F = 278.4 Hz, CF3), 111.78 (s, C-6), 55.51 (s, OMe), 14.90 (s, Me) ppm. HRMS (ESI+) for C10H11F3NO [M + H]: calcd 218.0787, found 218.0773.
3-Methoxy-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1c.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 1.47 mL, 16.4 mmol) and 3-methoxyaniline (1 equiv., 0.91 mL, 8.12 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 24 h at room temperature. 3-Methoxy-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1c was provided as a brown oil (1.62 g, 92%, estimated). 1H NMR δH = 7.35–7.22 (1 H, m, C(5)H), 6.79–6.70 (1 H, m, C(4)H), 6.44–6.32 (2 H, m, C(2,6)H), 3.79 (3 H, s, OMe), 2.05 (3 H, s, Me) ppm. 19F NMR δF = −74.81 (3 F, s, CF3), −74.91 (3 F, s, CF3) ppm. 13C NMR δC = 160.50 (s, C-3), 157.56 (q, 2JC–F = 33.9 Hz, CCF3), 148.95 (s, C-1), 130.08 (s, C-5), 119.78 (q, 1JC–F = 278.2 Hz, CF3), 110.60 (2 C, s, C-4,6), 104.49 (s, C-2), 54.91 (s, OMe), 14.01 (s, Me) ppm. HRMS (ESI+) for C10H11F3NO [M + H]: calcd 218.0787, found 218.0818.
4-Methoxy-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1d.
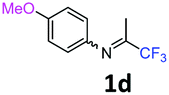
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 1.47 mL, 16.4 mmol) and 4-methoxyaniline (1 equiv., 1.01 g, 8.2 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 20 h at room temperature. 4-Methoxy-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1d was provided as a brown oil (1.65 g, 93%). 1H NMR δH = 6.86 (2 H, m, AA′, C(2,6)H), 6.73 (2 H, m, BB′, C(3,5)H), 3.75–3.71 (3 H, m, OMe), 2.03–2.00 (3 H, m, Me) ppm. 19F NMR δF = −74.58 to −74.63 (3 F, m, CF3) ppm. 13C NMR δC = 157.51 (s, C-4), 156.53 (q, 2JC–F = 33.5 Hz, CCF3), 140.36 (s, C-1), 120.85 (2 C, s, C-3,5), 119.96 (q, 1JC–F = 278.1 Hz, CF3), 114.31 (2 C, s, C-2,6), 55.09 (s, OMe), 13.99 (s, Me) ppm. HRMS (ESI+) for C10H11F3NO [M + H]: calcd 218.0787, found 218.0815.
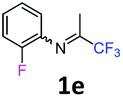
2-Fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1e.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 3.23 mL, 36 mmol) and 2-fluoroaniline (1 equiv., 1.74 mL, 18 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 16 h at room temperature. 2-Fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1e was provided as a brown oil (2.58 g, 70%). 1H NMR δH = 7.21–7.07 (3 H, m, C(4,5,6)H), 6.93–6.86 (1 H, m, C(3)H), 2.02 (3 H, s, Me) ppm. 19F NMR δF = −74.65 (3 F, s, CF3), −126.51 to −126.58 (1 F, m, F) ppm. 13C NMR δC = 160.40 (qd, 2JC–F = 34.1, 4JC–F = 0.5 Hz, CCF3), 151.01 (d, 1JC–F = 246.3 Hz, C-2), 134.91 (d, 2JC–F = 12.8 Hz, C-3), 126.57 (d, 3JC–F = 7.3 Hz, C-4), 124.59 (d, 4JC–F = 3.8 Hz, C-5), 121.68 (d, 3JC–F = 1.1 Hz, C-6), 119.53 (q, 1JC–F = 278.2 Hz, CF3), 116.18 (d, 2JC–F = 19.7 Hz, C-3), 15.01 (s, Me) ppm. HRMS (ESI+) for C9H8F4N [M + H]: calcd 206.0587, found 206.0590.
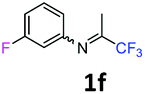
3-Fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1f.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 1.61 mL, 18 mmol) and 3-fluoroaniline (1 equiv., 0.87 mL, 9 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 48 h at room temperature. 3-Fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1f was provided as a brown oil (1.14 g, 62%). 1H NMR δH = 7.36–7.28 (1 H, m, C(5)H), 6.90–6.82 (1 H, m, C(4)H), 6.57–6.49 (2 H, m, C(2,6)H), 2.02 (3 H, s, Me) ppm. 19F NMR δF = −73.12 to −77.79 (3 F, m, CF3), −109.19 to −115.09 (1 F, m, F) ppm. 13C NMR δC = 163.44 (d, 1JC–F = 247.2 Hz, C-3), 158.64 (q, 2JC–F = 34.3 Hz, CCF3), 149.33 (d, 3JC–F = 9.1 Hz, C-1), 130.70 (d, 3JC–F = 9.2 Hz, C-5), 119.56 (q, 1JC–F = 278.3 Hz, CF3), 114.45 (d, 4JC–F = 2.9 Hz, C-6), 112 (d, 2JC–F = 21.3 Hz, C-4), 106.38 (d, 2JC–F = 24.1 Hz, C-2), 14.36 (s, Me) ppm. HRMS (ESI+) for C9H8F4N [M + H]: calcd 206.0587, found 206.0572.
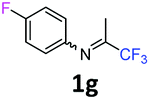
4-Fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1g.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 1.87 mL, 20.8 mmol) and 4-fluoroaniline (1 equiv., 1 mL, 10.4 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 13 h at room temperature. 4-Fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1g was provided as a brown oil (1.97 g, 93%). 1H NMR δH = 7.10–7.03 (2 H, m, C(3,5)H), 6.79–6.73 (2 H, m, C(2,6)H), 2.03 (3 H, s, Me) ppm. 19F NMR δF = −74.77 to −74.79 (3 F, m, CF3), −117.79 to −118.66 (1 F, m, F) ppm. 13C NMR δC = 160.39 (d, 1JC–F = 244.1 Hz, C-4), 158.01 (qd, 2JC–F = 33.9, 6JC–F = 1.1 Hz, CCF3), 143.59 (d, 4JC–F = 2.7 Hz, C-1), 120.68 (2 C, d, 3JC–F = 8.2 Hz, C-2,6), 119.68 (q, 1JC–F = 278.2 Hz, CF3), 115.98 (2 C, d, 2JC–F = 22.8 Hz, C-3,5), 14.38 (s, Me) ppm. LCMS (ESI−) for C9H6F4N [M − H] m/z 223 (M + H2O, 100%), 153 (15).
3-(Trifluoromethoxy)-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1h.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 1.01 mL, 11.3 mmol) and 3-(trifluoromethoxy)aniline (1 equiv., 0.76 mL, 5.65 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 48 h at room temperature. 3-(Trifluoromethoxy)-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1h was provided as a brown oil (1.25 g, 82%, estimated). 1H NMR δH = 7.40 (1 H, t, 3JH–H = 8.1 Hz, C(5)H), 7.04 (1 H, d, 3JH–H = 8.3 Hz, C(4)H), 6.71 (1 H, d, 3JH–H = 7.9 Hz, C(6)H), 6.68 (1 H, s, C(2)H), 2.03 (s, Me) ppm. 19F NMR δF = −58.08 (3 F, s, OCF3), −74.91 (3 F, s, CF3) ppm. 13C NMR δC = 158.85 (q, 2JC–F = 34.3 Hz, CCF3), 150.05 (q, 3JC–F = 2.02 Hz, C-3), 149.12 (s, C-1), 130.75 (s, C-5), 120.41 (q, 1JC–F = 258.2 Hz, OCF3), 119.49 (q, 1JC–F = 278.3 Hz, CF3), 117.27 (s, C-4), 116.96 (s, C-6), 111.58 (s, C-2), 14.52 (s, Me) ppm. HRMS (ESI+) for C10H8F6NO [M + H]: calcd 272.0505, found 272.0488.
4-(Trifluoromethoxy)-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1i.
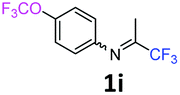
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 2.02 mL, 22.6 mmol) and 4-(trifluoromethoxy)aniline (1 equiv., 1.53 mL, 11.3 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 48 h at room temperature. 4-(Trifluoromethoxy)-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1i was provided as a brown oil (2.85 g, 93%, estimated), isomer ratio 87:13. 1H NMR δH = 7.17 (2 H, d, 3JH–H = 8.8 Hz, C(2,6)H), 6.95 (2 H, d, 3JH–H = 8.8 Hz, C(2′,6′)H), 6.73 (2 H, d, 3JH–H = 8.8 Hz, C(3,5)H), 6.61 (2 H, d, 3JH–H = 8.8 Hz, C(3′,5′)H), 1.96 (3 H, s, Me), 1.96 (3 H, s, Me′) ppm. 19F NMR δF = −58.40 to −58.67 (3 F, m, OCF3), −74.89 to −75.12 (3 F, m, CF3) ppm. 13C NMR δC = 158.94 (q, 2JC–F = 34.3 Hz, CCF3), 146.84 (q, 3JC–F = 2.02 Hz, C-4), 146.15 (s, C-1), 122.26 (2 C, s, C-2,6), 120.31 (2 C, s, C-3,5), 120.76 (q, 1JC–F = 256.8 Hz, OCF3), 119.79 (q, 1JC–F = 278.2 Hz, CF3), 14.46 (s, Me) ppm. HRMS (ESI+) for C10H8F6NO [M + H]: calcd 272.0505, found 272.0496.
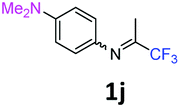
1-N,N-Dimethyl-4-N-(1,1,1-trifluoropropan-2-ylidene)benzene-1,4-diamine 1j.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 1.32 mL, 14.8 mmol) and N,N-dimethyl-1,4-benzenediamine (1 equiv., 0.92 mL, 7.39 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 24 h at room temperature. 1-N,N-Dimethyl-4-N-(1,1,1-trifluoropropan-2-ylidene)benzene-1,4-diamine 1j was provided as a brown oil (1.36 g, 80%). 1H NMR δH = 6.83 (2 H, m, AA′, C(2,6)H), 6.74 (2 H, m, BB′, C(3,5)H), 2.96 (6 H, s, NMe2), 2.11 (3 H, s, Me) ppm. 19F NMR δF = −74.19 (3 F, s, CF3) ppm. 13C NMR δC = 154.86 (q, 2JC–F = 33.3 Hz, CCF3), 148.94 (s, C-4), 136.65 (s, C-1), 121.94 (2 C, s, C-2,6), 120.15 (q, 1JC–F = 278.0 Hz, CF3), 112.86 (2 C, s, C-3,5), 40.84 (2 C, s, NMe2), 14.67 (s, Me) ppm. HRMS (ESI+) for C11H14F3N2 [M + H]: calcd 231.1104, found 231.1125.
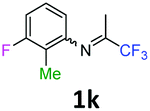
3-Fluoro-2-methyl-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1k.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 2.87 mL, 32 mmol) and 3-fluoro-2-methylaniline (1 equiv., 1.82 mL, 16 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 26 h at room temperature. 3-Fluoro-2-methyl-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1k was provided as a brown oil (2.01 g, 57%, estimated). 1H NMR δH = 7.17 (1 H, dd, 3JH–F = 14.3, 3JH–H = 7.8 Hz, C(4)H), 6.97 (1 H, dd, 4JH–F = 14.8, 3JH–H = 7.8 Hz, C(5)H), 6.43 (1 H, d, 3JH–H = 7.9 Hz, C(6)H), 2.01 (3 H, s, C(2)HMe), 1.99 (3 H, s, Me) ppm. 19F NMR δF = −74.56 (3 F, s, CF3), −115.94 (1 F, s, F) ppm. HRMS (ESI+) for C10H10F4N [M + H]: calcd 220.0744, found 220.0746.
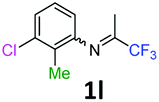
3-Chloro-2-methyl-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1l.
The product was prepared according to the general procedure, starting from 1,1,1-trifluoroacetone (2 equiv., 2.53 mL, 28.2 mmol) and 3-chloro-2-methylaniline (1 equiv. 1.71 mL, 14.1 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 26 h at room temperature. 3-Chloro-2-methyl-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1l was provided as a brown oil (1.67 g, 50%, estimated). 1H NMR δH = 7.22 (1 H, dd, 3JH–H = 8.0, 4JH–H = 1.0 Hz, C(4)H), 7.14 (1 H, t, 3JH–H = 7.9 Hz, C(5)H), 6.53 (1 H, dd, 3JH–H = 7.8, 4JH–H = 0.8 Hz, C(6)H), 2.14 (3 H, s, C(2)HMe), 1.98 (3 H, s, Me) ppm. 19F NMR δF = −74.51 (3 F, s, CF3) ppm. HRMS (ESI+) for C10H10F3NCl [M + H]: calcd 236.0448, found 236.0462.
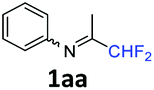
N-(1,1-Difluoropropan-2-ylidene)aniline 1aa.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (1.07 equiv., 5 mL, 58.2 mmol) and aniline (1 equiv., 5 mL, 54.6 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 3 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)aniline 1aa was provided as a brown oil (8.72 g, 96%). 1H NMR δH = 7.37 (2 H, t, 3JH–H = 7.8 Hz, C(3,5)H), 7.16 (1 H, t, 3JH–H = 7.2 Hz, C(4)H), 6.78 (2 H, d, 3JH–H = 7.4 Hz, C(2,6)H), 6.07 (1 H, t, 2JH–F = 55.6 Hz, CHF2), 1.95 (3 H, s, Me) ppm. 19F NMR δF = −121.27 (2 F, d, 2JF–H = 55.5 Hz, CHF2) ppm. 13C NMR δC = 163.54 (t, 2JC–F = 28.8 Hz, CCHF2), 148.47 (s, C-1), 129.12 (2 C, s, C-3,5), 124.86 (s, C-4), 119.01 (2 C, s, C-2,6), 114.96 (t, 1JC–F = 243.0 Hz, CHF2), 12.65 (s, Me) ppm. HRMS (ESI+) for C9H10F2N [M + H]: calcd 170.0774, found 170.0776.
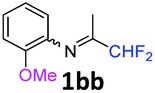
N-(1,1-Difluoropropan-2-ylidene)-2-methoxyaniline 1bb.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 1.32 mL, 16.2 mmol) and 2-methoxyaniline (1 equiv., 0.92 mL, 8.12 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 5 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-2-methoxyaniline 1bb was provided as a brown oil (1.59 g, 98%). 1H NMR δH = 7.14 (1 H, td, 3JH–H = 7.8, 4JH–H = 1.6 Hz, C(4)H), 6.99–6.92 (2 H, m, C(5,6)H), 6.74 (1 H, dd, 3JH–H = 7.6, 4JH–H = 1.6 Hz, C(3)H), 6.12 (1 H, t, 2JH–F = 55.6 Hz, CHF2), 3.80 (3 H, s, OMe), 1.89 (3 H, s, Me) ppm. 19F NMR δF = −120.86 (2 F, d, 2JF–H = 55.5 Hz, CHF2) ppm. 13C NMR δC = 164.88 (t, 2JC–F = 28.6 Hz, CCHF2), 148.64 (s, C-2), 137.30 (s, C-1), 125.75 (s, C-4), 120.87 (s, C-5), 120.17 (s, C-3), 114.83 (t, 1JC–F = 242.8 Hz, CHF2), 111.65 (s, C-6), 55.56 (s, OMe), 13.11 (s, Me) ppm. HRMS (ESI+) for C10H12F2NO [M + H]: calcd 200.0881, found 200.0875.
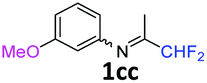
N-(1,1-Difluoropropan-2-ylidene)-3-methoxyaniline 1cc.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 1.32 mL, 16.2 mmol) and 3-methoxyaniline (1 equiv., 0.91 mL, 8.12 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 4 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-3-methoxyaniline 1cc was provided as a brown oil (1.58 g, 98%). 1H NMR (δH = 7.30–7.26) (1 H, m, C(5)H), 6.75–6.73 (1 H, m, C(4)H), 6.39–6.38 (2 H, m, C(2,6)H), 6.10 (1 H, t, 2JH–F = 55.6 Hz, CHF2), 3.83–3.80 (3 H, m, OMe), 1.99–1.97 (3 H, m, Me) ppm. 19F NMR δF = −121.26 to −121.44 (2 F, m, 2JF–H = 55.6 Hz, CHF2) ppm. 13C NMR δC = 163.41 (t, 2JC–F = 28.7 Hz, CCHF2), 160.41 (s, C-3), 149.81 (s, C-1), 129.95 (s, C-5), 114.89 (t, 1JC–F = 242.9 Hz, CHF2), 110.92 (s, C-6), 110.25 (s, C-4), 104.65 (s, C-2), 55.01 (s, OMe), 12.35 (s, Me) ppm. HRMS (ESI+) for C10H12F2NO [M + H]: calcd 200.0881, found 200.0884.
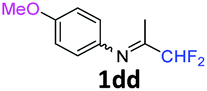
N-(1,1-Difluoropropan-2-ylidene)-4-methoxyaniline 1dd.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 1.44 mL, 17.8 mmol) and 4-methoxyaniline (1 equiv., 8.91 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 5 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-4-methoxyaniline 1dd was provided as a brown oil (1.74 g, 98%, estimated). 1H NMR δH = 6.87 (2 H, m, AA′, C(2,6)H), 6.73 (2 H, m, BB′, C(3,5)H), 6.02 (1 H, t, 2JH–F = 55.7 Hz, CHF2), 3.77–3.69 (3 H, m, OMe), 1.94–1.93 (3 H, m, Me) ppm. 19F NMR δF = −121.11 (2 F, dd, 2JF–H = 55.8, 4JF–H = 4.0 Hz, CHF2) ppm. 13C NMR δC = 162.77 (t, 2JC–F = 29.4 Hz, CCHF2), 157.11 (s, C-4), 141.22 (s, C-1), 120.80 (2 C, s, C-3,5), 115.23 (t, 1JC–F = 242.7 Hz, CHF2), 114.21 (2 C, s, C-2,6), 55.11 (s, OMe), 12.26 (s, Me) ppm. HRMS (ESI+) for C10H12F2NO [M + H]: calcd 200.0881, found 200.0893.
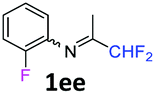
N-(1,1-Difluoropropan-2-ylidene)-2-fluoroaniline 1ee.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 2.92 mL, 36 mmol) and 2-fluoroaniline (1 equiv., 1.74 mL, 18 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 4 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-2-fluoroaniline 1ee was provided as a brown oil (2.62 g, 78%, estimated). 1H NMR δH = 7.19–7.08 (3 H, m, C(4,5,6)H), 6.90–6.82 (1 H, m, C(3)H), 6.10 (1 H, t, 2JH–F = 55.5 Hz, CHF2), 1.99–1.91 (3 H, m, Me) ppm. 19F NMR δF = −121.24 (2 F, d, 2JF–H = 55.5 Hz, CHF2), −126.63 (1 F, s, F) ppm. 13C NMR δC = 166.37 (t, 2JC–F = 28.9 Hz, CCHF2), 151.51 (d, 1JC–F = 245.8 Hz, C-2), 135.82 (d, 2JC–F = 13.0 Hz, C-1), 126.07 (d, 3JC–F = 7.3 Hz, C-4), 124.48 (d, 4JC–F = 3.8 Hz, C-5), 121.72 (s, C-6), 116.11 (d, 2JC–F = 19.8 Hz, C-3), 114.49 (t, 1JC–F = 243.0 Hz, CHF2), 13.14 (s, Me) ppm. HRMS (ESI+) for C9H9F3N [M + H]: calcd 188.0682, found 188.0687.
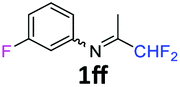
N-(1,1-Difluoropropan-2-ylidene)-3-fluoroaniline 1ff.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 1.46 mL, 18 mmol) and 3-fluoroaniline (1 equiv., 0.87 mL, 9 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 5 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-3-fluoroaniline 1ff was provided as a brown oil (1.53 g, 91%). 1H NMR δH = 7.33–7.28 (1 H, m, C(5)H), 6.84 (1 H, td, 3JH–H = 8.5, 4JH–H = 2.5 Hz, C(4)H), 6.55–6.49 (2 H, m, C(2,6)H), 6.03 (1 H, t, 2JH–F = 55.5 Hz, CHF2), 1.94 (3 H, s, Me) ppm. 19F NMR δF = −111.96 to −112.07 (1 F, m, F), −121.51 (2 F, d, 2JF–H = 55.4 Hz, CHF2) ppm. 13C NMR δC = 164.38 (t, 2JC–F = 28.9 Hz, CCHF2), 163.35 (d, 1JC–F = 247.45 Hz, C-3), 150.28 (d, 3JC–F = 9.2 Hz, C-1), 130.63 (d, 3JC–F = 9.2 Hz, C-5), 114.57 (t, 1JC–F = 242.4 Hz, CHF2), 114.48 (d, 4JC–F = 2.9 Hz, C-6), 111.38 (d, 2JC–F = 21.3 Hz, C-4), 106.36 (d, 2JC–F = 23.7 Hz, C-2), 12.65 (s, Me) ppm. HRMS (ESI+) for C9H9F3N [M + H]: calcd 188.0682, found 188.0672.
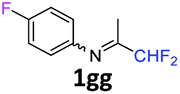
N-(1,1-Difluoropropan-2-ylidene)-4-fluoroaniline 1gg.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 1.35 mL, 16.7 mmol) and 4-fluoroaniline (1 equiv., 0.8 mL, 8.33 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 3 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-4-fluoroaniline 1gg was provided as a brown oil (1.29 g, 83%). 1H NMR δH = 7.04 (2 H, t, 3JH–H = 8.7 Hz, C(2,6)H), 6.75–6.72 (2 H, m, C(3,5)H), 6.02 (1 H, t, 2JH–F = 55.6 Hz, CHF2), 1.94 (3 H, s, Me) ppm. 19F NMR δF = −118.69 to −119.06 (1 F, m, F), −121.38 (2 F, dd, 2JF–H = 55.6, 4JF–H = 2.6 Hz, CHF2) ppm. 13C NMR δC = 164.10 (t, 2JC–F = 28.2 Hz, CCHF2), 160.27 (d, 1JC–F = 243.3 Hz, C-4), 144.44 (d, 4JC–F = 1.2, C-1), 120.60 (2 C, d, 3JC–F = 8.1 Hz, C-2,6), 115.86 (2 C, d, 2JC–F = 22.6 Hz, C-3,5), 114.82 (t, 1JC–F = 243.0 Hz, CHF2), 12.57 (s, Me) ppm. HRMS (ESI−) for C9H7F3N [M − H]: calcd 186.0525, found 186.0544.
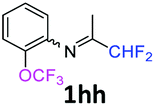
N-(1,1-Difluoropropan-2-ylidene)-2-(trifluoromethoxy)aniline 1hh.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 1.83 mL, 22.6 mmol) and 2-(trifluoromethoxy)aniline (1 equiv., 1.54 mL, 11.3 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 5 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-2-(trifluoromethoxy)aniline 1hh was provided as a brown oil (2.13 g, 75%). 1H NMR δH = 7.27–7.18 (2 H, m, C(4,6)H), 7.12–7.06 (1 H, m, C(5)H), 6.78–6.73 (1 H, m, C(3)H), 6.03 (1 H, t, 2JH–F = 55.5 Hz, CHF2), 1.85 (3 H, s, Me) ppm. 19F NMR δF = −58.46 (3 F, s, OCF3), −121.95 (2 F, d, 2JF–H = 55.4 Hz, CHF2) ppm. 13C NMR δC = 166.44 (t, 2JC–F = 29.1 Hz, CCHF2), 141.52 (s, C-2), 138.31 (s, C-1), 127.75 (s, C-4), 125.82 (s, C-5), 122.38 (s, C-6), 122.14 (q, 1JC–F = 234.3 Hz, OCF3), 120.96 (s, C-3), 114.62 (t, 1JC–F = 242.9 Hz, CHF2), 12.96 (s, Me) ppm. HRMS (ESI positive) for C10H9F5NO [M + H]: calcd 254.0599, found 254.0622.
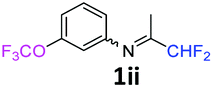
N-(1,1-Difluoropropan-2-ylidene)-3-(trifluoromethoxy)aniline 1ii.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 0.92 mL, 11.3 mmol) and 3-(trifluoromethoxy)aniline (1 equiv., 0.76 mL, 5.65 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 7 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-3-(trifluoromethoxy)aniline 1ii was provided as a brown oil (1.32 g, 93%). 1H NMR δH = 7.37 (1 H, t, 3JH–H = 8.1 Hz, C(5)H), 7.04–6.99 (1 H, m, C(4)H), 6.69 (1 H, d, 3JH–H = 7.9 Hz, C(6)H), 6.66 (1 H, s, C(2)H), 6.03 (1 H, t, 2JH–F = 55.5 Hz, CHF2), 1.94 (3 H, s, Me) ppm. 19F NMR δF = −58.10 (3 F, s, OCF3), −121.65 (2 F, d, 2JF–H = 55.6 Hz, CHF2). 13C NMR δC = 164.80 (t, 2JC–F = 29.0 Hz, CCHF2), 150.07–150.02 (2 C, m, C-1,3), 130.57 (s, C-5), 121.72 (q, 1JC–F = 258.6 Hz, OCF3), 117.37 (s, C-4), 117.00 (s, C-6), 114.68 (t, 1JC–F = 243.2 Hz, CHF2), 111.92 (s, C-2), 12.74 (s, Me) ppm. HRMS (ESI+) for C10H9F5NO [M + H]: calcd 254.0599, found 254.0583.
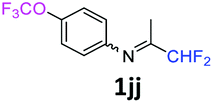
N-(1,1-Difluoropropan-2-ylidene)-4-(trifluoromethoxy)aniline 1jj.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 1.83 mL, 22.6 mmol) and 4-(trifluoromethoxy)aniline (1 equiv., 1.53 mL, 11.3 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 3 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-4-(trifluoromethoxy)aniline 1jj was provided as a brown oil (2.59 g, 91%). 1H NMR δH = 7.22 (2 H, d, 3JH–H = 8.6 Hz, C(2,6)H), 6.78 (2 H, d, 3JH–H = 8.8 Hz, C(3,5)H), 6.03 (1 H, t, 2JH–F = 55.6 Hz, CHF2), 1.94 (3 H, s, Me) ppm. 19F NMR δF = −58.44 (3 F, s, OCF3), −121.67 (2 F, dd, 2JF–H = 55.6, 4JF–H = 2 Hz, CHF2) ppm. 13C NMR δC = 164.54 (t, 2JC–F = 28.9 Hz, CCHF2), 147.21 (s, C-1), 146.39 (q, 3JC–F = 2 Hz, C-4), 122.10 (2 C, s, C-2,6), 120.76 (q, 1JC–F = 256.7 Hz, OCF3), 120.34 (2 C, s, C-3,5), 114.84 (t, 1JC–F = 244.4 Hz, CHF2), 12.38 (s, Me) ppm. HRMS (ESI+) for C10H9F5NO [M + H]: calcd 254.0599, found 254.0622.
3-N-(1,1-Difluoropropan-2-ylidene)-1-N,N-dimethylbenzene-1,3-diamine 1kk.
Under an argon atmosphere, an excess of cold 1,1-difluoroacetone (2 equiv., 0.83 mL, 10.2 mmol) was added to 1-N,N-dimethyl-1,3-phenylenediamine dihydrochloride (1 equiv., 695 mg, 5.1 mmol) and sodium hydride (2.1 equiv., 257.2 mg, 10.72 mmol) in anhydrous DCM (15 mL) in the presence of 4 Å MS. Then the reaction mixture was stirred for 24 h at room temperature. 4 Å MS were then filtered off on Celite and washed with ether. The filtrate was concentrated under reduced pressure to give 3-N-(1,1-difluoropropan-2-ylidene)-1-N,N-dimethylbenzene-1,3-diamine 1kk as a black paste (1.26 g). 1H NMR δH = 7.21 (1 H, t, 3JH–H = 8.2 Hz, C(5)H), 6.56–6.51 (1 H, m, C(4)H), 6.13–6.08 (2 H, m, C(2,6)H), 6.06 (1 H, t, 2JH–F = 55.5 Hz, CHF2), 2.95 (6 H, s, NMe2), 1.96 (3 H, s, Me) ppm. 19F NMR δF = −121.31 (2 F, d, 2JF–H = 55.5 Hz, CHF2) ppm. HRMS (ESI+) for C11H15F2N2 [M + H]: calcd 213.1198, found 213.1209.
4-N-(1,1-Difluoropropan-2-ylidene)-1-N,N-dimethylbenzene-1,4-diamine 1ll.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 1.19 mL, 14.7 mmol) and N,N-dimethyl-1,4-benzenediamine (1 equiv., 0.92 mL, 7.35 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 20 h at room temperature. 4-N-(1,1-Difluoropropan-2-ylidene)-1-N,N-dimethylbenzene-1,4-diamine 1ll was provided as a yellow solid (1.46 g, 94%). 1H NMR δH = 6.77 (4 H, A2B2, 3JH–H = 8.9 Hz, Δν = 27.76 Hz, C(2,6/3,5)H), 6.05 (1 H, t, 2JH–F = 55.9 Hz, CHF2), 2.96 (6 H, s, NMe2), 2.03 (3 H, s, Me) ppm. 19F NMR δF = −120.62 (2 F, d, 2JF–H = 55.9 Hz, CHF2) ppm. 13C NMR δC = 161.26 (t, 2JC–F = 28.4 Hz, CCHF2), 148.61 (s, C-2), 137.60 (s, C-1), 121.72 (2 C, s, C-2,6), 115.70 (t, 1JC–F = 242.6 Hz, CHF2), 112.84 (2 C, s, C-3,5), 40.83 (2 C, s, NMe2), 12.74 (s, Me) ppm. HRMS (ESI+) for C11H15F2N2 [M + H]: calcd 213.1198, found 213.1214. C11H14F2N2 (212): calcd (%) N 13.19, C 62.19, H 6.60, found N 13.22, C 61.99, H 6.68. MP: 80.5–82 °C.
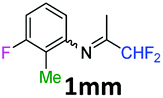
N-(1,1-Difluoropropan-2-ylidene)-3-fluoro-2-methylaniline 1mm.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 2.59 mL, 32 mmol) and 3-fluoro-2-methylaniline (1 equiv., 1.82 mL, 16 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 18 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-3-fluoro-2-methylaniline 1mm was provided as a brown oil (2.88 g, 90%, estimated). 1H NMR δH = 7.16–7.13 (1 H, m, C(4)H), 6.83 (1 H, t, 3JH–H = 8.8 Hz, C(5)H), 6.41 (1 H, d, 3JH–H = 7.9 Hz, C(6)H), 6.10 (1 H, t, 2JH–F = 55.5 Hz, CHF2), 1.99 (3 H, d, 4JH–F = 2.0 Hz, C(2Me)H), 1.89 (3 H, s, Me) ppm. 19F NMR δF = −116.18 to −116.24 (1 F, m, F), −121.21 (2 F, dd, 2JF–H = 55.5, 4JF–H = 2.4 Hz, CHF2) ppm. 13C NMR δC = 164.50 (t, 2JC–F = 29.0 Hz, CCHF2), 161.79 (d, 1JC–F = 244.3 Hz, C-3), 148.91 (d, 3JC–F = 7.8 Hz, C-1), 126.98 (d, 2JC–F = 9.8 Hz, C-4), 114.68 (s, C-2), 114.55 (t, 1JC–F = 243.0 Hz, CHF2), 113.50 (d, 4JC–F = 3.1 Hz, C-6), 111.17 (d, 3JC–F = 23.0 Hz, C-5), 12.83 (s, Me), 9.02 (d, 3JC–F = 4.8 Hz, C(2)Me) ppm. HRMS (ESI+) for C10H11F3N [M + H]: calcd 202.0838, found 202.0866.
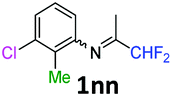
3-Chloro-N-(1,1-difluoropropan-2-ylidene)-2-methylaniline 1nn.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 2.29 mL, 28.2 mmol) and 3-chloro-2-methylaniline (1 equiv., 1.71 mL, 14.1 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 18 h at room temperature. 3-Chloro-N-(1,1-difluoropropan-2-ylidene)-2-methylaniline 1nn was provided as a brown oil (2.79 g, 91%, estimated). 1H NMR δH = 7.17 (1 H, dd, 3JH–H = 8.0, 4JH–H = 1.1 Hz, C(4)H), 7.10 (1 H, t, 3JH–H = 7.7 Hz, C(5)H), 6.52 (1 H, dd, 3JH–H = 7.7, 4JH–H = 0.9 Hz, C(6)H), 6.11 (1 H, t, 2JH–F = 55.5 Hz, CHF2), 2.12 (3 H, s, C(2Me)H), 1.88 (3 H, s, Me) ppm. 19F NMR δF = −121.15 (2 F, d, 2JF–H = 55.5 Hz, CHF2) ppm. 13C NMR δC = 164.43 (t, 2JC–F = 28.9 Hz, CCHF2), 148.44 (s, C-1), 135.57 (s, C-3), 127.06 (s, C-5), 125.48 (s, C-4), 116.39 (s, C-6), 114.63 (t, 1JC–F = 243.1 Hz, CHF2), 113.56 (s, C-2), 14.26 (s, C(2)Me), 12.89 (s, Me) ppm. HRMS (ESI+) for C10H11F2NCl [M + H]: calcd 218.0543, found 218.0557.
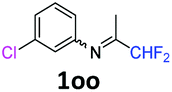
3-Chloro-N-(1,1-difluoropropan-2-ylidene)aniline 1oo.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 5.08 mL, 62.7 mmol) and 3-chloroaniline (1 equiv., 3.33 mL, 31.4 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 18 h at room temperature. 3-Chloro-N-(1,1-difluoropropan-2-ylidene)-aniline 1oo was provided as a brown oil (9.68 g). 1H NMR δH = 7.31 (1 H, t, 3JH–H = 8.0 Hz, C(5)H), 7.16 (1 H, d, 3JH–H = 8.1 Hz, C(4)H), 6.81 (1 H, t, 4JH–H = 1.9 Hz, C(2)H), 6.68 (1 H, d, 3JH–H = 7.9 Hz, C(6)H), 6.06 (1 H, t, 2JH–F = 55.4 Hz, CHF2), 1.97 (3 H, s, Me) ppm. 19F NMR δF = −121.46 (2 F, d, 2JF–H = 55.4 Hz, CHF2) ppm. 13C NMR δC = 164.76 (t, 2JC–F = 29.0 Hz, CCHF2), 149.64 (s, C-1), 134.99 (s, C-3), 130.44 (s, C-5), 124.96 (s, C-4), 119.18 (s, C-2), 117.24 (s, C-6), 114.55 (t, 1JC–F = 243.2 Hz, CHF2), 12.87 (s, Me) ppm. HRMS (ESI+) for C9H9F2NCl [M + H]: calcd 204.0386, found 204.0367.
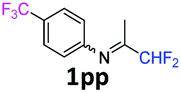
N-(1,1-Difluoropropan-2-ylidene)-4-(trifluoromethyl)aniline 1pp.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 0.503 mL, 6.21 mmol) and 4-(trifluoromethyl)aniline (1 equiv., 0.386 mL, 3.1 mmol) in the presence of anhydrous MgSO4. The reaction mixture was stirred for 16 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-4-(trifluoromethyl)aniline 1pp was provided as a brown oil (212 mg, 29%, estimated). 1H δH = 7.63 (2 H, d, 3JH–H = 8.3 Hz, C(3,5)H), 6.85 (2 H, d, 3JH–H = 8.2 Hz, C(2,6)H), 6.05 (1 H, t, 2JH–F = 55.4 Hz, CHF2), 1.93 (3 H, s, Me) ppm. 19F NMR δF = −62.13 (3 F, s, CF3), −121.34 to −121.59 (2 F, m, CHF2) ppm. 13C NMR δC = 164.59 (t, 2JC–F = 29.0 Hz, CCHF2), 151.64 (s, C-1), 126.64 (2 C, q, 3JC–F = 3.8 Hz, C-3,5), 124.30 (q, 1JC–F = 271.6 Hz, CF3), 120.30 (q, 2JC–F = 32.6 Hz, C-4), 119.03 (2 C, s, C-2,6), 114.49 (t, 1JC–F = 243.3 Hz, CHF2), 12.97 (s, Me) ppm. HRMS (ESI+) for C10H9F5N [M + H]: calcd 238.0650, found 238.0658.
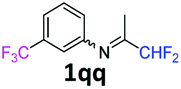
N-(1,1-Difluoropropan-2-ylidene)-3-(trifluoromethyl)aniline 1qq.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 1.01 mL, 12.4 mmol) and 3-(trifluoromethyl)aniline (1 equiv., 0.767 mL, 6.21 mmol) in the presence of 4 Å MS. The reaction mixture was stirred for 5 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-3-(trifluoromethyl)aniline 1qq was provided as a brown oil (1.47 g, 93%, estimated). 1H NMR δH = 7.49 (1 H, t, 3JH–H = 7.8 Hz, C(5)H), 7.42 (1 H, d, 3JH–H = 7.8 Hz, C(4)H), 7.03 (1 H, s, C(2)H), 6.94 (1 H, d, 3JH–H = 7.2 Hz, C(4)H), 6.05 (1 H, t, 2JH–F = 55.5 Hz, CHF2), 1.95 (3 H, s, Me) ppm. 19F NMR δF = −62.80 (3 F, s, CF3), −121.44 (2 F, d, 2JF–H = 55.4 Hz, CHF2) ppm. 13C NMR δC = 165.21 (t, 2JC–F = 28.9 Hz, CCHF2), 148.91 (s, C-1), 131.84 (q, 2JC–F = 32.5 Hz, C-3), 129.96 (s, C-5), 124.03 (q, 1JC–F = 272.3 Hz, CF3), 122.38 (s, C-6), 121.68 (q, 3JC–F = 3.8 Hz, C-4), 116.01 (q, 3JC–F = 4.0 Hz, C-2), 114.53 (t, 1JC–F = 243.2 Hz, CHF2), 12.78 (s, Me) ppm. HRMS (ESI+) for C10H9F5N [M + H]: calcd 238.0650, found 238.0660.
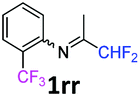
N-(1,1-Difluoropropan-2-ylidene)-2-(trifluoromethyl)aniline 1rr.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 0.503 mL, 6.21 mmol) and 2-(trifluoromethyl)aniline (1 equiv., 0.386 mL, 3.1 mmol) in the presence of anhydrous MgSO4. The reaction mixture was stirred for 16 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-2-(trifluoromethyl)aniline 1rr was provided as a brown oil (291.5 mg, 40%, estimated). 1H NMR δH = 7.67 (1 H, d, 3JH–H = 7.9 Hz, C(3)H), 7.52 (1 H, t, 3JH–H = 7.7 Hz, C(5)H), 7.23 (1 H, t, 3JH–H = 7.7 Hz, C(5)H), 6.72 (1 H, d, 3JH–H = 7.9 Hz, C(6)H), 6.08 (1 H, t, 2JH–F = 55.4 Hz, CHF2), 1.90 (3 H, s, Me) ppm. 19F NMR δF = −61.85 (3 F, s, CF3), −121.82 (2 F, d, 2JF–H = 55.4 Hz, CHF2) ppm. 13C NMR δC = 165.58 (t, 2JC–F = 29.4 Hz, CCHF2), 146.98 (q, 3JC–F = 2.02 Hz, C-1), 132.87 (s, C-5), 126.77 (q, 3JC–F = 5.05 Hz, C-3), 124.50 (s, C-4), 123.67 (q, 1JC–F = 273.0 Hz, CF3), 119.65 (q, 2JC–F = 31.3 Hz, C-2), 119.18 (s, C-6), 114.27 (t, 1JC–F = 243.3 Hz, CHF2), 13.42 (s, Me) ppm. HRMS (ESI+) for C10H9F5N [M + H]: calcd 238.0650, found 238.0654.
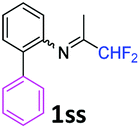
N-(1,1-Difluoropropan-2-ylidene)-2-phenylaniline 1ss.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 0.485 mL, 5.98 mmol) and 2-aminodiphenyl (1 equiv., 505 mg, 2.99 mmol) in the presence of anhydrous MgSO4. The reaction mixture was stirred for 14 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-2-phenylaniline 1ss was provided as a brown oil (953.7 mg). 1H NMR δH = 7.42–7.23 (8 H, m, C(3,4,5,2′,3′,4′,5′,6′)H), 6.71 (1 H, dd, 3JH–H = 7.8, 4JH–H = 1.0 Hz, C(6)H), 5.88 (1 H, t, 2JH–F = 55.6 Hz, CHF2), 1.64 (3 H, s, Me) ppm. 19F NMR δF = −121.61 (2 F, d, 2JF–H = 55.5 Hz, CHF2) ppm. 13C NMR δC = 163.53 (t, 2JC–F = 28.8 Hz, CCHF2), 146.07 (s, C-1), 139.00 (s, C-1′), 132.02 (s, C-2), 131.44–123.83 (8 C, m, C-3,4,5,2′,3′,4′,5′,6′), 119.16 (s, C-6), 114.67 (t, 1JC–F = 243.2 Hz, CHF2), 13.40 (s, Me) ppm. HRMS (ESI positive) for C15H14F2N [M + H]: calcd 246.1089, found 246.1112.
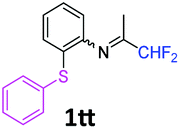
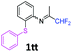
N-(1,1-Difluoropropan-2-ylidene)-2-(phenylsulfanyl)aniline 1tt.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 0.422 mL, 5.2 mmol) and 2-(phenylsulfanyl)aniline (1 equiv., 523 mg, 2.6 mmol) in the presence of anhydrous MgSO4. The reaction mixture was stirred for 12 h at room temperature. N-(1,1-Difluoropropan-2-ylidene)-2-(phenylsulfanyl)aniline 1tt was provided as a brown oil (796.1 mg). 1H NMR δH = 7.34–7.20 (6 H, m, C(3,2′,3′,4′,5′,6′)H), 7.09 (2 H, td, 3JH–H = 7.65, 4JH–H = 1.3 Hz, C(4,5)H), 6.69 (1 H, dd, 3JH–H = 7.8, 4JH–H = 1.1 Hz, C(6)H), 6.01 (1 H, t, 2JH–F = 55.5 Hz, CHF2), 1.78 (3 H, s, Me) ppm. 19F NMR δF = −121.06 (2 F, d, 2JF–H = 55.4 Hz, CHF2) ppm. 13C NMR δC = 164.78 (t, 2JC–F = 28.9 Hz, CCHF2), 148.85 (s, C-1), 136.88 (s, C-1′), 134.64 (s, C-2), 132.37–127.42 (6 C, m, C-3,2′,3′,4′,5′,6′), 126.57 (s, C-5), 125.43 (s, C-4), 118.87 (s, C-6), 114.45 (t, 1JC–F = 243.3 Hz, CHF2), 13.04 (s, Me) ppm. HRMS (ESI+) for C15H14F2NS [M + H]: calcd 278.0810, found 278.0781.
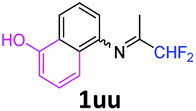
5-[(1,1-Difluoropropan-2-ylidene)amino]-1-naphthol 1uu.
The product was prepared according to the general procedure, starting from 1,1-difluoroacetone (2 equiv., 0.509 mL, 6.28 mmol) and 5-amino-1-naphthol (1 equiv., 500 mg, 3.14 mmol) in the presence of anhydrous MgSO4. The reaction mixture was stirred for 14 h at room temperature. 5-[(1,1-Difluoropropan-2-ylidene)amino]-1-naphthol 1uu was provided as a purple solid (670 mg, 91%, estimated). 1H NMR δH = 8.01 (1 H, d, 3JH–H = 8.5 Hz, C(8)H), 7.45 (1 H, t, 3JH–H = 8.0 Hz, C(7)H), 7.35–7.17 (2 H, m, C(3,4)H), 6.84 (1 H, d, 3JH–H = 7.3 Hz, C(2)H), 6.78 (1 H, d, 3JH–H = 7.2 Hz, C(6)H), 6.24 (1 H, t, 2JH–F = 55.6 Hz, CHF2), 5.45 (1 H, br s, OH), 1.91 (3 H, s, Me) ppm. 19F NMR δF = −120.84 (2 F, d, 2JF–H = 55.5 Hz, CHF2) ppm. 13C NMR δC = 165.18 (t, 2JC–F = 28.9 Hz, CCHF2), 151.81 (s, C-1), 144.46 (s, C-5), 126.46 (s, C-1-C-C-8), 126.24 (s, C-3), 125.23 (s, C-4-C-C-5), 125.00 (s, C-7), 118.80 (s, C-8), 115.48 (s, C-4), 114.83 (t, 1JC–F = 243.3 Hz, CHF2), 114.07 (s, C-6), 109.46 (s, C-2), 13.18 (s, Me) ppm. HRMS (ESI+) for C13H12F2NO [M + H]: calcd 236.0881, found 236.0909.
General procedure for the synthesis of quinoline derivatives 2 and 3
Under an argon atmosphere, a solution of the desired FAR (1,1,2,2-tetrafluoro-N,N-dimethylethan-1-amine (4a; TFEDMA), 2-chloro-N,N-diethyl-1,1,2-trifluoroethan-1-amine (4b; Yarovenko reagent), and N,N-diethyl-1,1,2,3,3,3-hexafluoropropan-1-amine (4c; Ishikawa reagent)) (1.2 equiv.) was activated by adding boron trifluoride diethyl etherate (BF3·Et2O) (1.2 equiv.) in dry acetonitrile (3.6 mmol per 5 mL) and stirred for 15 min. Then a solution of the desired ketimine derivative 1 (1 equiv.) in dry acetonitrile (3 mmol per 5 mL) was slowly added to this mixture via syringe. After 15 min at room temperature, the mixture was heated at 50 °C for 19 h. Acetonitrile was removed under reduced pressure and the reaction mixture was purified by flash chromatography using a gradient of ethyl acetate in pentane to provide the final compounds 2 or 3.
4-(Difluoromethyl)-2-(trifluoromethyl)quinoline 2ai.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.42 mL, 3.61 mmol) by (BF3·Et2O) (1.2 equiv., 0.46 mL, 3.61 mmol) and N-(1,1,1-trifluoropropan-2-ylidene)aniline 1 (1 equiv., 562 mg, 3.01 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-2-(trifluoromethyl)quinoline 2ai was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (458 mg, 62%). 1H NMR δH = 8.33 (1 H, d, 3JH–H = 8.5 Hz, C(8)H), 8.16 (1 H, d, 3JH–H = 8.5 Hz, C(5)H), 7.93 (1 H, s, C(3)H), 7.92–7.87 (1 H, m, C(7)H), 7.80 (1 H, t, 3JH–H = 7.7 Hz, C(6)H), 7.22 (1 H, t, 2JH–F = 54.2 Hz, C(4-CHF2)H) ppm. 19F NMR δF = −67.66 (3 F, s, CF3), −115.53 (2 F, d, 2JF–H = 54.1 Hz, CHF2) ppm. 13C NMR δC = 147.94 (q, 2JC–F = 35.3 Hz, C-2), 147.87 (t, C-8-C-N), 140.33 (t, 2JC–F = 22.3 Hz, C-4), 131.39 (s, C-7), 131.29 (s, C-8), 130.06 (s, C-6), 125.09 (s, C-5-C-C-4), 123.44 (s, C-5), 121.33 (q, 1JC–F = 275.73 Hz, C(2)CF3), 114.14 (td, 3JC–F = 7.9, 3JC–F = 2.1 Hz, C-3), 112.74 (t, 1JC–F = 241.5 Hz, C(4)CHF2) ppm. HRMS (ESI+) for C11H7F5N [M + H]: calcd 248.0493, found 248.0520. C11H6F5N (247): calcd (%) N 5.66, C 53.40, H 2.43, found N 5.73, C 53.83, H 2.58. MP: 64–65.1 °C.
4-[Chloro(fluoro)methyl]-2-(trifluoromethyl)quinoline 2aii.
The product was prepared according to the general procedure, starting from an activated solution of Yarovenko's reagent 4b (1.2 equiv., 0.73 mL, 3.21 mmol) by (BF3·Et2O) (1.2 equiv., 0.41 mL, 3.21 mmol) and N-(1,1,1-trifluoropropan-2-ylidene)aniline 1a (1 equiv., 500 mg, 2.67 mmol) in the presence of anhydrous acetonitrile. 4-[Chloro(fluoro)methyl]-2-(trifluoromethyl)quinoline 2aii was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a light yellow solid (402 mg, 57%). 1H NMR δH = 8.32 (1 H, d, 3JH–H = 8.4 Hz, C(8)H), 8.13 (1 H, d, 3JH–H = 8.5 Hz, C(5)H), 7.95 (1 H, s, C(3)H), 7.92–7.88 (1 H, m, C(7)H), 7.81–7.77 (1 H, m, C(3)H), 7.66 (1 H, d, 2JH–F = 48.9 Hz, C(4-CHFCl)H) ppm. 19F NMR δF = −67.65 (3 F, s, CF3), −138.75 (1 F, d, 2JF–H = 48.9 Hz, C4-CHFCl) ppm. 13C NMR δC = 148.02 (q, 2JC–F = 35.4 Hz, C-2), 147.88 (s, C-8-C-N), 143.82 (d, 2JC–F = 21.1 Hz, C-4), 131.40 (s, C-7), 131.31 (s, C-8), 129.82 (s, C-6), 124.03 (d, 2JC–F = 3.6 Hz, C-5-C-C-4), 123.08 (s, C-5), 121.34 (q, 1JC–F = 275.4 Hz, C(2)CF3), 113.04 (dq, 3JC–F = 9.9, 3JC–F = 2.0 Hz, C-3), 96.75 (d, 1JC–F = 244.6 Hz, C(4)CHFCl) ppm. C11H6F4NCl (263): calcd (%) N 5.31, C 50.07, H 2.27, found N 5.11, C 50.19, H 2.65. HRMS (ESI+) for C11H7F4NCl [M + H]: calcd 264.0198, found 264.0231. MP: 49.4–50.4 °C.
4-(1,2,2,2-Tetrafluoroethyl)-2-(trifluoromethyl)quinoline 2aiii.
The product was prepared according to the general procedure, starting from an activated solution of Ishikawa's reagent 4c (1.2 equiv., 0.95 mL, 3.26 mmol) by (BF3·Et2O) (1.2 equiv., 0.41 mL, 3.26 mmol) and N-(1,1,1-trifluoropropan-2-ylidene)aniline 1a (1 equiv., 508 mg, 2.71 mmol) in the presence of anhydrous acetonitrile. 4-(1,2,2,2-Tetrafluoroethyl)-2-(trifluoromethyl)quinoline 2aiii was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a light brown solid (112 mg, 14%, estimated). 1H NMR δH = 8.32 (1 H, d, 3JH–H = 8.4 Hz, C(8)H), 7.99–7.97 (2 H, m, C(3,5)H), 7.89 (1 H, t, 3JH–H = 8.4 Hz, C(7)H), 7.78 (1 H, t, 3JH–H = 8.3 Hz, C(6)H), 6.46 (dq, 2JH–F = 43.9, 3JH–F = 5.5 Hz, C(4-CHFCF3)H) ppm. 19F NMR δF = −67.72 (3 F, s, C2-CF3), −77.25 (3 F, dd, 3JF–F = 13.1, 3JF–H = 5.8 Hz, C4-CHFCF3), −198.87 (1 F, dq, 2JF–H = 44.3, 3JF–F = 12.8 Hz, C4-CHFCF3) ppm. 13C NMR δC = 147.72 (q, 2JC–F = 35.4 Hz, C-2), 147.51 (s, C-8-C-N), 137.94 (d, 2JC–F = 19.1 Hz, C-4), 131.34 (s, C-7), 131.16 (s, C-8), 130.06 (s, C-6), 125.89 (d, C-5-C-C-4, 2JC–F = 3.7 Hz), 122.72 (s, C-5), 121.91 (q, 1JC–F = 282.6 Hz, C(4)CHCF3), 121.68 (q, 1JC–F = 282.6 Hz, C(2)CF3), 115.77 (d, 3JC–F = 10.4 Hz, C-3), 85.41 (dq, 1JC–F = 189.9, 2JC–F = 36.1 Hz, C(4)CHFCF3) ppm. C12H6F7N (297): calcd (%) N 4.71, C 48.45, H 2.02, found N 4.71, C 48.80, H 2.32. HRMS (ESI+) for C11H7F7N [M + H]: calcd 298.0461, found 298.0462. MP: 58–58.8 °C.
4-(Difluoromethyl)-8-methoxy-2-(trifluoromethyl)quinoline 2b.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.33 mL, 2.82 mmol) by (BF3·Et2O) (1.2 equiv., 0.357 mL, 2.82 mmol) and 2-methoxy-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1b (1 equiv., 510 mg, 2.35 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-8-methoxy-2-(trifluoromethyl)quinoline 2b was provided after purification using a gradient of ethyl acetate in pentane (10–25%) as a yellow solid (575 mg, 88%). 1H NMR δH = 7.97 (1 H, s, C(5)H), 7.74–7.63 (2 H, m, C(6,7)H), 7.21 (1 H, d, 4JH–F = 7.8 Hz, C(3)H), 7.20 (1 H, t, 2JH–F = 54.3 Hz, C(4-CHF2)H), 4.12 (3 H, s, Me) ppm. 19F NMR δF = −67.16 (3 F, s, CF3), −116.17 (2 F, d, 2JF–H = 54.4 Hz, CHF2) ppm. 13C NMR δC = 156.58 (s, C-8), 146.50 (q, 2JC–F = 35.7 Hz, C-2), 140.11 (t, 2JC–F = 22.2 Hz, C-4), 139.95 (s, C-8-C-N), 130.74 (s, C-6), 126.43 (s, C-5-C-C-4), 121.40 (q, 1JC–F = 275.2 Hz, C(2)CF3), 114.70–114.16 (2 C, m, C-5, 7), 112.55 (t, 1JC–F = 241.5 Hz, C(4)CHF2), 109.56 (s, C-3), 56.58 (s, Me) ppm. C12H8F5NO (277): calcd (%) N 5.05, C 51.95, H 2.88, found N 5.16, C 51.92, H 2.74. MP: 72.5–76.8 °C.
4-(Difluoromethyl)-7-methoxy-2-(trifluoromethyl)quinoline 2c.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.324 mL, 2.77 mmol) by (BF3·Et2O) (1.2 equiv., 0.35 mL, 2.77 mmol) and 3-methoxy-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1c (1 equiv., 500 mg, 2.3 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-7-methoxy-2-(trifluoromethyl)quinoline 2c was provided after purification using a gradient of ethyl acetate in pentane (10–25%) as a light brown solid (407 mg, 64%). 1H NMR δH = 8.00 (1 H, d, 3JH–H = 9.3 Hz, C(5)H), 7.76 (1 H, s, C(3)H), 7.57 (1 H, d, 4JH–H = 2.6 Hz, C(8)H), 7.39 (1 H, dd, 3JH–H = 9.3, 4JH–H = 2.6 Hz, C(6)H), 7.14 (1 H, t, 2JH–F = 54.3 Hz, C(4-CHF2)H), 3.97 (3 H, s, Me) ppm. 19F NMR δF = −67.74 (3 F, s, CF3), −115.04 (2 F, d, 2JF–H = 54.4 Hz, C4-CHF2) ppm. 13C NMR δC = 161.92 (s, C-7), 150.09 (s, C-8-C-N), 148.04 (q, 2JC–F = 35.2 Hz, C-2), 140.01 (t, 2JC–F = 22.4 Hz, C-4), 124.36 (s, C-5), 123.61 (s, C-6), 121.26 (q, 1JC–F = 275.2 Hz, C(2)CF3), 120.32 (t, 3JC–F = 2.8 Hz, C-5-C-C-4), 112.86 (t, 1JC–F = 241.4 Hz, C(4)CHF2), 111.81 (td, 3JC–F = 8.0, 3JC–F = 2.3 Hz, C-3), 108.56 (s, C-8), 55.91 (s, Me) ppm. C12H8F5NO (277): calcd (%) N 5.05, C 51.95, H 2.88, found N 5.14, C 51.71, H 2.87. MP: 83.4–87.2 °C.
4-(Difluoromethyl)-6-methoxy-2-(trifluoromethyl)quinoline 2d.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.325 mL, 2.78 mmol) by (BF3·Et2O) (1.2 equiv., 0.352 mL, 2.78 mmol) and 4-methoxy-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1d (1 equiv., 502 mg, 2.31 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-6-methoxy-2-(trifluoromethyl)quinoline 2d was provided after purification using a gradient of ethyl acetate in pentane (10–25%) as a brown solid (543 mg, 85%). 1H NMR δH = 8.16 (1 H, d, 3JH–H = 9.3 Hz, C(8)H), 7.85 (1 H, s, C(3)H), 7.50 (1 H, dd, 3JH–H = 9.3, 4JH–H = 2.6 Hz, C(7)H), 7.29 (1 H, s, C(5)H), 7.11 (1 H, t, 2JH–F = 54.3 Hz, C(4-CHF2)H), 3.97 (3 H, s, Me) ppm. 19F NMR δF = −67.30 (3 F, s, CF3), −115.95 (2 F, d, 2JF–H = 54.4 Hz, C4-CHF2) ppm. 13C NMR δC = 160.35 (s, C-6), 144.95 (q, 2JC–F = 35.4 Hz, C-2), 144.11 (s, C-8-C-N), 138.21 (t, 2JC–F = 22.1 Hz, C-4), 132.56 (s, C-8), 126.63 (s, C-5-C-C-4), 124.40 (s, C-7), 121.43 (q, 1JC–F = 274.7 Hz, C(2)CF3), 114.59 (td, 3JC–F = 8.1, 3JC–F = 2.3 Hz, C-3), 113.17 (t, 1JC–F = 241.1 Hz, C(4)CHF2), 101.06 (s, C-3), 55.88 (s, Me) ppm. C12H8F5NO (277): calcd (%) N 5.05, C 51.95, H 2.88, found N 5.03, C 51.64, H 2.80. MP: 105.9–108.2 °C.
4-(Difluoromethyl)-8-fluoro-2-(trifluoromethyl)quinoline 2e.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.347 mL, 2.97 mmol) by (BF3·Et2O) (1.2 equiv., 0.376 mL, 2.97 mmol) and 2-fluoro-N-(1,1,1- trifluoropropan-2-ylidene)aniline 1e (1 equiv., 507 mg, 2.47 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-8-fluoro-2-(trifluoromethyl)quinoline 2e was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as an orange solid (466 mg, 70%). 1H NMR δH = 7.99 (1 H, s, C(3)H), 7.94 (1 H, d, 3JH–H = 8.6 Hz, C(5)H), 7.76 (1 H, td, 3JH–H = 8.2, 4JH–F = 5.0 Hz, C(6)H), 7.63–7.55 (1 H, m, C(7)H), 7.20 (1 H, t, 2JH–F = 54.1 Hz, C(4-CHF2)H) ppm. 19F NMR δF = −67.64 (3 F, s, CF3), −115.74 (2 F, d, 2JF–H = 54.2 Hz, C4-CHF2), −119.97 (1 F, dd, 3JF–H = 10.5, 4JF–H = 5.4 Hz, F) ppm. 13C NMR δC = 158.51 (d, 1JC–F = 261.9 Hz, C-8), 147.96 (qd, 2JC–F = 34.4 Hz, 4JC–F = 1.7 Hz, C-2), 140.33 (td, 2JC–F = 22.6, 4JC–F = 2.9 Hz, C-4), 138.40 (d, 2JC–F = 12.2 Hz, C-8-C-N), 130.35 (d, 3JC–F = 8.1 Hz, C-6), 126.50 (s, C-5-C-C-4), 120.95 (q, 1JC–F = 275.5 Hz, C(2)CF3), 119.25 (d, 4JC–F = 5.3 Hz, C-5), 115.90 (d, 2JC–F = 18.6 Hz, C-7), 115.20 (t, 3JC–F = 7.5 Hz, C-3), 112.48 (t, 1JC–F = 241.9 Hz, C(4)CHF2) ppm. C11H5F6N (265): calcd (%) N 5.28, C 49.78, H 1.88, found N 5.44, C 50.00, H 1.95. MP: 49.7–51.3 °C.
4-(Difluoromethyl)-7-fluoro-2-(trifluoromethyl)quinoline 2f.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.349 mL, 2.98 mmol) by (BF3·Et2O) (1.2 equiv., 0.378 mL, 2.98 mmol) and 3-fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1f (1 equiv., 509 mg, 2.48 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-7-fluoro-2-(trifluoromethyl)quinoline 2f was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (405 mg, 62%). 1H NMR δ = 8.19 (1 H, dd, 4JH–H = 9.2, 3JH–F = 5.7 Hz, C(8)H), 7.93 (1 H, dd, 3JH–H = 9.3, 4JH–F = 2.3 Hz, C(5)H), 7.88 (1 H, s, C(3)H), 7.61–7.54 (1 H, m, C(6)H), 7.17 (1 H, 2JH–F = 54.1 Hz, C(4-CHF2)H) ppm. 19F NMR δF = −67.89 (3 F, s, CF3), −105.57 to −105.66 (1 F, m, F), −114.89 (2 F, d, 2JF–H = 54.0 Hz, C4-CHF2) ppm. 13C NMR δC = 163.84 (d, 1JC–F = 254.8 Hz, C-7), 149.36 (d, 3JC–F = 13.1 Hz, C 8-C-N), 149.09 (q, 2JC–F = 35.6 Hz, C-2), 140.60 (td, 2JC–F = 22.5, 5JC–F = 1.4 Hz, C-4), 125.92 (d, 3JC–F = 9.8 Hz, C-5), 122.08 (s, C-5-C-C-4), 121.04 (q, 1JC–F = 275.4 Hz, C(2)CF3), 120.77 (d, 2JC–F = 25.6 Hz, C-6), 114.89 (d, 2JC–F = 20.7 Hz, C-8), 113.92–113.16 (m, C-3), 112.81 (t, 1JC–F = 241.8 Hz, C(4)CHF2) ppm. C11H5F6N (265): calcd (%) N 5.28, C 49.78, H 1.88, found N 5.36, C 49.88, H 1.77. MP: 61.6–62.8 °C.
4-(Difluoromethyl)-6-fluoro-2-(trifluoromethyl)quinoline 2gi.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.346 mL, 2.96 mmol) by (BF3·Et2O) (1.2 equiv., 0.374 mL, 2.96 mmol) and 4-fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1g (1 equiv., 505 mg, 2.46 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-6-fluoro-2-(trifluoromethyl)quinoline 2gi was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (434 mg, 67%). 1H NMR δH = 8.32 (1 H, dd, 3JH–H = 9.3, 4JH–F = 5.5 Hz, C(8)H), 7.93 (1 H, s, C(3)H), 7.81–7.73 (1 H, m, C(5)H), 7.67–7.66 (1 H, m, C(7)H), 7.12 (1 H, t, 2JH–F = 54.1 Hz, C(4-CHF2)H) ppm. 19F NMR δF = −67.69 (3 F, s, CF3), −105.84 to −105.77 (1 F, m, F), −115.61 (2 F, d, 2JF–H = 54.1 Hz, C4-CHF2) ppm. 13C NMR δC = 162.40 (d, 1JC–F = 254.3 Hz, C-6), 147.31 (qd, 2JC–F = 35.7, 6JC–F = 3.2 Hz, C-2), 145.05 (s, C-8-C-N), 139.92 (td, 2JC–F = 22.5, 4JC–F = 6.2 Hz, C-4), 133.96 (d, 3JC–F = 9.8 Hz, C-8), 126.06 (d, 3JC–F = 10.7 Hz, C-5-C-C-4), 122.01 (d, 2JC–F = 26.0 Hz, C-7), 121.24 (q, 1JC–F = 275.1 Hz, C(2)CF3), 115.17–115.02 (m, C-3), 112.74 (t, 1JC–F = 237.4 Hz, C(4)CHF2), 107.68 (d, 2JC–F = 24.1 Hz, C-5) ppm. HRMS (ESI+) for C11H6F6N [M + H]: calcd 266.0399, found 266.0387. MP: 68.2–69.8 °C.
4-[Chloro(fluoro)methyl]-6-fluoro-2-(trifluoromethyl)quinoline 2gii.
The product was prepared according to the general procedure, starting from an activated solution of Yarovenko's reagent 4b (1.2 equiv., 0.675 mL, 2.97 mmol) by (BF3·Et2O) (1.2 equiv., 0.376 mL, 2.97 mmol) and 4-fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1g (1 equiv., 507 mg, 2.47 mmol) in the presence of anhydrous acetonitrile. 4-[Chloro(fluoro)methyl]-6-fluoro-2-(trifluoromethyl)quinoline 2gii was provided after purification using a gradient of ethyl acetate in pentane (0–5%) combining with the non-cyclised compound, the 1-chloro-1,5,5,5-tetrafluoro-4-((4-fluorophenyl)amino)pent-3-en-2-one (6′g) with a ratio of 7:1 (35% by 19F NMR). 1H δH = 8.32 (1 H, dd, 3JH–H = 9.3, 4JH–F = 5.5 Hz, C(8)H), 7.94 (1 H, s, C(3)H), 7.75 (1 H, dd, 3JH–F = 9.4, 4JH–H = 2.7 Hz, C(5)H), 7.65 (1 H, ddd, 3JH–F = 9.4, 4JH–F = 7.9, 4JH–H = 2.7 Hz, C(7)H), 7.54 (1 H, d, 2JH–F = 48.7 Hz, C(4-CHFCl)H) ppm.
6-Fluoro-4-(1,2,2,2-tetrafluoroethyl)-2-(trifluoromethyl)quinoline 2giii.
The product was prepared according to the general procedure, starting from an activated solution of Ishikawa's reagent 4c (1.2 equiv., 0.854 mL, 2.92 mmol) by (BF3·Et2O) (1.21 equiv., 0.372 mL, 2.93 mmol) and 4-fluoro-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1g (1 equiv., 498 mg, 2.43 mmol) in the presence of anhydrous acetonitrile. 6-Fluoro-4-(1,2,2,2-tetrafluoroethyl)-2-(trifluoromethyl)quinoline 2giii was provided after purification using a gradient of ethyl acetate in pentane (0–5%) in mixture with the non-cyclized compound, the 1,1,1,2,6,6,6-heptafluoro-5-((4-fluorophenyl)amino)hex-4-en-3-one (6′giii) with a ratio of 1:3. as a brown oil (64.7 mg). 1H NMR δH = 8.35 (1 H, dd, 3JH–H = 9.3, 4JH–F = 5.5 Hz, C(8)H), 7.97 (1 H, s, C(3)H), 7.72–7.65 (1 H, m, C(5)H), 7.64–7.62 (1 H, m, C(7)H), 6.33 (1 H, dq, 2JH–F = 43.8, 3JH–F = 5.4 Hz, C(4-CHFCF3)H) ppm. HRMS (ESI+) for C12H6F8N [M + H]: calcd 316.0367, found 316.0375.
4-(Difluoromethyl)-7-(trifluoromethoxy)-2-(trifluoromethyl)quinoline 2h.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.259 mL, 2.21 mmol) by (BF3·Et2O) (1.2 equiv., 0.28 mL, 2.21 mmol) and 3-(trifluoromethoxy)-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1h (1 equiv., 500 mg, 1.84 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-7-(trifluoromethoxy)-2-(trifluoromethyl)quinoline 2h was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (219 mg, 36%). 1H NMR δH = 8.24 (1 H, d, 3JH–H = 9.3 Hz, C(5)H), 8.16 (1 H, s, C(8)H), 7.94 (1 H, s, C(3)H), 7.65 (1 H, dd, 3JH–H = 9.3, 4JH–H = 2.4 Hz, C(6)H), 7.18 (1 H, t, 2JH–F = 54.1 Hz, C(4-CHF2)H) ppm. 19F NMR δF = −57.88 (3 F, s, OCF3), −67.89 (3 F, s, CF3), −114.87 (2 F, d, 2JF–H = 54.1 Hz, C4-CHF2) ppm. 13C NMR δC = 151 (s, C-7), 149.37 (q, 2JC–F = 35.8 Hz, C-2), 148.63 (s, C-8-C-N), 140.65 (t, 2JC–F = 22.6 Hz, C-4), 125.80 (s, C-5), 123.99 (s, C-6), 123.23–123.19 (m, C-5-C-C-4), 121.07 (q, 1JC–F = 276.7 Hz, C(2)CF3), 120.55 (q, 1JC–F = 260.0 Hz, OCF3), 120.25 (s, C-8), 114.60 (td, 3JC–F = 8.0, 3JC–F = 2.0 Hz, C-3), 112.72 (t, 1JC–F = 242.0 Hz, C(4)CHF2) ppm. C12H5F8NO (331): calcd (%) N 4.23, C 43.48, H 1.51, found N 4.28, C 43.94, H 1.35. MP: 36.3–37.8 °C.
4-(Difluoromethyl)-6-(trifluoromethoxy)-2-(trifluoromethyl)quinoline 2i.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.278 mL, 2.38 mmol) by (BF3·Et2O) (1.2 equiv., 0.282 mL, 2.22 mmol) and of 4-(trifluoromethoxy)-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1i (1 equiv., 584 mg, 1.85 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-6-(trifluoromethoxy)-2-(trifluoromethyl)quinoline 2i was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (300.3 mg, 49%). 1H NMR δH = 8.39 (1 H, d, 3JH–H = 9.3 Hz, C(8)H), 7.97 (2 H, s, C(3,5)H), 7.77 (1 H, dd, 3JH–H = 9.3, 4JH–H = 1.9 Hz, C(7)H), 7.15 (1 H, t, 2JH–F = 54.0 Hz, C(4-CHF2)H) ppm. 19F NMR δF = −57.72 (3 F, s, OCF3), −67.79 (3 F, s, CF3), −115.31 (2 F, d, 2JF–H = 53.9 Hz, C4-CHF2) ppm. 13C NMR δC = 149.50 (s, C-6), 148.41 (q, 2JC–F = 35.9 Hz, C-2), 146.02 (s, C-8-C-N), 140.46 (t, 2JC–F = 22.6 Hz, C-4), 133.66 (s, C-8), 125.52 (s, C-5-C-C-4), 125.35 (s, C-7), 121.12 (q, 1JC–F = 276.4 Hz, C(2)CF3), 120.74 (q, 1JC–F = 260.6 Hz, OCF3), 115.36 (td, 3JC–F = 8.0, 3JC–F = 2.1 Hz, C-3), 113.97 (s, C-5), 112.67 (t, 1JC–F = 242.0 Hz, C(4)CHF2) ppm. C12H5F8NO (331): calcd (%) N 4.23, C 43.48, H 1.51, found N 4.20, C 43.77, H 1.83. MP: 42.2–43.8 °C.
4-(Difluoromethyl)-N,N-dimethyl-2-(trifluoromethyl)quinolin-6-amine 2j.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.305 mL, 2.61 mmol) by (BF3·Et2O) (1.2 equiv., 0.33 mL, 2.61 mmol) and of 1-N,N-dimethyl-4-N-(1,1,1-trifluoropropan-2-ylidene)benzene-1,4-diamine 1j (1 equiv., 500 mg, 2.17 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-N,N-dimethyl-2-(trifluoromethyl)quinolin-6-amine 2j was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (235 mg, 37%). 1H NMR δH = 8.07 (1 H, d, 3JH–H = 9.5 Hz, C(8)H), 7.75 (1 H, s, C(3)H), 7.43 (1 H, dd, 3JH–H = 9.5, 4JH–H = 2.8 Hz, C(7)H), 7.07 (1 H, t, 2JH–F = 54.5 Hz, C(4-CHF2)H), 6.84 (1 H, s, C(5)H), 3.14 (1 H, s, NMe2) ppm. 19F NMR δF = −66.90 (3 F, s, CF3), −117.13 (2 F, d, 2JF–H = 54.7 Hz, C4-CHF2) ppm. 13C NMR δC = 150.28 (s, C-6), 143.01–141.69 (2 C, m, C-2/C-8-C-N), 136.12 (t, 2JC–F = 21.6 Hz, C-4), 131.80 (s, C-8), 127.25 (t, 3JC–F = 2.5 Hz, C-5-C-C-4), 121.98 (q, 1JC–F = 274.0 Hz, C(2)CF3), 120.46 (s, C-7), 114.48 (td, 3JC–F = 8.2, 3JC–F = 2.3 Hz, C-3), 113.29 (t, 1JC–F = 240.4 Hz, C(4)CHF2), 98.92 (s, C-5), 40.46 (2 C, s, NMe2) ppm. C13H11F5N2 (290): calcd (%) N 9.65, C 53.75, H 3.79, found N 9.42, C 53.55, H 3.81. MP: 107.5–108.4 °C.
4-(Difluoromethyl)-7-fluoro-8-methyl-2-(trifluoromethyl)quinoline 2k.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.644 mL, 5.5 mmol) by (BF3·Et2O) (1.2 equiv., 0.698 mL, 5.5 mmol) and 3-fluoro-2-methyl-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1k (1 equiv., 1.76 g, 4.59 mmol) in the presence of anhydrous acetonitrile. 4-(Difluoromethyl)-7-fluoro-8-methyl-2-(trifluoromethyl)quinoline 2k was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (488.4 mg, 38%). 1H NMR δH = 7.99 (1 H, dd, 3JH–H = 9.0, 4JH–F = 5.9 Hz, C(5)H), 7.88 (1 H, s, C(3)H), 7.54 (1 H, t, 3JH–H = 9.0 Hz, C(6)H), 7.17 (1 H, t, 2JH–F = 54.2 Hz, C(4-CHF2)H), 2.75 (3 H, d, 4JH–F = 2.6 Hz, Me) ppm. 19F NMR δF = −67.78 (3 F, s, CF3), −108.31 to −108.46 (1 F, m, F), −114.94 (2 F, d, 2JF–H = 54.2 Hz, C4-CHF2) ppm. 13C NMR δC = 161.60 (d, 1JC–F = 250.2 Hz, C-7), 148.47–147.14 (2 C, m, C-2/C-8-C-N), 140.54 (td, 2JC–F = 22.3, 5JC–F = 1.8 Hz, C-4), 123.79 (d, 2JC–F = 16.2 Hz, C-8), 122.26 (2 C, d, 3JC–F = 10.3 Hz, C-5/C-5-C-C-4), 121.32 (q, 1JC–F = 275.3 Hz, C(2)CF3), 120.25 (d, 2JC–F = 27.2 Hz, C-6), 113.49–112.94 (m, C-3), 112.84 (t, 1JC–F = 241.6 Hz, C(4)CHF2), 9.38 (d, 3JC–F = 3.7 Hz, Me) ppm. HRMS (ESI+) for C12H8F6N [M + H]: calcd 280.0555, found 280.0570. MP: 78.7–79.4 °C.
7-Chloro-4-(difluoromethyl)-8-methyl-2-(trifluoromethyl)quinoline 2l.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.599 mL, 5.12 mmol) by (BF3·Et2O) (1.2 equiv., 0.648 mL, 5.12 mmol) and 3-chloro-2-methyl-N-(1,1,1-trifluoropropan-2-ylidene)aniline 1l (1 equiv., 2.01 g, 4.26 mmol) in the presence of anhydrous acetonitrile. 7-Chloro-4-(difluoromethyl)-8-methyl-2-(trifluoromethyl)quinoline 2l was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a beige amorphous solid (485 mg, 38%). 1H NMR δH = 7.93–7.91 (2 H, m, C(3,5)H), 7.74 (1 H, d, 3JH–H = 9.1 Hz, C(6)H), 7.17 (1 H, t, 2JH–F = 54.2 Hz, C(4-CHF2)H), 2.93 (3 H, s, Me) ppm. 19F δF = −67.78 (3 F, s, CF3), −115.12 (2 F, d, 2JF–H = 54.1 Hz, C4-CHF2) ppm. 13C NMR δC = 147.53 (q, 2JC–F = 35.8 Hz, C-2), 147.44 (s, C-8-C-N), 140.63 (t, 2JC–F = 22.3 Hz, C-4), 137.44 (s, C-7), 137.10 (s, C-8), 131.35 (s, C-6), 121.50 (s, C-5), 121.27 (q, 1JC–F = 275.3 Hz, C(2)CF3), 119.32 (s, C-5-C-C-4), 113.93–113.73 (m, C-3), 112.70 (t, 1JC–F = 241.7 Hz, C(4)CHF2), 14.91 (s, Me) ppm. C12H7F5NCl (295): calcd (%) N 4.73, C 48.70, H 2.36, found N 4.62, C 48.82, H 2.55.
2,4-Bis(difluoromethyl)quinoline 3ai.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.427 mL, 3.65 mmol) by (BF3·Et2O) (1.2 equiv., 0.463 mL, 3.65 mmol) and N-(1,1-difluoropropan-2-ylidene)aniline 1aa (1 equiv., 515 mg, 3.04 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)quinoline 3ai was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (537 mg, 77%). 1H NMR δH = 8.23 (1 H, d, 3JH–H = 8.5 Hz, C(8)H), 8.14 (1 H, d, 3JH–H = 8.5 Hz, C(5)H), 7.92 (1 H, s, C(3)H), 7.86 (1 H, t, 3JH–H = 7.7 Hz, C(7)H), 7.74 (1 H, t, 3JH–H = 7.7 Hz, C(6)H), 7.19 (1 H, t, 2JH–F = 54.3 Hz, C(4-CHF2)H), 6.81 (1 H, t, 2JH–F = 55.1 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −114.46 (2 F, d, 2JF–H = 55.1 Hz, C2-CHF2), −115.16 (2 F, d, 2JF–H = 54.3 Hz, C4-CHF2) ppm. 13C NMR δC = 152.70 (t, 2JC–F = 27.1 Hz, C-2), 147.80 (s, C-8-C-N), 139.93 (t, 2JC–F = 22.2 Hz, C-4), 130.91 (s, C-7), 130.83 (s, C-8), 129.33 (s, C-6), 124.84 (s, C-5-C-C-4), 123.55 (s, C-5), 114.37 (t, 1JC–F = 242.0 Hz, C(4)CHF2), 114.32–114.17 (m, C-3), 113.13 (t, 1JC–F = 241.2 Hz, C(2)CHF2) ppm. C11H7F4N (229): calcd (%) N 6.10, C 57.60, H 3.05, found N 6.20, C 57.56, H 2.96. MP: 48.2–49.7 °C.
4-[Chloro(fluoro)methyl]-2-(difluoromethyl)quinoline 3aii.
The product was prepared according to the general procedure, starting from an activated solution of Yarovenko's reagent 4b (1.2 equiv., 0.56 mL, 3.55 mmol) by (BF3·Et2O) (1.2 equiv., 0.45 mL, 3.55 mmol) and N-(1,1-difluoropropan-2-ylidene)aniline 1aa (1 equiv., 500 mg, 2.96 mmol) in the presence of anhydrous acetonitrile. 4-[Chloro(fluoro)methyl]-2-(difluoromethyl)quinoline 3aii was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a brown solid (567 mg, 78%). 1H NMR δH = 8.02 (1 H, d, 3JH–H = 8.5 Hz, C(8)H), 7.88 (1 H, d, 3JH–H = 8.5 Hz, C(5)H), 7.75 (1 H, s, C(3)H), 7.62 (1 H, t, 3JH–H = 7.7 Hz, C(7)H), 7.51–7.42 (2 H, m, C(7)H/C(4-CHFCl)H), 6.65 (1 H, t, 2JH–F = 55.1 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −114.5 (2 F, dd, 2JF–H = 55.2 Hz, 4JF–H = 3 Hz, C2-CHF2), −138.0 (1 F, d, 2JF–H = 49 Hz, C4-CHFCl) ppm. 13C NMR δC = 152.63 (t, 2JC–F = 27 Hz, C-2), 147.69 (s, C-8-C-N), 143.25 (d, 2JC–F = 21 Hz, C-4), 130.80 (s, C-7), 130.70 (s, C-8), 128.96 (s, C-6), 123.68 (d, 3JC–F = 3.6 Hz, C-5-C-C-4), 123.06 (s, C-5), 114.33 (t, 1JC–F = 241.3 Hz, C(2)CHF2), 113.0 (dt, 3JC–F = 9.6, 3JC–F = 1.9 Hz, C-3), 97.08 (d, 1JC–F = 244.3 Hz, C(4)CHFCl) ppm. HRMS (ESI+) for C11H8F3NCl [M + H]: calcd 246.0292, found 246.0312.
2,4-Bis(difluoromethyl)-8-methoxyquinoline 3b.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.354 mL, 3.02 mmol) by (BF3·Et2O) (1.2 equiv., 0.383 mL, 3.02 mmol) and N-(1,1-difluoropropan-2-ylidene)-2-methoxyaniline 1bb (1 equiv., 502 mg, 2.52 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-8-methoxyquinoline 3b was provided after purification using a gradient of ethyl acetate in pentane (10–25%) as a yellow solid (473 mg, 72%). 1H NMR δH = 7.94 (1 H, s, C(5)H), 7.62 (2 H, d, 3JH–H = 4.2 Hz, C(6,7)H), 7.31–7.02 (2 H, m, C(3)H/C(4-CHF2)H), 6.88 (1 H, t, 2JH–F = 51.4 Hz, C(2-CHF2)H), 4.09 (3 H, s, Me) ppm. 19F NMR δF = −113.42 (2 F, d, 2JF–H = 54.9 Hz, C2-CHF2), −115.88 (d, 2JF–H = 54.3 Hz, C4-CHF2) ppm. 13C NMR δC = 156.17 (s, C-8), 151.14 (t, 2JC–F = 27.7 Hz, C-2), 140.04–139.19 (2 C, m, C-4/C-8-C-N), 129.84 (s, C-6), 126.13 (s, C-5-C-C-4), 114.92 (s, C-5), 114.72 (t, J = 7.9 Hz, C-7), 114.48 (t, 1JC–F = 241.9 Hz, C(2)CHF2), 112.85 (t, 1JC–F = 241.1 Hz, C(4)CHF2), 109.17 (s, C-3), 56.41 (s, Me) ppm. C12H9F4NO (259): calcd (%) N 5.40, C 55.55, H 3.47, found N 5.35, C 55.30, H 3.51. MP: 96.8–98.6 °C.
2,4-Bis(difluoromethyl)-7-methoxyquinoline 3c.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.356 mL, 3.04 mmol) by (BF3·Et2O) (1.2 equiv., 0.386 mL, 3.04 mmol) and N-(1,1-difluoropropan-2-ylidene)-3-methoxyaniline 1cc (1 equiv., 505 mg, 2.54 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-7-methoxyquinoline 3c was provided after purification using a gradient of ethyl acetate in pentane (10–25%) as a yellow solid (515 mg, 78%). 1H NMR δH = 7.79 (1 H, d, 3JH–H = 9.3 Hz, C(5)H), 7.57 (1 H, s, C(3)H), 7.30 (1 H, d, 4JH–H = 2.6 Hz, C(8)H), 7.14 (1 H, dd, 3JH–H = 9.3, 4JH–H = 2.6 Hz, C(6)H), 6.94 (1 H, t, 2JH–F = 54.4 Hz, C(4-CHF2)H), 6.62 (1 H, t, 2JH–F = 55.2 Hz, C(2-CHF2)H), 3.79 (3 H, s, Me) ppm. 19F NMR δF = −114.79 (2 F, d, 2JF–H = 54.4 Hz, C2-CHF2), −114.95 (2 F, d, 2JF–H = 55.3 Hz, C4-CHF2) ppm. 13C NMR δC = 161.45 (s, C-7), 152.86 (t, 2JC–F = 26.7 Hz, C-2), 149.83 (s, C-8-C-N), 139.56 (t, 2JC–F = 22.2 Hz, C-4), 124.33 (s, C-5), 122.45 (s, C-6), 119.77 (t, 3JC–F = 2.8 Hz, C-5-C-C-4), 114.25 (t, 1JC–F = 242.4 Hz, C(2)CHF2), 113.18 (t, 1JC–F = 241.0 Hz, C(4)CHF2), 111.89–111.70 (m, C-3), 108.24 (s, C-8), 55.60 (s, Me) ppm. C12H9F4NO (259): calcd (%) N 5.40, C 55.55, H 3.47, found N 5.55, C 55.57, H 3.67. MP: 55.4–56.9 °C.
2,4-Bis(difluoromethyl)-6-methoxyquinoline 3d.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.357 mL, 3.05 mmol) by (BF3·Et2O) (1.2 equiv., 0.386 mL, 3.05 mmol) and N-(1,1-difluoropropan-2-ylidene)-4-methoxyaniline 1dd (1 equiv., 506 mg, 2.54 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-6-methoxyquinoline 3d was provided after purification using a gradient of ethyl acetate in pentane (10–25%) as an orange solid (468 mg, 71%). 1H NMR δH = 8.15 (1 H, d, 3JH–H = 9.3 Hz, C(8)H), 7.90 (1 H, s, C(3)H), 7.53 (1 H, dd, 3JH–H = 9.3, 4JH–H = 2.5 Hz, C(7)H), 7.36 (1 H, s, C(5)H), 7.16 (1 H, t, 2JH–F = 54.4 Hz, C(4-CHF2)H), 6.85 (1 H, t, 2JH–F = 55.2 Hz, C(2-CHF2)H), 4.03 (3 H, s, Me) ppm. 19F NMR δF = −113.99 (2 F, d, 2JF–H = 55.3 Hz, C2-CHF2), −115.52 (2 F, d, 2JF–H = 54.4 Hz, C4-CHF2) ppm. 13C NMR δC = 159.77 (s, C-6), 149.93 (t, 2JC–F = 27.0 Hz, C-2), 143.966 (s, C-8-C-N), 138.02 (t, 2JC–F = 22.0 Hz, C-4), 132.11 (s, C-8), 126.21 (s, C-5-C-C-4), 123.71 (s, C-7), 114.70 (tt, 3JC–F = 8.0, 3JC–F = 1.8 Hz, C-3), 114.51 (t, C(2)CHF2, 1JC–F = 241.9 Hz), 113.57 (t, C(4)CHF2, 1JC–F = 240.8 Hz), 101.35 (s, C-5), 55.79 (s, Me) ppm. C12H9F4NO (259): calcd (%) N 5.40, C 55.55, H 3.47, found N 5.43, C 55.24, H 3.30. MP: 93.5–97.2 °C.
2,4-Bis(difluoromethyl)-8-fluoroquinoline 3e.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.377 mL, 3.22 mmol) by (BF3·Et2O) (1.2 equiv., 0.408 mL, 3.22 mmol) and N-(1,1-difluoropropan-2-ylidene)-2-fluoroaniline 1ee (1 equiv., 502 mg, 2.68 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-8-fluoroquinoline 3e was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (466 mg, 39%) in combination (3:1) with the 1,1,5,5-tetrafluoro-4-((2-fluorophenyl)amino)pent-3-en-2-one after purification (6′e). 1H NMR δH = 8.00 (1 H, s, C(3)H), 7.93 (1 H, d, 3JH–H = 8.6 Hz, C(5)H), 7.73–7.68 (1 H, m, C(6)H), 7.60–7.52 (1 H, m, C(7)H), 7.17 (1 H, t, 2JH–F = 54.2 Hz, C(4-CHF2)H), 6.86 (1 H, t, 2JH–F = 54.8 Hz, C(2-CHF2)H) ppm. HRMS (ESI+) for C11H7F5N [M + H]: calcd 248.0493, found 248.0499.
2,4-Bis(difluoromethyl)-7-fluoroquinoline 3f.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.376 mL, 3.22 mmol) by (BF3·Et2O) (1.2 equiv., 0.408 mL, 3.22 mmol) and N-(1,1-difluoropropan-2-ylidene)-3-fluoroaniline 1ff (1 equiv., 501 mg, 2.68 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-7-fluoroquinoline 3f was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (464 mg, 70%). 1H NMR δH = 8.17 (1 H, dd, 4JH–H = 9.3, 3JH–F = 5.7 Hz, C(8)H), 7.86 (1 H, s, C(3)H), 7.84 (1 H, s, C(5)H), 7.57–7.49 (1 H, m, C(6)H), 7.14 (1 H, t, 2JH–F = 54.2 Hz, C(4-CHF2)H), 6.78 (1 H, t, 2JH–F = 55.0 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −106.41 to −106.96 (1 F, m, F), −114.43 (2 F, d, 2JF–H = 54.2 Hz, C2-CHF2), −114.83 (2 F, d, 2JF–H = 55.0 Hz, C4-CHF2) ppm. 13C NMR δC = 163.60 (d, 1JC–F = 253.6 Hz, C-7), 153.93 (t, 2JC–F = 27.3 Hz, C-2), 149.21 (d, 3JC–F = 12.7 Hz, C-8-C-N), 140.17 (td, 2JC–F = 22.4, 5JC–F = 1.1 Hz, C-4), 126.00 (d, 3JC–F = 9.8 Hz, C-5), 121.79 (d, 4JC–F = 1.0 Hz, C-5-C-C-4), 119.91 (d, 2JC–F = 25.39 Hz, C-6), 114.54 (d, 2JC–F = 20.56 Hz, C-8), 114.12 (t, 1JC–F = 242.9 Hz, C(2)CHF2), 113.86 (m, C-3), 113.23 (t, 1JC–F = 241.5 Hz, C(4)CHF2) ppm. C11H6F5N (247): calcd (%) N 5.66, C 53.41, H 2.43, found N 5.79, C 53.54, H 2.69. MP: 73.2–74.6 °C.
2,4-Bis(difluoromethyl)-6-fluoroquinoline 3gi.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.5 equiv., 0.375 mL, 3.2 mmol) by (BF3·Et2O) (1.51 equiv., 0.408 mL, 3.22 mmol) and N-(1,1-difluoropropan-2-ylidene)-4-fluoroaniline 1gg (1 equiv., 400 mg, 2.14 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-6-fluoroquinoline 3gi was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (205 mg, 39%). 1H NMR δH = 8.24 (1 H, dd, 3JH–H = 9.3, 4JH–F = 5.5 Hz, C(8)H), 7.92 (1 H, s, C(3)H), 7.80–7.74 (1 H, m, C(5)H), 7.65–7.60 (1 H, m, C(7)H), 7.09 (1 H, t, 2JH–F = 54.2 Hz, C(4-CHF2)H), 6.79 (1 H, t, 2JH–F = 55.0 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −107.18 to −107.24 (1 F, m, F), −114.45 (2 F, d, 2JF–H = 55.0 Hz, C2-CHF2), −115.17 (2 F, d, 2JF–H = 54.2 Hz, C4-CHF2) ppm. 13C NMR δC = 162.01 (d, 1JC–F = 252.9 Hz, C-6), 152.09 (td, 2JC–F = 27.3, 6JC–F = 3.1 Hz, C-2), 144.98 (s, C-8-C-N), 139.56 (td, 2JC–F = 22.4, 4JC–F = 6.1 Hz, C-4), 133.46 (d, 3JC–F = 9.6 Hz, C-8), 125.76 (d, 3JC–F = 10.4 Hz, C-5-C-C-4), 121.43 (d, 2JC–F = 25.8 Hz, C-7), 115.28 (t, 3JC–F = 7.8 Hz, C-3), 114.21 (t, 1JC–F = 241.3 Hz, C(2)CHF2), 110.78 (t, 1JC–F = 242.0 Hz, C(4)CHF2), 107.84 (d, 2JC–F = 24.0 Hz, C-5) ppm. C11H6F5N (247): calcd (%) N 5.66, C 53.41, H 2.43, found N 5.67, C 53.42, H 2.57. HRMS (ESI+) for C11H7F5N [M + H]: calcd 248.0493, found 248.0497. MP: 68.7–71.2 °C.
4-[Chloro(fluoro)methyl]-2-(difluoromethyl)-6-fluoroquinoline 3gii.
The product was prepared according to the general procedure, starting from an activated solution of Yarovenko's reagent 4b (1.2 equiv., 0.597 mL, 2.62 mmol) by (BF3·Et2O) (1.2 equiv., 0.332 mL, 2.62 mmol) and N-(1,1-difluoropropan-2-ylidene)-4-fluoroaniline 1gg (1 equiv., 409 mg, 2.19 mmol) in the presence of anhydrous acetonitrile. 4-[Chloro(fluoro)methyl]-2-(difluoromethyl)-6-fluoroquinoline 3gii was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a brown liquid (17 mg, 3% estimated). 1H NMR δH = 8.26 (1 H, dd, 3JH–H = 9.3, 4JH–F = 5.5 Hz, C(8)H), 7.94 (1 H, s, C(3)H), 7.77 (1 H, dd, 3JH–F = 9.6, 4JH–H = 2.7 Hz, C(5)H), 7.63 (1 H, ddd, 3JH–H = 9.3, 4JH–F = 8.0, 4JH–H = 2.7 Hz, C(7)H), 7.53 (1 H, d, 2JH–F = 48.9 Hz, C(4-CHFCl)H), 6.80 (1 H, t, 2JH–F = 55.0 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −106.78 to −106.84 (1 F, m, F), −114.49 (2 F, dd, 2JF–H = 55.0, 4JF–H = 2.9 Hz, C2-CHF2), −137.65 (1 F, d, 2JF–H = 48.7 Hz, C4-CHFCl) ppm. 13C NMR δC = 161.61 (d, 1JC–F = 253.2 Hz, C-6), 152.11 (t, 2JC–F = 27.1 Hz, C-2), 144.94 (s, C-8-C-N), 143.01 (dd, 2JC–F = 20.9, 4JC–F = 6.2 Hz, C-4), 133.56 (d, 3JC–F = 9.7 Hz, C-8), 128.42 (s, C-5-C-C-4), 121.43 (d, 2JC–F = 25.8 Hz, C-7), 114.11 (t, 1JC–F = 241.0 Hz, C(2)CHF2), 114.10 (d, 3JC–F = 8.8 Hz, C-3), 107.65 (d, 2JC–F = 24.1 Hz, C-5), 97.09 (d, 1JC–F = 244.8 Hz, C(4)CHFCl) ppm. HRMS (ESI positive) for C11H7F4NCl [M + H]: calcd 264.0198, found 264.0198.
2,4-Bis(difluoromethyl)-8-(trifluoromethoxy)quinoline 3h.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.44 equiv., 0.34 mL, 2.91 mmol) by (BF3·Et2O) (1.44 equiv., 0.37 mL, 2.92 mmol) and N-(1,1-difluoropropan-2-ylidene)-2-(trifluoromethoxy)aniline 1hh (1 equiv., 512 mg, 2.02 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-8-(trifluoromethoxy)quinoline 3h was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (244 mg, 39%). 1H NMR δH = 8.11 (1 H, dd, 3JH–H = 8.1, 4JH–H = 1.4 Hz, C(7)H), 8.01 (1 H, s, C(3)H), 7.76–7.75 (2 H, m, C(5,6)H), 7.18 (1 H, t, 2JH–F = 54.1 Hz, C(4-CHF2)H), 6.85 (1 H, t, 2JH–F = 54.8 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −57.55 (3 F, s, OCF3), −114.12 (2 F, d, 2JF–H = 54.9 Hz, C2-CHF2), −114.91 (2 F, d, 2JF–H = 54.3 Hz, C4-CHF2) ppm. 13C NMR δC = 153.55 (t, 2JC–F = 28.0 Hz, C-2), 146.06 (s, C-8), 141.11 (s, C-8-C-N), 140.23 (t, 2JC–F = 22.5 Hz, C-4), 128.98 (s, C-5), 126.25 (s, C-5-C-C-4), 122.56 (s, C-7), 122.14 (s, C-6), 120.47 (q, 1JC–F = 259.6 Hz, OCF3), 115.59 (t, 3JC–F = 7.9 Hz, C-3), 114.29 (t, 1JC–F = 242.4 Hz, C(2)CHF2), 112.98 (t, 1JC–F = 241.7 Hz, C(4)CHF2) ppm. C12H6F7NO (313): calcd (%) N 4.47, C 45.98, H 1.92, found N 4.57, C 46.23, H 1.98. MP: 59.4–60 °C.
2,4-Bis(difluoromethyl)-7-(trifluoromethoxy)quinoline 3i.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.278 mL, 2.38 mmol) by (BF3·Et2O) (1.2 equiv., 0.301 mL, 2.38 mmol) and N-(1,1-difluoropropan-2-ylidene)-3-(trifluoromethoxy)aniline 1ii (1 equiv., 501 mg, 1.98 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-7-(trifluoromethoxy)quinoline 3i was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a light brown liquid (440 mg, 71%). 1H NMR δH = 8.08 (1 H, d, 3JH–H = 9.2 Hz, C(5)H), 7.93 (1 H, s, C(8)H), 7.79 (1 H, s, C(3)H), 7.46 (1 H, d, 3JH–H = 9.2 Hz, C(6)H), 7.03 (1 H, t, 2JH–F = 54.2 Hz, C(4-CHF2)H), 6.69 (1 H, t, 2JH–F = 55.0 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −58.14 (3 F, s, OCF3), −114.72 (2 F, d, 2JF–H = 54.1 Hz, C2-CHF2), −115.17 (2 F, d, 2JF–H = 54.9 Hz, C4-CHF2) ppm. 13C NMR δC = 154.12 (t, 2JC–F = 27.3 Hz, C-2), 150.58 (s, C-7), 148.46 (s, C-8-C-N), 140.14 (t, 2JC–F = 22.5 Hz, C-4), 125.76 (s, C-5), 123.13 (s, C-6), 122.95 (s, C-5-C-C-4), 120.58 (q, 1JC–F = 259.4 Hz, OCF3), 120.02 (s, C-8), 114.72 (tt, 3JC–F = 8, 3JC–F = 1.8 Hz, C-3), 114.04 (t, 1JC–F = 242.4 Hz, C(2)CHF2), 113.12 (t, 1JC–F = 241.4 Hz, C(4)CHF2) ppm. C12H6F7NO (313): calcd (%) N 4.47, C 45.98, H 1.92, found N 4.49, C 46.13, H 2.15.
2,4-Bis(difluoromethyl)-6-(trifluoromethoxy)quinoline 3j.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.5 equiv., 0.347 mL, 2.96 mmol) by (BF3·Et2O) (1.51 equiv., 0.378 mL, 2.98 mmol) and N-(1,1-difluoropropan-2-ylidene)-4-(trifluoromethoxy)aniline 1jj (1 equiv., 500 mg, 1.98 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-6-(trifluoromethoxy)quinoline 3j was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (361 mg, 58%). 1H NMR δH = 8.30 (1 H, d, 3JH–H = 9.3 Hz, C(8)H), 7.96–7.95 (2 H, m, C(3,5)H), 7.73 (1 H, dd, 3JH–H = 9.3, 4JH–H = 1.8 Hz, C(7)H), 7.13 (1 H, t, 2JH–F = 56.0 Hz, C(4-CHF2)H), 6.80 (1 H, t, 2JH–F = 55.0 Hz, C(2-CHF2)H) ppm. 19F δF = −57.71 (3 F, s, OCF3), −114.67 (2 F, d, 2JF–H = 55.0 Hz, C2-CHF2), −114.88 (2 F, d, 2JF–H = 54.1 Hz, C4-CHF2) ppm. 13C NMR δC = 153.23 (t, 2JC–F = 27.5 Hz, C-2), 148.97 (s, C-6), 146.01 (s, C-8-C-N), 140.06 (t, 2JC–F = 22.5 Hz, C-4), 133.17 (s, C-8), 125.23 (s, C-5-C-C-4), 124.89 (s, C-7), 120.59 (q, 1JC–F = 259.2 Hz, OCF3), 115.64–115.49 (m, C-3), 114.32 (s, C-5), 114.08 (t, 1JC–F = 241.6 Hz, C(2)CHF2), 113.09 (t, 1JC–F = 240.0 Hz, C(4)CHF2) ppm. C12H6F7NO (313): calcd (%) N 4.47, C 45.98, H 1.92, found N 4.49, C 46.35, H 2.07. MP: 47.5–48.2 °C.
2,4-Bis(difluoromethyl)-N,N-dimethylquinolin-7-amine 3k.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.331 mL, 2.83 mmol) by (BF3·Et2O) (1.2 equiv., 0.358 mL, 2.83 mmol) and 3-N-(1,1-difluoropropan-2-ylidene)-1-N,N-dimethylbenzene-1,3-diamine 1kk (1 equiv., 500 mg, 2.36 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-N,N-dimethylquinolin-7-amine 3k was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a brown solid (98.2 mg, 15%). 1H NMR δH = 7.94 (1 H, d, 3JH–H = 9.4 Hz, C(5)H), 7.55 (1 H, s, C(8)H), 7.29 (1 H, dd, 3JH–H = 9.4, 4JH–H = 2.7 Hz, C(6)H), 7.21 (1 H, d, J = 2.7 Hz, C(3)H), 7.08 (1 H, t, 2JH–F = 54.6 Hz, C(4-CHF2)H), 6.72 (1 H, t, 2JH–F = 55.4 Hz, C(2-CHF2)H), 3.13 (6 H, s, NMe2) ppm. 19F NMR δF = −114.59 (2 F, d, 2JF–H = 54.6 Hz, C2-CHF2), −114.89 (2 F, d, 2JF–H = 55.4 Hz, C4-CHF2) ppm. 13C NMR δC = 152.95 (t, 2JC–F = 26.5 Hz, C-2), 151.69 (s, C-7), 150.03 (s, C-8-C-N), 139.30 (t, 2JC–F = 23.9 Hz, C-4), 124.08 (s, C-5), 118.49 (s, C-8), 116.98 (s, C-5-C-C-4), 114.56 (t, 1JC–F = 264.0 Hz, C(2)CHF2), 113.54 (t, 1JC–F = 240.8 Hz, C(4)CHF2), 109.73 (tt, 3JC–F = 8.0, 3JC–F = 2.2 Hz, C-3), 107.26 (s, C-6), 40.38 (2 C, s, NMe2) ppm. C13H12F4N2 (272): calcd (%) N 10.20, C 57.30, H 4.41, found N 10.06, C 57.26, H 4.41. MP: 83.7–84.7 °C.
2,4-Bis(difluoromethyl)-N,N-dimethylquinolin-6-amine 3l.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.331 mL, 2.83 mmol) by (BF3·Et2O) (1.2 equiv., 0.358 mL, 2.83 mmol) and 4-N-(1,1-difluoropropan-2-ylidene)-1-N,N-dimethylbenzene-1,4-diamine 1ll (1 equiv., 500 mg, 2.36 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-N,N-dimethylquinolin-6-amine 3l was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as an orange solid (91.9 mg, 14%). 1H NMR δH = 8.01 (1 H, d, 3JH–H = 9.4 Hz, C(8)H), 7.76 (1 H, s, C(3)H), 7.42 (1 H, dd, 3JH–H = 9.5, 4JH–H = 2.7 Hz, C(7)H), 7.07 (1 H, t, 2JH–F = 54.6 Hz, C(4-CHF2)H), 6.90 (1 H, s, C(5)H), 6.75 (1 H, t, 2JH–F = 55.5 Hz, C(2-CHF2)H), 3.14 (1 H, s, NMe2) ppm. 19F NMR δF = −113.38 (2 F, d, 2JF–H = 55.5 Hz, C2-CHF2), −116.67 (2 F, d, 2JF–H = 54.6 Hz, C4-CHF2) ppm. 13C NMR δC = 149.96 (s, C-6), 147.42 (t, 2JC–F = 26.8 Hz, C-2), 141.73 (s, C-8-C-N), 136.29 (t, 2JC–F = 21.5 Hz, C-4), 131.36 (s, C-8), 126.89 (s, C-5-C-C-4), 120.03 (s, C-7), 114.90 (t, 1JC–F = 239.9 Hz, C(2)CHF2), 114.64 (t, 3JC–F = 8.1 Hz, C-3), 113.69 (t, 1JC–F = 241.4 Hz, C(4)CHF2), 99.55 (s, C-5), 40.53 (s, NMe2) ppm. C13H12F4N2 (272): calcd (%) N 10.20, C 57.30, H 4.41, found N 10.09, C 56.86, H 4.40. MP: 115.6–116.9 °C.
2,4-Bis(difluoromethyl)-7-fluoro-8-methylquinoline 3m.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.698 mL, 5.96 mmol) by (BF3·Et2O) (1.2 equiv., 0.756 mL, 5.96 mmol) and N-(1,1-difluoropropan-2-ylidene)-3-fluoro-2-methylaniline 1mm (1 equiv., 1 g, 4.97 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-7-fluoro-8-methylquinoline 3m was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a yellow solid (990 mg, 76%). 1H NMR δH = 7.96 (1 H, dd, 3JH–H = 9.0, 4JH–F = 5.9 Hz, C(5)H), 7.86 (1 H, s, C(3)H), 7.47 (1 H, t, 3JH–H = 9.0 Hz, C(6)H), 7.13 (1 H, t, 2JH–F = 54.3 Hz, C(4-CHF2)H), 6.81 (1 H, t, 2JH–F = 55.1 Hz, C(2-CHF2)H), 2.71 (3 H, d, 4JH–F = 2.5 Hz, Me) ppm. 19F NMR δF = −109.41 (1 F, ddd, 3JH–F = 8.4, 4JH–F = 5.6, 4JCH3–F = 2.7 Hz, F), −114.20 (2 F, d, 2JH–F = 55.1 Hz, C2-CHF2), −114.56 (2 F, d, 2JH–F = 54.3 Hz, C4-CHF2) ppm. 13C NMR δC = 161.37 (d, 1JC–F = 249.1 Hz, C-7), 152.53 (t, 2JC–F = 27.5 Hz, C-2), 148.08 (d, 3JC–F = 10.1 Hz, C-8-C-N), 140.19 (t, 2JC–F = 21.5 Hz, C-4), 123.23 (d, 2JC–F = 16.1 Hz, C-8), 122.36 (d, 3JC–F = 10.2 Hz, C-5), 121.85 (s, C-5-C-C-4), 119.38 (d, 2JC–F = 27.0 Hz, C-6), 114.58 (t, 1JC–F = 241.2 Hz, C(2)CHF2), 113.12 (t, 1JC–F = 242.4 Hz, C(4)CHF2), 113.12 (s, C-3), 9.34 (d, 4JH–F = 3.8 Hz, Me) ppm. C12H8F5N (261): calcd (%) N 5.36, C 55.13, H 3.06, found N 5.26, C 55.13, H 3.22. MP: 87.7–89 °C.
7-Chloro-2,4-bis(difluoromethyl)-8-methylquinoline 3n.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.662 mL, 5.66 mmol) by (BF3·Et2O) (1.2 equiv., 0.717 mL, 5.66 mmol) and 3-chloro-N-(1,1-difluoropropan-2-ylidene)-2-methylaniline 1nn (1 equiv., 1.03 g, 4.72 mmol) in the presence of anhydrous acetonitrile. 7-Chloro-2,4-bis(difluoromethyl)-8-methylquinoline 3n was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a colourless solid (977 mg, 75%). 1H NMR δH = 7.89–7.88 (2 H, m, C(3,5)H), 7.67 (1 H, d, 3JH–H = 9.1 Hz, C(6)H), 7.13 (1 H, t, 2JH–F = 54.3 Hz, C(4-CHF2)H), 6.80 (1 H, t, 2JH–F = 55.1 Hz, C(2-CHF2)H), 2.88 (3 H, s, Me) ppm. 19F NMR δF = −114.17 (2 F, d, 2JF–H = 55.1 Hz, C2-CHF2), −114.75 (2 F, d, 2JF–H = 54.3 Hz, C4-CHF2) ppm. 13C NMR δC = 152.34 (t, 2JC–F = 27.6 Hz, C-2), 147.27 (s, C-8-C-N), 140.27 (t, 2JC–F = 22.2 Hz, C-4), 136.83 (s, C-7), 136.57 (s, C-8), 130.56 (s, C-6), 123.48 (s, C-5-C-C-4), 121.65 (s, C-5), 114.52 (t, 1JC–F = 241.3 Hz, C(2)CHF2), 113.97 (t, 3JC–F = 7.9 Hz, C-3), 113.08 (t, 1JC–F = 242.4 Hz, C(4)CHF2), 14.84 (s, Me) ppm. C12H8F4NCl (277): calcd (%) N 5.04, C 51.86, H 2.88, found N 4.92, C 52.02, H 3.03. MP: 58.9–59.5 °C.
7-Chloro-2,4-bis(difluoromethyl)quinoline 3o.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 3.45 mL, 29.5 mmol) by (BF3·Et2O) (1.2 equiv., 3.73 mL, 29.5 mmol) and 3-chloro-N-(1,1-difluoropropan-2-ylidene)aniline 1oo (1 equiv., 5 g, 24.6 mmol) in the presence of anhydrous acetonitrile. 7-Chloro-2,4-bis(difluoromethyl)quinoline 3o was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a colourless solid (2.93 g, 45%). 1H NMR δH = 8.20 (1 H, d, 4JH–H = 2.1 Hz, C(8)H), 8.07 (1 H, d, 3JH–H = 9.0 Hz, C(5)H), 7.88 (1 H, s, C(3)H), 7.67 (1 H, dd, 3JH–H = 9.0, 4JH–H = 2.1 Hz, C(6)H), 7.12 (1 H, t, 2JH–F = 54.2 Hz, C(4-CHF2)H), 6.77 (1 H, t, 2JH–F = 55.0 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −114.73 (4 F, t, 2JF–H = 54.4 Hz, C2-CHF2/C4-CHF2) ppm. 13C NMR δC = 153.83 (t, 2JC–F = 27.3 Hz, C-2), 148.27 (s, C8-C-N), 140.12 (t, 2JC–F = 22.4 Hz, C-4), 137.12 (s, C-7), 130.33 (s, C-6), 129.71 (s, C-8), 124.95 (s, C-5), 123.13 (s, C-5-C-C-4), 114.58 (tt, 3JC–F = 8, 3JC–F = 1.8 Hz, C-3), 114.09 (t, 1JC–F = 242.9 Hz, C(2)CHF2), 113.08 (t, 1JC–F = 241.5 Hz, C(4)CHF2) ppm. C11H6F4NCl (263): calcd (%) N 5.31, C 50.12, H 2.29, found N 5.23, C 50.00, H 2.29. MP: 90.2–91 °C.
2,4-Bis(difluoromethyl)-6-(trifluoromethyl)quinoline 3p.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.195 mL, 1.66 mmol) by (BF3·Et2O) (1.2 equiv., 0.211 mL, 1.66 mmol) and N-(1,1-difluoropropan-2-ylidene)-4-(trifluoromethyl)aniline 1pp (1 equiv., 328 mg, 1.39 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-6-(trifluoromethyl)quinoline 3p was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a grey solid (93.9 mg, 23%). 1H NMR δH = 8.46 (1 H, s, C(5)H), 8.37 (1 H, d, 3JH–H = 8.9 Hz, C(8)H), 8.04 (1 H, dd, 3JH–H = 8.9, 4JH–H = 1.8 Hz, C(7)H), 8.00 (1 H, s, C(3)H), 7.20 (1 H, t, 2JH–F = 54.0 Hz, C(4-CHF2)H), 6.82 (1 H, t, 2JH–F = 54.9 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −62.70 (3 F, s, CF3), −114.48 (2 F, d, 2JF–H = 54.0 Hz, C2-CHF2), −114.98 (2 F, d, 2JF–H = 54.9 Hz, C4-CHF2) ppm. 13C NMR δC = 154.90 (t, 2JC–F = 27.4 Hz, C-2), 148.77 (s, C-8-C-N), 141.09 (t, 2JC–F = 22.5 Hz, C-4), 132.14 (s, C-8), 131.11 (q, 2JC–F = 33.1 Hz, C-6), 126.76 (q, 3JC–F = 2.9 Hz, C-7), 123.95 (t, 3JC–F = 2.5 Hz, C-5-C-C-4), 123.65 (q, 1JC–F = 272.9 Hz, CF3), 121.81 (s, C-5), 115.71 (tt, 3JC–F = 7,6, 3JC–F = 2,2 Hz, C-3), 113.94 (t, 1JC–F = 243.4 Hz, C(2)CHF2), 112.93 (t, 1JC–F = 241.9 Hz, C(4)CHF2) ppm. C12H6F7N (297): calcd (%) N 4.71, C 48.50, H 2.04, found N 4.78, C 48.51, H 2.17. MP: 57.7–58.3 °C.
2,4-Bis(difluoromethyl)-7-(trifluoromethyl)quinoline 3q.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.56 mL, 4.79 mmol) by (BF3·Et2O) (1.2 equiv., 0.606 mL, 4.79 mmol) and N-(1,1-difluoropropan-2-ylidene)-3-(trifluoromethyl)aniline 1qq (1 equiv., 0.946 g, 3.99 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-7-(trifluoromethyl)quinoline 3q was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a colourless solid (328.5 mg, 28%). 1H NMR δH = 8.56 (1 H, s, C(8)H), 8.31 (1 H, d, 3JH–H = 8.9 Hz, C(5)H), 8.02 (1 H, s, C(3)H), 7.93 (1 H, d, 3JH–H = 8.7 Hz, C(6)H), 7.19 (1 H, t, 2JH–F = 54.1 Hz, C(4-CHF2)H), 6.82 (1 H, t, 2JH–F = 54.9 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −63.10 (3 F, s, CF3), −114.71 (2 F, d, 2JF–H = 56.4 Hz, C2-CHF2), −114.81 (2 F, d, 2JF–H = 54.8 Hz, C4-CHF2) ppm. 13C NMR δC = 154.19 (t, 2JC–F = 27.5 Hz, C-2), 146.86 (s, C-8-C-N), 140.20 (t, 2JC–F = 22.6 Hz, C-4), 132.69 (q, 2JC–F = 33.7 Hz, C-7), 128.43 (s, C-8), 126.15 (s, C-5-C-C-4), 125.08 (s, C-5), 124.85 (s, C-6), 123.54 (q, 1JC–F = 272.4 Hz, CF3), 116.20 (t, 3JC–F = 7.6 Hz, C-3), 114.02 (t, 1JC–F = 242.3 Hz, C(2)CHF2), 112.98 (t, 1JC–F = 241.5 Hz, C(4)CHF2) ppm. C12H6F7N (297): calcd (%) N 4.71, C 48.50, H 2.04, found N 4.74, C 48.35, H 2.10. MP: 39.7–40 °C.
2,4-Bis(difluoromethyl)-8-(trifluoromethyl)quinoline 3r.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.178 mL, 1.52 mmol) by (BF3·Et2O) (1.2 equiv., 0.192 mL, 1.52 mmol) and N-(1,1-difluoropropan-2-ylidene)-2-(trifluoromethyl)aniline 1rr (1 equiv., 0.5 g, 1.26 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-8-(trifluoromethyl)quinoline 3r was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a colourless solid (traces).1H NMR δH = 8.37 (1 H, d, 2JH–H = 8.4 Hz, C(7)H), 8.22 (1 H, d, 2JH–H = 7.3 Hz, C(5)H), 8.02 (1 H, s, C(3)H), 7.81 (1 H, t, 3JH–H = 7.9 Hz, C(6)H), 7.18 (1 H, t, 2JH–F = 54.1 Hz, C(4-CHF2)H), 6.84 (1H, t, 2JH–F = 54.9 Hz, C(2-CHF2)H). 19F NMR δF = −60.17 (3 F, s, CF3), −114.25 (2 F, d, 2JF–H = 56.4 Hz, C2-CHF2), −114.36 (2 F, d, 2JF–H = 54.8 Hz, C4-CHF2) ppm.
2,4-Bis(difluoromethyl)-8-phenylquinoline 3s.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.403 mL, 3.45 mmol) by (BF3·Et2O) (1.2 equiv., 0.437 mL, 3.45 mmol) and N-(1,1-difluoropropan-2-ylidene)-2-phenylaniline 1ss (1 equiv., 704 mg, 2.87 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-8-phenylquinoline 3s was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a colourless solid (248.9 mg, 28%). 1H NMR δH = 8.16 (1 H, dd, 3JH–H = 8.5, 4JH–H = 1.5 Hz, C(5)H), 8.00 (1 H, s, C(3)H), 7.92 (1 H, dd, 3JH–H = 7.2, 4JH–H = 1.3 Hz, C(7)H), 7.85–7.77 (1 H, m, C(6)H), 7.76–7.70 (2 H, m, C(5′)H), 7.58–7.45 (3 H, m, C(6′)H), 7.24 (1 H, t, 2JH–F = 54.3 Hz, C(4-CHF2)H), 6.74 (1 H, t, 2JH–F = 55.1 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −113.70 (2 F, d, 2JF–H = 55.1 Hz, C2-CHF2), −114.97 (2 F, d, 2JF–H = 54.3 Hz, C4-CHF2) ppm. 13C NMR δC = 152.18 (t, 2JC–F = 27.8 Hz, C-2), 145.30 (s, C-8-C-N), 142.28 (s, C-1′), 140.04 (t, 2JC–F = 22.0 Hz, C-4), 138.65 (s, C-8), 131.61 (s, C-7), 131.02 (2 C, s, C-3′,5′), 129.09 (s, C-6), 128.06 (2 C, s, C-2′,6′), 127.84 (s, C-4′), 125.29 (s, C-5-C-C-4), 122.83 (s, C-5), 114.68 (t, 1JC–F = 240.9 Hz, C(2)CHF2), 114.17 (t, 3JC–F = 8.0 Hz, C-3), 113.25 (t, 1JC–F = 242.4 Hz, C(4)CHF2) ppm. C1H11F4N (305): calcd (%) N 4.59, C 66.89, H 3.63, found N 4.71, C 66.99, H 3.70. MP: 58.6–59.5 °C.
2,4-Bis(difluoromethyl)-8-(phenylsulfanyl)quinoline 3t.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.253 mL, 2.16 mmol) by (BF3·Et2O) (1.2 equiv., 0.274 mL, 2.16 mmol) and N-(1,1-difluoropropan-2-ylidene)-2-(phenylsulfanyl)aniline 1tt (1 equiv., 500 mg, 1.8 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)-8-(phenylsulfanyl)quinoline 3t was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a colourless solid (308.7 mg, 52%). 1H NMR δH = 7.99 (1 H, s, C(3)H), 7.82 (1 H, d, 3JH–H = 8.4 Hz, C(5)H), 7.66 (2 H, dd, 3JH–H = 6.4, 4JH–H = 3.0 Hz, C(2′,6′)H), 7.53–7.49 (3 H, m, C(3′,4′,5′)H), 7.47 (1 H, t, 3JH–H = 8.0 Hz, C(6)H), 7.17 (1 H, t, 2JH–F = 54.3 Hz, C(4-CHF2)H), 7.06 (1 H, d, 3JH–H = 7.6 Hz, C(7)H), 6.89 (1 H, t, 2JH–F = 54.9 Hz, C(2-CHF2)H) ppm. 19F NMR δF = −113.66 (2 F, d, 2JF–H = 55.0 Hz, C2-CHF2), −115.22 (2 F, d, 2JF–H = 54.2 Hz, C4-CHF2) ppm. 13C NMR δC = 151.47 (t, 2JC–F = 27.7 Hz, C-2), 143.85 (s, C-8-C-N), 143.02 (s, C-8), 140.20 (t, 2JC–F = 22.2 Hz, C-4), 136.16 (2 C, s, C-2′,6′), 130.89 (s, C-1′), 130.11 (2 C, s, C-3′,5′), 129.64 (s, C-4′), 129.33 (s, C-6), 126.16 (s, C-7), 125.13 (s, C-5-C-C-4), 119.37 (s, C-5), 114.86 (t, 3JC–F = 7.9 Hz, C-3), 114.31 (t, 1JC–F = 242.4 Hz, C(2)CHF2), 112.87 (t, 1JC–F = 241.4 Hz, C(4)CHF2). C17H11F4NS (337): calcd (%) N 4.15, C 60.53, H 3.29, found N 4.22, C 60.60, H 3.36. MP: 109.9–110.7 °C.
2,4-Bis(difluoromethyl)benzo[h]quinolin-7-ol 3u.
The product was prepared according to the general procedure, starting from an activated solution of TFEDMA 4a (1.2 equiv., 0.298 mL, 2.55 mmol) by (BF3·Et2O) (1.2 equiv., 0.323 mL, 2.55 mmol) and 5-[(1,1-difluoropropan-2-ylidene)amino]-1-naphthol 1uu (1 equiv., 500 mg, 2.13 mmol) in the presence of anhydrous acetonitrile. 2,4-Bis(difluoromethyl)benzo[h]quinolin-7-ol 3u was provided after purification using a gradient of ethyl acetate in pentane (0–5%) as a brown solid (321.7 mg, 51%). 1H NMR δH = 8.90 (1 H, d, 3JH–H = 8.3 Hz, C(10)H), 8.39 (1 H, d, 3JH–H = 9.4 Hz, C(5)H), 8.04 (1 H, s, C(3)H), 7.93 (1 H, d, 3JH–H = 9.4 Hz, C(6)H), 7.61 (1 H, t, 3JH–H = 8.0 Hz, C(9)H), 7.25 (1 H, t, 2JH–F = 54.4 Hz, C(4-CHF2)H), 7.11 (1 H, d, 3JH–H = 7.6 Hz, C(8)H), 6.92 (1 H, t, 2JH–F = 55.3 Hz, C(2-CHF2)H), 5.43 (1 H, br s, OH) ppm. 19F NMR δF = −113.93 (2 F, d, 2JF–H = 55.3 Hz, C2-CHF2), −114.96 (2 F, d, 2JF–H = 54.4 Hz, C4-CHF2) ppm. 13C NMR δC = 151.71 (s, C-7), 151.27 (t, 2JC–F = 27.2 Hz, C-2), 146.29 (s, C-10-C-C-N), 139.47 (t, 2JC–F = 22.2 Hz, C-4), 132.81 (2 C, s, C-5-C-C-4/C-10-C-CN), 128.23 (s, C-9), 124.51 (s, C-5), 123.57 (s, C-7-C-C-6), 118.94 (s, C-6), 117.77 (s, C-10), 114.73 (tt, 3JC–F = 7.6, 3JC–F = 2.2 Hz, C-3), 114.60 (t, 1JC–F = 241.0 Hz, C(2)CHF2), 113.24 (s, C-5), 113.06 (t, 1JC–F = 242.4 Hz, C(4)CHF2) ppm. C15H9F4NO (295): calcd (%) N 4.74, C 61.02, H 3.07, found N 4.82, C 60.87, H 3.14. MP: 139.5–141.2 °C.
Functionalization in position 8 of quinoline derivatives
8-(Bromomethyl)-2,4-bis(difluoromethyl)-7-fluoroquinoline 7.
Method A to a solution of 2,4-bis(difluoromethyl)-7-fluoro-8-methylquinoline 3m (1 equiv., 300 mg, 1.15 mmol) and oxone (2 equiv., 349 mg, 2.3 mmol) in a mixture of DCM (2.7 mL) and H2O (0.3 mL) (9:1) was added KBr (2 equiv., 273 mg, 2.3 mmol) at room temperature, and stirred under visible light irradiation for 14 h. Upon addition of KBr, the reaction mixture became brown, and then turned into a colourless suspension after 10 min under light. A saturated aq. solution of Na2S2O3 was added to the reaction mixture, and the product was extracted with ethyl acetate. The combined extracts were washed with brine and dried over Na2SO4. The organic phase was concentrated under reduced pressure.
Method B to a mixture of 2,4-bis(difluoromethyl)-7-fluoro-8-methylquinoline 3m (1 equiv., 300 mg, 1.15 mmol), NaIO4 (1.5 equiv., 368 mg, 1.72 mmol) and LiBr (1.5 equiv., 149 mg, 1.72 mmol) was added aq. H2SO4 (2%) (6 mL). The reaction mixture was heated at 95 °C for 6 h. It was then cooled to room temperature and extracted with ethyl acetate. The combined organic layers were washed with a saturated aq. solution of Na2S2O3 and then water, dried over anhydrous Na2SO4 and concentrated under reduced pressure. In both cases the crude reaction mixture was purified by column chromatography using a gradient of ethyl acetate in cyclohexane (0–15%).
8-(bromomethyl)-2,4-bis(difluoromethyl)-7-fluoroquinoline 7 was obtained as a colourless solid (194 mg) (Method A: 63%/Method B: 50%). 1H NMR δH = 8.17 (1H, dd, 3JH–H = 9.4, 4JH–F = 5.7 Hz, C(5)H), 7.93 (1 H, s, C(3)H), 7.55 (1 H, t, 3JH–H = 9.1 Hz, C(6)H), 7.14 (1 H, t, 2JH–F = 54.1 Hz, C(4-CHF2)H), 6.86 (1 H, t, 2JH–F = 55.0 Hz, C(2-CHF2)H), 5.22 (2 H, d, 4JH–F = 1.6 Hz, C(8-CH2Br)H). 19F NMR δF = −106.81 to −106.85 (1 F, m, F), −114.12 (2 F, d, 2JH–F = 54.1 Hz, C2-CHF2), −114.52 (2 F, d, 2JH–F = 55.0 Hz, C4-CHF2). 13C NMR δC = 161.55 (d, 1JC–F = 257.1 Hz, C-7), 153.60 (t, 2JC–F = 27.7 Hz, C-2), 146.39 (d, 3JC–F = 8.3 Hz, C-8-C-N), 140.59 (td, 2JC–F = 22.5, 5JC–F = 1.5 Hz, C-4), 125.95 (d, 2JC–F = 10.7 Hz, C-8), 122.95 (d, 3JC–F = 13.7 Hz, C-5), 122.14 (s, C-5-C-C-4), 119.74 (d, 2JC–F = 25.7 Hz, C-6), 114.26 (t, 1JC–F = 242.4 Hz, C(2)CHF2), 114.36–114.18 (m, C-3), 113.20 (t, 1JC–F = 241.7 Hz, C(4)CHF2), 20.07 (d, 3JC–F = 4.6 Hz, C(8)CH2Br). C12H7F5NBr (340): calcd (%) N 4.12, C 42.38, H 2.07, found N 4.22, C 42.33, H 2.68.
8-(Bromomethyl)-7-chloro-2,4-bis(difluoromethyl)quinoline 8.
Method A the same previous procedure was used on 7-chloro-2,4-bis(difluoromethyl)-8-methylquinoline 3n (1 equiv., 300 mg, 1.08 mmol).
Method B the same previous procedure was used on 7-chloro-2,4-bis(difluoromethyl)-8-methylquinoline 3n (1 equiv., 302 mg, 1.09 mmol). In both cases the crude reaction mixture was purified by column chromatography using a gradient of ethyl acetate in cyclohexane (0–15%).
8-(Bromomethyl)-7-chloro-2,4-bis(difluoromethyl)quinoline 8 was obtained as a colourless solid (263 mg) (Method A: 68%/Method B: 55%). 1H NMR δH = 8.08 (1 H, d, 3JH–H = 9.1 Hz, C(5)H), 7.96 (1 H, s, C(3)H), 7.75 (1 H, d, 3JH–H = 9.1 Hz, C(6)H), 7.14 (1 H, t, 2JH–F = 54.1 Hz, C(4-CHF2)H), 6.85 (1 H, t, 2JH–F = 54.9 Hz, C(2-CHF2)H), 5.36 (1 H, s, C(8-CH2Br)H). 19F NMR δF = −114.27 to −114.56 (4 F, m, C2-CHF2/C4-CHF2). 13C NMR δC = 153.37 (t, 2JC–F = 27.8 Hz, C-2), 145.97 (s, C-8-C-N), 140.63 (t, 2JC–F = 22.4 Hz, C-4), 137.78 (s, C-7), 135.37 (s, C-8), 131.05 (s, C-6), 124.80 (s, C-5), 123.76 (s, C-5-C-C-4), 115.04–114.85 (m, C-3), 114.21 (t, 1JC–F = 242.4 Hz, C(2)CHF2), 113.05 (t, 1JC–F = 241.8 Hz, C(4)CHF2), 25.08 (s, C(8)CH2Br). C12H7F4NBrCl (356): calcd (%) N 3.93, C 40.42, H 1.98 found N 3.78, C 40.78, H 2.18. MP: 113.3–114 °C.
7-Chloro-2,4-bis(difluoromethyl)quinoline-8-carboxylic acid 9.
To a solution of 8-(bromomethyl)-7-chloro-2,4-bis(difluoromethyl)quinoline 8 (1 equiv., 370 mg, 1.04 mmol) in aq. H2SO4 (70%) (3962 mg) at 110 °C was added aq. HNO3 (65%) (321 mg) dropwise. CAUTION: a release of brown toxic gas was observed. The reaction mixture was stirred for 17 h, then it was cooled down and poured into ice. The precipitate was filtered off, washed with water and dried under vacuum. The crude was triturated into pentane to remove all impurities. 7-Chloro-2,4-bis(difluoromethyl)quinoline-8-carboxylic acid 9 was obtained as a beige solid (77.7 mg, 64%). 1H NMR δH = 9.55 (1 H, br s, OH), 8.22 (1 H, d, 3JH–H = 9.0 Hz, C(5)H), 8.00 (1 H, s, C(3)H), 7.84 (1 H, d, 3JH–H = 9.1 Hz, C(6)H), 7.18 (1 H, t, 2JH–F = 53.9 Hz, C(4-CHF2)H), 6.84 (1 H, t, 2JH–F = 54.7 Hz, C(2-CHF2)H). 19F NMR δF = −114.47 (2 F, d, 2JH–F = 53.9 Hz, C2-CHF2), −115.22 (2 F, d, 2JH–F = 54.7 Hz, C4-CHF2). 13C NMR δC = 167.41 (s, C(8)CO2H), 153.70 (t, 2JC–F = 27.8 Hz, C-2), 145.41 (s, C-8-C-N), 141.30 (t, 2JC–F = 22.7 Hz, C-4), 137.17 (s, C-7), 131.93 (s, C-6), 130.18 (s, C-8), 126.57 (s, C-5), 123.41 (s, C-5-C-C-4), 115.48 (t, 3JC–F = 7.9 Hz, C-3), 113.30 (t, 1JC–F = 243.4 Hz, C(2)CHF2), 112.74 (t, 1JC–F = 242.3 Hz, C(4)CHF2). C12H7F4NBrCl (356): calcd (%) N 4.55, C 46.80, H 1.95, found N 4.56, C 46.41, H 2.06. MP: 158–159 °C.
7-Chloro-2,4-bis(difluoromethyl)quinoline-8-carbonitrile 10 and tris({[7-chloro-2,4-bis(difluoromethyl)quinolin-8-yl]methyl})amine 11.
To a solution of 7-chloro-2,4-bis(difluoromethyl)-8-methylquinoline 3n (1 equiv., 300 mg, 1.08 mmol) and aq. HBr (48%) (3 equiv., 0.368 mL, 3.24 mmol) in CCl4 (4.5 mL) was added dropwise aq. H2O2 (30%) (2 equiv., 0.17 mL, 2.16 mmol) at room temperature. The suspension became red and the reaction mixture was stirred under a sunlamp for 1 h. Then, acetonitrile (2 mL), aq. NH3 (35%) (50.2 equiv., 3 mL, 54.3 mmol), and I2 (2.5 equiv., 685 mg, 2.7 mmol) were added to the mixture at room temperature, and stirred overnight at 60 °C. The reaction mixture was cooled down to room temperature and quenched by a saturated aq. solution of Na2S2O3. The mixture was extracted with DCM and the combined organic layers were washed with water and brine, then dried over Na2SO4 and concentrated under vacuum. 7-Chloro-2,4-bis(difluoromethyl)quinoline-8-carbonitrile 10 was isolated after purification using ethyl acetate in cyclohexane (0–15%) as a yellow solid (91 mg, 29%). 1H NMR δH = 8.36 (1 H, d, 3JH–H = 9.1 Hz, C(5)H), 8.03 (1 H, s, C(3)H), 7.85 (1 H, d, 3JH–H = 9.1 Hz, C(6)H), 7.14 (1 H, t, 2JH–F = 51.0 Hz, C(4-CHF2)H), 6.88 (1 H, t, 2JH–F = 51.7 Hz, C(2-CHF2)H). 19F NMR δF = −113.56 (2 F, d, 2JH–F = 53.8 Hz, C2-CHF2), −114.48 (2 F, d, 2JH–F = 54.6 Hz, C4-CHF2). 13C NMR δC = 155.77 (t, 2JC–F = 28.3 Hz, C-2), 147.88 (s, C-8-C-N), 142.63 (s, C-7), 141.07 (t, 2JC–F = 22.9 Hz, C-4), 130.33 (s, C-6), 129.10 (s, C-5), 123.23 (s, C-5-C-C-4), 116.48 (t, 3JC–F = 7.9 Hz, C-3), 114.62 (s, C(8)CN), 113.66 (t, 1JC–F = 242.9 Hz, C(2)CHF2), 113.55 (s C-8), 112.89 (t, 1JC–F = 242.3 Hz, C(4)CHF2). HRMS (ESI+) for C12H6F4N2Cl [M + H]: calcd 289.0150, found 289.0164. MP: 116–116.5 °C.
Tris({[7-chloro-2,4-bis(difluoromethyl)quinolin-8-yl]methyl})amine 11 was isolated during the purification of 10 as a yellow solid (94.5 mg, 10%). 1H NMR δH = 7.95 (3 H, d, 3JH–H = 9.1 Hz, C(5)H), 7.82 (3 H, s, C(3)H), 7.57 (3 H, d, 3JH–H = 9.1 Hz, C(6)H), 7.11 (3 H, t, 2JH–F = 54.2 Hz, C(4-CHF2)H), 6.39 (3 H, t, 2JH–F = 55.1 Hz, C(2-CHF2)H), 4.49 (6 H, s, C(8-CH2)H). 19F NMR δF = −114.15 (6 F, d, 2JH–F = 55.1 Hz, C2-CHF2), −114.67 (6 F, d, 2JH–F = 54.2 Hz, C4-CHF2).13C NMR δC = 152.45 (3 C, t, 2JC–F = 27.7 Hz, C-2), 147.95 (3 C, s, C-8-C-N), 140.13 (3 C, t, 2JC–F = 22.3 Hz, C-4), 139.25 (3 C, s, C-7), 136.72 (3 C, s, C-8), 130.92 (3 C, s, C-6), 123.33 (3 C, s, C-5), 123.01 (3 C, s, C-5-C-C-4), 114.28 (3 C, t, 1JC–F = 242.4 Hz, C(2)CHF2), 113.96 (3 C, t, 3JC–F = 7.9 Hz, C-3), 113.08 (3 C, t, 1JC–F = 241.4 Hz, C(4)CHF2), 50.30 (3 C, s, C(8)CH2). HRMS (ESI+) for C36H22F12N4Cl3 [M + H]: calcd 843.0713, found 843.0716. MP: 120–121 °C.
8-(Benzenesulfinyl)-2,4-bis(difluoromethyl)quinoline 13.
FeCl3 (0.03 equiv., 1.59 mg, 0.00979 mmol) and 2,4-bis(difluoromethyl)-8-(phenylsulfanyl)quinoline 3t (1 equiv., 110 mg, 0.326 mmol) were dissolved in acetonitrile (1 mL) and stirred for 5 min. To this solution was added H5IO6 (1.1 equiv., 81.8 mg, 0.359 mmol) at once. The yellow suspension was stirred at room temperature for 3 h. The reaction was quenched by the addition of a saturated aq. solution of Na2S2O3 and extracted with DCM. Organic layers were dried over Na2SO4 and concentrated under vacuum. 8-(Benzenesulfinyl)-2,4-bis(difluoromethyl)quinoline 13 was obtained as a sticky brown solid (91.2 mg, 79%). 1H NMR δH = 8.61 (1 H, dd, 3JH–H = 7.3, 4JH–H = 1.2 Hz, C(7)H), 8.22 (1 H, dd, 3JH–H = 8.5, 4JH–H = 1.3 Hz, C(6)H), 7.96 (1 H, d, 3JH–H = 7.5 Hz, C(5)H), 7.93 (1 H, s, C(3)H), 7.91–7.87 (2 H, m, C(2′,6′)H), 7.41–7.31 (3 H, m, C(3′,4′,5′)H), 7.14 (1 H, t, 2JH–F = 54.1 Hz, C(4-CHF2)H), 6.81 (1 H, t, 2JH–F = 55.0 Hz, C(2-CHF2)H). 19F NMR δF = −113.77 to −115.99 (2 F, m, C4-CHF2), −113.36 to −117.48 (2 F, m, C4-CHF2). 13C NMR δC = 152.39 (dd, 2JC–F = 28.2, 26.2 Hz, C-2), 145.60 (s, C-8-C-N), 145.42 (s, C-1′), 143.42 (s, C-8), 140.52 (t, 2JC–F = 22.5 Hz, C-4), 131.28 (s, C-4′), 129.60 (s, C-5), 129.09 (2 C, s, C-3′,5′), 126.66 (s, C-7), 125.93 (2 C, s, C-2′,6′), 124.93 (s, C-5-C-C-4), 115.39–115.08 (m, C-3), 113.45 (t, 1JC–F = 243.4 Hz, C(4)CHF2), 112.79 (t, 1JC–F = 241.9 Hz, C(2)CHF2). C17H11F4NOS (353): calcd (%) N 3.96, C 57.79, H 3.14, found N 3.94, C 57.64, H 3.32.
Acknowledgements
The CNRS (Centre National de la Recherche Scientifique), Bayer Cropscience AG (Monheim) and Bayer S.A.S (Lyon) are deeply acknowledged for their financial support. The analytical platform of the University of Strasbourg is warmly thanked for help with mass spectroscopy, elemental analysis and crystallography.
Notes and references
-
J. P. Bégué and D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, John Wiley & Sons, Hoboken, New Jersey, 2008 Search PubMed.
-
I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, 2009 Search PubMed.
-
P. Jeschke, in Modern Methods in Crop Protection Research, ed. P. Jeschke, W. Krämer, U. Schirmer and M. Witschel, Wiley-VCH, Weinheim, Germany, 2012, ch. 4, pp. 73–128 Search PubMed.
- P. Jeschke, ChemBioChem, 2004, 5, 571–589 CrossRef PubMed.
- P. Jeschke, E. Baston and F. R. Leroux, Mini-Rev. Med. Chem., 2007, 7, 1027–1034 CrossRef CAS PubMed.
- K. L. Kirk, Org. Process Res. Dev., 2008, 12, 305–321 CrossRef CAS.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- D. O′Hagan and D. B. Harper, J. Fluorine Chem., 1999, 100, 127–133 CrossRef.
-
K. Uneyama, Fundamentals in Organic Fluorine Chemistry, Blackwell, Oxford, 2006 Search PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- P. Jeschke, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS PubMed.
-
G. Theodoridis, in Advances in Fluorine Science, ed. A. Tressaud, Elsevier B.V., 2006, vol. 2, ch. 4, pp. 120–175 Search PubMed.
-
J.-P. Bégué, in Chimie bioorganique et médicinale du fluor, E. C. ÉDITIONS, 2005, p. 384 Search PubMed.
- M. M. Ghorab, F. A. Ragab, H. I. Heiba, R. K. Arafa and E. M. El-Hossary, Med. Chem. Res., 2010, 20, 388–400 CrossRef.
- F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827–856 CrossRef CAS PubMed.
-
P. V. Reddy, in Organofluorine Compounds in Biology and Medicine, Elsevier, 2015, pp. 1–27 Search PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- A. M. Thayer, Chem. Eng. News, 2006, 84, 15–24 Search PubMed.
-
The Pesticide Manual: A World Compendium, British Crop Protection Council, Alton edn, 2012 Search PubMed.
- S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchal and H. D. Patel, RSC Adv., 2014, 4, 24463–24476 RSC.
- Y. Li and W. Gao, Beilstein J. Org. Chem., 2010, 6, 966–972 CrossRef CAS PubMed.
- H. Kumar, A. Chattopadhyay, R. Prasath, V. Devaraji, R. Joshi, P. Bhavana, P. Saini and S. K. Ghosh, J. Phys. Chem. B, 2014, 118, 7257–7266 CrossRef CAS PubMed.
- G. Shan, X. Sun, Q. Xia and Y. Rao, Org. Lett., 2011, 13, 5770–5773 CrossRef CAS PubMed.
- R. E. Lutz, C. J. Ohnmacht and A. R. Patel, J. Med. Chem., 1971, 14, 926–928 CrossRef CAS.
- J. Mulero, G. Martinez, J. Oliva, S. Cermeno, J. M. Cayuela, P. Zafrilla, A. Martinez-Cacha and A. Barba, Food Chem., 2015, 180, 25–31 CrossRef CAS PubMed.
- M. Kidwai, K. R. Bhushan, P. Sapra, R. K. Saxena and R. Gupta, Bioorg. Med. Chem., 2000, 8, 69–72 CrossRef CAS PubMed.
- Y.-M. Wu, Y. Li and J. Deng, J. Fluorine Chem., 2006, 127, 223–228 CrossRef CAS.
- A. R. Surrey and H. F. Hammer, J. Am. Chem. Soc., 1946, 68, 113–116 CrossRef CAS PubMed.
- W. S. Johnson and B. G. Buell, J. Am. Chem. Soc., 1952, 74, 4513–4516 CrossRef CAS.
-
V. Nenajdenko, in Fluorine in Heterocyclic Chemistry, ed. V. Nenajdenko, Springer International Publishing, 2014, vol. 2 Search PubMed.
- V. A. Petrov, S. Swearingen, W. Hong and W. Chris Petersen, J. Fluorine Chem., 2001, 109, 25–31 CrossRef CAS.
- N. N. Yarovenko and M. A. Raksha, Zh. Obshch. Khim., 1959, 29, 2159–2163 CAS.
- N. N. Yarovenko and M. A. Raksha, J. Gen. Chem. USSR (Engl. Transl.), 1959, 29, 2125–2128 Search PubMed.
- A. Takaoka, H. Iwakiri and N. Ishikawa, Bull. Chem. Soc. Jpn., 1979, 52, 3377–3380 CrossRef CAS.
- V. A. Petrov, Adv. Org. Synth., 2006, 2, 269–290 CAS.
- A. Vilsmeier and A. Haack, Ber. Dtsch. Chem. Ges., 1927, 60, 119–122 CrossRef.
- C. Wakselman and M. Tordeux, J. Chem. Soc., Chem. Commun., 1975, 956–956 RSC.
-
S. Pazenok, N. Lui and A. Neeff, (Bayer CropScience AG), WO Pat., 2008022777, 2008 Search PubMed.
- A. Takaoka, K. Iwamoto, T. Kitazume and N. Ishikawa, J. Fluorine Chem., 1979, 14, 421–428 CrossRef CAS.
- E. Schmitt, B. Rugeri, A. Panossian, J.-P. Vors, S. Pazenok and F. R. Leroux, Org. Lett., 2015, 17, 4510–4513 CrossRef CAS PubMed.
- F. Giornal, G. Landelle, N. Lui, J.-P. Vors, S. Pazenok and F. R. Leroux, Org. Process Res. Dev., 2014, 18, 1002–1009 CrossRef CAS.
- F. Giornal, S. Pazenok, L. Rodefeld, N. Lui, J.-P. Vors and F. R. Leroux, J. Fluorine Chem., 2013, 152, 2–11 CrossRef CAS.
-
S. Pazenok, N. Lui, A. Neeff, W. Etzel, J.-P. Vors, F. Leroux, G. Landelle and M. J. Ford, (Bayer CropScience AG), WO Pat., 2014187773, 2014 Search PubMed.
- E. Schmitt, G. Landelle, J.-P. Vors, N. Lui, S. Pazenok and F. R. Leroux, Eur. J. Org. Chem., 2015, 6052–6060 CrossRef CAS.
-
S. Pazenok, N. Lui, C. Funke, W. Etzel and A. Neeff, (Bayer CropScience AG), WO Pat., 2015144578, 2015 Search PubMed.
- E. Schmitt, A. Panossian, J.-P. Vors, C. Funke, N. Lui, S. Pazenok and F. R. Leroux, Chem. – Eur. J., 2016, 22, 11239–11244 CrossRef CAS PubMed.
-
K. T. Finley, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2000, p. 32 Search PubMed.
- T. L. Gilchrist, J. Chem. Soc., Perkin Trans. 1, 2001, 2491–2515 RSC.
- A. Roe and G. F. Hawkins, J. Am. Chem. Soc., 1949, 71, 1785–1786 CrossRef CAS.
- J. Hamer, W. J. Link, A. Jurjevich and T. L. Vigo, Recl. Trav. Chim. Pays-Bas, 1962, 81, 1058–1060 CrossRef CAS.
- R. D. Chambers, D. Holling, G. Sandford, H. Puschmann and J. A. K. Howard, J. Fluorine Chem., 2002, 117, 99–101 CrossRef CAS.
- R. D. Chambers, D. Holling, G. Sandford, A. S. Batsanov and J. A. K. Howard, J. Fluorine Chem., 2004, 125, 661–671 CrossRef CAS.
- M. Oishi, H. Kondo and H. Amii, Chem. Commun., 2009, 1909–1911 RSC.
- H. Yanai, H. Mimura, K. Kawada and T. Taguchi, Tetrahedron, 2007, 63, 2153–2160 CrossRef CAS.
- H. Keller and M. Schlosser, Tetrahedron, 1996, 52, 4637–4644 CrossRef CAS.
- D. M. Volochnyuk, A. O. Pushechnikov, D. G. Krotko, D. A. Sibgatulin, S. A. Kovalyova and A. A. Tolmachev, Synthesis, 2003, 1531–1540 CrossRef CAS.
- N. S. Boltacheva, V. I. Filyakova and V. N. Charushin, Russ. J. Org. Chem., 2005, 41, 1452–1457 CrossRef CAS.
- K. Uneyama, J. Fluorine Chem., 1999, 97, 11–25 CrossRef CAS.
- M. G. Mormino, P. S. Fier and J. F. Hartwig, Org. Lett., 2014, 16, 1744–1747 CrossRef CAS PubMed.
- B. Duda, S. N. Tverdomed, B. S. Bassil and G.-V. Röschenthaler, Tetrahedron, 2014, 70, 8084–8096 CrossRef CAS.
- J. C. Sloop, C. L. Bumgardner and W. D. Loehle, J. Fluorine Chem., 2002, 118, 135–147 CrossRef CAS.
- J. C. Sloop, J. Phys. Org. Chem., 2009, 22, 110–117 CrossRef CAS.
- F. Zhao, X. Yang and J. Liu, Tetrahedron, 2004, 60, 9945–9951 CrossRef CAS.
- S. Perrone, F. Rosato, A. Salomone and L. Troisi, Tetrahedron, 2013, 69, 3878–3884 CrossRef CAS.
- A. Combes, Bull. Soc. Chim. Fr., 1888, 49, 89–92 Search PubMed.
- CCDC 1487974 (3aiii) and 1487975 (9) contain the supplementary crystallographic data for this paper.
-
B. Wuerzer and R. Berghaus, presented in part at the 10th Asian-Pacific Weed Science Society Conference, Part. 1, Thailand, 1985.
-
H. Hagen, R.-D. Kohler, J. Markert and B. Wuerzer, (BASF AG), DE Pat., 3233089, 1984 Search PubMed.
- K. Moriyama, M. Takemura and H. Togo, Org. Lett., 2012, 14, 2414–2417 CrossRef CAS PubMed.
- T. M. A. Shaikh, L. Emmanuvel and A. Sudalai, J. Org. Chem., 2006, 71, 5043–5046 CrossRef CAS PubMed.
- Y. Kawagoe, K. Moriyama and H. Togo, Eur. J. Org. Chem., 2014, 4115–4122 CrossRef CAS.
- S. Kim, K. Nehru, S. S. Kim, D. W. Kim and H. C. Jung, Synthesis, 2002, 2484–2486 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1487974 and 1487975. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qo00319b |
|
| This journal is © the Partner Organisations 2016 |

 *ad
*ad