
Supramolecular radical polymers self-assembled from the stacking of radical cations of rod-like viologen di- and trimers†
Tian-Guang
Zhan
ac,
Tian-You
Zhou
a,
Feng
Lin
a,
Liang
Zhang
b,
Cen
Zhou
a,
Qiao-Yan
Qi
a,
Zhan-Ting
Li
b and
Xin
Zhao
*a
aKey Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai, 200032, China. E-mail: xzhao@mail.sioc.ac.cn
bDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China
cCollege of Chemistry and Life Science, Zhejiang Normal University, Jinhua 321004, P. R. China
First published on 6th September 2016
Abstract
A series of π-conjugated oligomeric viologens (COVs) whose viologen segments are connected by phenyl or biphenyl units have been designed and synthesized. They exhibited interesting electrochemical and optical properties which are different from isolated viologen, as revealed by cyclic voltammetric and spectroscopic studies. The self-assembly behavior of the COV radical cations generated by treating COVs with sodium dithionite in aqueous media was studied systematically with UV-vis-NIR, electron paramagnetic resonance (EPR), dynamic light scattering (DLS), and cryo-transmission electron microscopy (cryo-TEM), which revealed that they self-assembled into linear supramolecular radical polymers driven by the dimerization of radical cations. Comprehensive DFT calculations for the COVs and their radical cations were also performed and their structure–property relationships were revealed.
Introduction
1,1′-Disubstituted-4,4′-bipyridylium (BIPY) salts, also known as “viologens” (V2+), have attracted a great deal of attention over the past few decades because of their highly electron-deficient character and excellent electrochemical properties, which have been widely utilized in the fields of supramolecular chemistry and materials science. Using viologen-based building blocks, a myriad of systems including interlocked supramolecules,1 molecular machines,2 host–guest complexes,3 sensors,4 solar cell ptototype,5 and organic electrochromic materials,6 as well as supramolecular polymers,7 supramolecular cross-linked networks,8 and supramolecular organic frameworks9 have been successfully constructed.On the other hand, distinctive structures bearing multiple viologen units such as viologen oligomers,10 dendrimers11 or polymers12 have also been reported recently. These multi-viologen-based structures possess multiple interaction sites and thus are very useful in fabricating highly complicated supramolecular architectures. However, since the viologen units in these compounds were connected by unconjugated spacers, such as alkyl or oligoglycol chains, it is not surprising that they all exhibited similar electrochemical and optical properties as that of pristine viologen. Replacing unconjugated spacers with conjugated ones will not only provide an efficient approach to tune the electrochemical and optical properties of viologen oligomers, but also bring them rigid and pre-organization structures that were benefit for further self-assembly.13 Very recently, a series of macrocyclic molecules, ExBox or Ex2Box,14 TVBox15 and Ex-cage,16 containing extended conjugated pyridylium units were reported by Stoddart and co-workers, which not only showed excellent application in encapsulating various electron rich guest molecules, but also displayed unique electron transfer properties,17 even in the field of induced-fit catalysis.18
It is well-known that a viologen unit could be reduced to a radical cation through a reversible one electron redox process. Radical cations of viologen derivatives have been known to undergo dimerization,19 also referred to as “pimerization”20 in aqueous media since the 1960s. It has been further demonstrated that the stacking interactions of radical cations, particularly those formed by bipyridinium or tetrathiafulvalene, could be enhanced remarkably when they were incorporated into preorganized frameworks or entrapped into a confined space.21 Stoddart et al. demonstrated that bipyridinium radical cations formed strong inclusion complexes with tetracationic cyclophane in its reduced diradical dicationic state (CBPQT˙2+) as a consequence of radical-paring interaction both in solution and in the solid state.22 In this context, dimerization or pimerization of BIPY˙+ has recently emerged as a new non-covalent force to construct various supramolecular architectures such as mechanical interlocked molecules,23 foldamers,24 and even semiconducting devices.25 Very recently Li and Zhao et al. have successfully utilized the dimerization of a viologen radical cation as the driving force to construct two- and three-dimensional supramolecular frameworks (SOFs) in aqueous media.26 However, to the best of our knowledge, the radical cation units involved in these building blocks are non-conjugated. We envisioned that building blocks containing conjugated multiple radical cations might bring novel stacking interactions and assembly properties.
To explore the features and applications of conjugated viologens, in this work, a series of conjugated oligomeric viologens (COVs) were constructed by connecting 4,4′-bipyridinium units by phenyl or biphenyl spacers (Scheme 1). Comprehensive studies on the properties of the COVs and DFT calculations indicated that the COVs not only showed different electrochemical and photophysical properties from the isolated viologen unit, but also exhibited interesting radical-dimerization-driven assembly behavior, on the basis of which linear supramolecular radical polymers have been fabricated in aqueous media. It should be noted that very recently Colquhoun, Greenland and co-workers also reported two linear oligomeric viologens in which viologen units were connected by methoxy-substituted biphenyl spacer(s).27 However, in those oligomers, the viologen units were electronically independent and the lack of conjugation was observed as a result of the significantly twisted conformations of the viologen oligomers and due to the steric hindrance between the methoxy groups and the α-hydrogen atoms of the pyridium segments. For the COVs reported herein, such steric hindrance is relieved and conjugation in the whole oligomers can be expected.
 | ||
| Scheme 1 The structures of the π-conjugated oligomeric viologens (COVs). | ||
Results and discussion
Synthetic procedures
The target compounds were prepared by the Zincke reaction, an effective method to synthesize 1,1′-diarylsubstituted-4,4′-bipyridylium compounds.28 As shown in Scheme 2, 1,4-benzenediamine was refluxed with excess Zincke salt 1 in ethanol to give compound 3 in 15% yield. The low yield might be attributed to the decreasing reactivity of the second amino group once the 4,4′-bipyridin-1-ium was generated. PV2 was then obtained by methylation of compound 3 with iodomethane in 70% yield after anion exchange with ammonium hexafluorophosphate. Following the same procedure but using excess 1,4-benzenediamine, monocation 4 could be obtained in 97% yield. Compound 4 further reacted with Zincke salt 2 in ethanol to generate compound 5, which was then refluxed with iodomethane in acetonitrile to give PV3. These procedures were also applied to the synthesis of BPV2 and BPV3 starting from 4,4′-biphenyldiamine. These compounds have been fully characterized by 1H and 13C NMR, and (HR) mass spectroscopy, on the basis of which their chemical structures were unambiguously assigned (see the ESI† for details).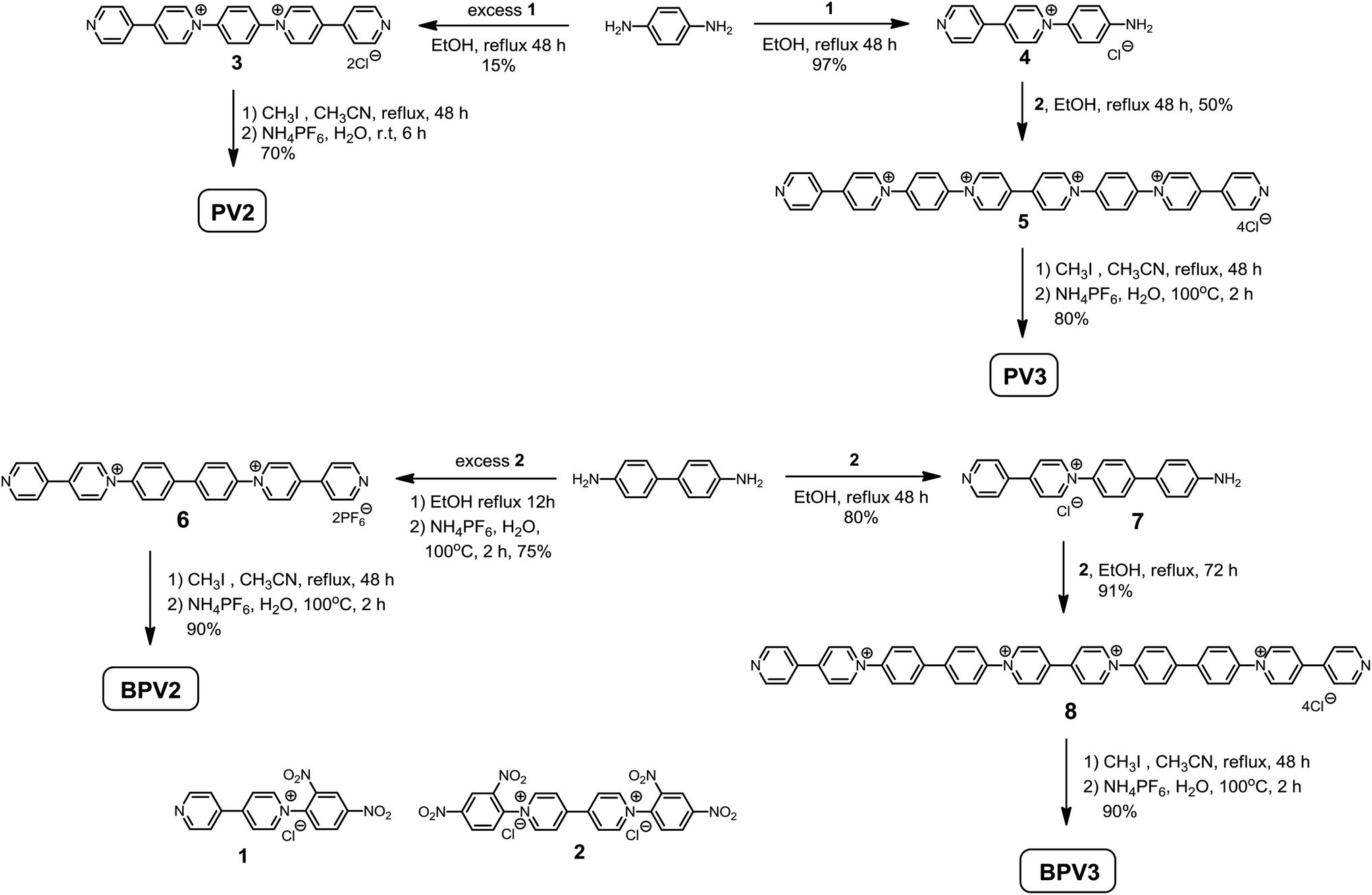 | ||
| Scheme 2 The synthetic routes for the COVs PV2, PV3, BPV2 and BPV3. | ||
Electrochemical properties
The electrochemical properties of these π-conjugated oligomeric viologens (COVs) were investigated by means of cyclic voltammetry (CV) both in CH3CN (using PF6 salts) and H2O (using chloride salts), respectively. n−Bu4NPF6 (0.1 M, for CH3CN solutions) or KCl (0.1 M, for aqueous solutions) were used as the supporting electrolyte and saturated calomel electrode (SCE) was used as a reference. A preliminary study revealed that no electrochemical process was observed in the range of positive potentials from 0 to +2.0 V for these COVs. Therefore, the subsequent CV experiments were conducted in the range of negative potentials from 0 to −2.0 V. The cyclic voltammograms are provided in Fig. 1, compared with the reference compound methylviologen (MV), the reduction waves of all the viologen oligomers and the oxidation peaks of their corresponding reduced species were considerably shifted to a less negative potential, suggesting an enhanced electron-accepting capability. These results could be attributed to the extended conjugation of the oligomers, which could stabilize the radical cations of the viologen units. This phenomenon is different from the viologen oligomers reported by Colquhoun et al.,27 in which the viologen units were electronically independent and thus they exhibited almost identical first reduction potentials. | ||
| Fig. 1 Cyclic voltammograms of MV, PV2, BPV2, PV3 and BPV3 recorded at a scan rate of 0.1 V s−1 (a) in CH3CN and (b) in H2O at 25 °C. The concentrations of the COVs and electrolyte were 1.0 mM and 0.1 M, respectively. | ||
Compared to MV, the cyclic voltammograms of the COVs in CH3CN were rather complicated and several irreversible redox processes were involved (Fig. 1a). It was also found that, the CVs of these COVs recorded in H2O (Fig. 1b) are simpler than those recorded in CH3CN to some extent, especially for the oligomers bearing a higher valence which are easier to be reduced to radical cations than those with a lower valence. For oligomers bearing the same valence, they displayed quite close first reduction potentials. For example, the first reduction peaks of PV2 and BPV2 in CH3CN are −0.30 and −0.32 V, respectively, suggesting that PV2 is easier to be reduced since it is more electron deficient than BPV2. They could be further reduced to neutral species at −0.60 V (for PV2) and −0.66 V (for BPV2), respectively. Moreover, in the CV spectrum of BPV2, four distinct one-electron processes were observed. In contrast, in the case of PV2, some of the reduction peaks merged into one. Hexavalent PV3 and BPV3 exhibited different behaviours. The first reduction peak of PV3 is −0.24 V and several reduction processes were observed. It could be further reduced to neutral species at −0.73 V. In the case of BPV3, its first reduction peak was −0.22 V and the reduction peaks merged into one at −0.64 V, at which BPV3 could be reduced to its neutral form. The values of their first reduction peaks suggested that BPV3 was easier to be reduced than PV3, although the latter exhibits a more electron-deficient structure. These results suggested that extending phenyl to biphenyl exerts influence on the first reduction potential of these viologen oligomers to a certain extent.
The CV performances of these oligomers under different scan rates (0.01 to 0.3 V s−1) were also investigated in CH3CN (Fig. S1†) and H2O (Fig. S2†), respectively. While the change of the scan rate exhibited little influence on reduction potentials, it exerted a considerable impact on the oxidation peaks of the reduced species in CH3CN. Interestingly, unlike in CH3CN, the reduction peaks of these compounds seem more sensitive to the scan rate in H2O. All the reduction peaks shifted to a more negative region when the scan rate increased. Two most distinct changes were observed. The first one is that the first reduction peak of PV3 shifted from −0.29 V to −0.41 V (Fig. S2b†); and the second one is that the two reduction peaks of BPV2 (−0.73 V and −0.78 V) integrated into one peak at −0.76 V, with the increase of the scan rate (Fig. S2c†).
Photophysical properties
The photophysical properties of these COV oligomers were investigated by UV-visible and fluorescence spectroscopies, also in CH3CN (using PF6 salts) and H2O (using chloride salts), respectively. When CH3CN was used as the solvent, PV2 exhibited a major absorption band centered at 287 nm (Fig. 2a). It was assigned to the absorption of a viologen unit which showed about 27 nm redshift compared to that of MV (Fig. S3a†). The absorption of the phenyl unit appeared as a shoulder around 250 nm. Interestingly, a weak absorption band centered at 420 nm was also observed (Fig. 2b). This broad peak was assigned to charge transfer (CT) absorption which should occur between the electron-deficient viologen and the relatively electron-rich phenyl unit. In order to ascertain whether it is an intramolecular or an intermolecular process, a UV-vis dilution experiment was performed. It was found that the absorbance of the CT band obeyed the Beer–Lambert law (Fig. S4a†), suggesting that the CT process occurred intramolecularly. PV3 displayed a similar UV-vis absorption feature but its maximum absorption peak was red-shifted by 5 nm relative to PV2, which was attributed to the extended conjugated backbone of PV3. It exhibited two weak absorption bands in the visible region, with one appearing at 420 nm and the other at 638 nm (Fig. 2c). These bands were also attributed to the intramolecular CT processes, as evidenced by UV-vis dilution experiments (Fig. S4b†). Similar absorptions were observed for PV2 and PV3 in the UV region when water was used as the solvent. However, there were no CT bands observed for PV2 and PV3 in H2O (Fig. S5a and b†), suggesting that no intramolecular charge transfer occurred in PV2 and PV3 when they are in aqueous media. | ||
| Fig. 2 (a) Normalized UV-vis spectra of PV2, PV3, BPV2 and BPV3, and UV-vis spectra of (b) PV2 and (c) PV3 at different concentrations in the visible region at 25 °C in CH3CN. (d) Fluorescence spectra of PV2, PV3, BPV2 and BPV3 in CH3CN at 25 °C. The concentration was 6.0 × 10−6 M, λex = 290 nm. | ||
Oligomers BPV2 and BPV3 displayed two absorption peaks in CH3CN. They exhibited the same peak at 260 nm which was assigned to the absorption of the biphenyl unit. Their peaks corresponding to the viologen segment were red-shifted relative to those of PV2 and PV3, being 340 nm for BPV2 and 350 nm for BPV3, respectively, suggesting an extended conjugation length of BPV series. Compared with BPV2, the absorption of the viologen units of BPV3 exhibited a red-shift of 10 nm. They displayed similar absorption in water. Interestingly, no CT absorption was observed for BPV2 and BPV3 both in CH3CN (Fig. S4c and d†) and H2O (Fig. S5c and d†), which might be attributed to the unmatched energy level between viologen and biphenyl units. The molar absorptive coefficients of the four oligomers were generated from UV-vis dilution experiments and listed in Table S1 (see the ESI†), which revealed that the value of absorptive coefficients increased with the extended conjugation.
A fluorescence spectroscopic study revealed that BPV2 and BPV3 were almost fluorescence silent in CH3CN (Fig. 2d). In contrast, a solution of PV2 in CH3CN exhibited a strong but broad emission peak centered at 436 nm. An emission band centered at 454 nm for PV3 was observed under the same conditions. Its emission was red-shifted by 18 nm relative to PV2. Such strong emission for PV2 and PV3 could be attributed to the twisted intramolecular charge transfer (TICT) process.29 However, no emission was observed for all the four COVs when water was used as the solvent (Fig. S6b†). This result is consistent with the UV-vis experiments above, which revealed that charge transfer existed in PV2 and PV3 but was absent in BPV2 and BPV3 when CH3CN was used as the solvent and in water no charge transfer in all of them.
Structural studies by DFT calculations
We have tried many times to grow single crystals of these conjugated viologen compounds, but unfortunately all we got were tiny needle-like crystals which were not suitable for X-ray crystallographic analysis. It might be attributed to their inherent highly positive charged nature. DFT calculations at the B3LYP/6-31G(d) level for PV2, PV3, BPV2 and BPV3, as well as their radical cations (PV2)˙2+, (PV3)˙3+, (BPV2)˙2+ and (BPV3)˙3+ were thus conducted to gain insights into the structural features of these novel π-conjugated oligomeric viologens.For PV2, PV3, BPV2 and BPV3, the aromatic rings in the conjugated backbones of their optimized structures twisted each other (Fig. S7a†). However, after the oligomers were reduced to the corresponding radical cations, the aromatic rings of BIPY units turned to adopt a coplanar conformation, while the twisted geometry still existed between the BIPY˙+ units and phenyl or biphenyl linkers (Fig. S7b†). Such coplanar conformation is favoured for the delocalization of the π electrons in the BIPY˙+ unit. The lengths of the conjugated backbones were estimated to extend from 2.28 nm for PV2 to 4.43 nm for BPV3, and almost remained unchanged after the BIPY units were reduced to BIPY˙+ radical cations.
The order of the frontier molecular orbitals (FMOs), HOMO–LUMO band gaps (Eg) and each energy level was estimated (Fig. 3). All the compounds showed lowed-lying FMO energy levels. For tetravalent compounds PV2 and BPV2, the LUMOs were found to localize on the BIPY unit and the HOMOs were on the phenyl or biphenyl rings. Meanwhile, BPV2 exhibited a higher FMO energy level and smaller Eg than that of PV2, as a result of the stronger electron-donating feature of the biphenyl unit. For hexavalent compounds PV3 and BPV3, there were two kinds of BIPY units and the LUMOs localized on the middle one. The HOMO of BPV3 was on the biphenyl segment. Different from the other three COVs, the HOMO of PV3 has a degenerate state, that is, HOMO−1 and HOMO−2 (Fig. 3c), which are localized on the terminal BIPY units, in contrast to BPV3, where HOMO is localized on the biphenyl units. On the other hand, the calculations also revealed that PV3 and BPV3 displayed lower FMO energy levels and smaller band gaps than those of the tetravalent oligomers, which could be explained by the effect of the higher valence and stronger electron-deficient properties of the latter. The electron-deficient order of these oligomers was estimated as PV3 > BPV3 > PV2 > BPV2 from their electron spin polarization (ESP) maps (Fig. S8†), and the backbone of PV3 possessed significant outstanding electron-deficient properties compared to the other three samples.
 | ||
| Fig. 3 Molecular orbital diagrams for the LUMOs and HOMOs of (a) PV2, (b) BPV2, (c) PV3 and (d) BPV3 generated by DFT calculations at the B3LYP/6-31G(d) level. | ||
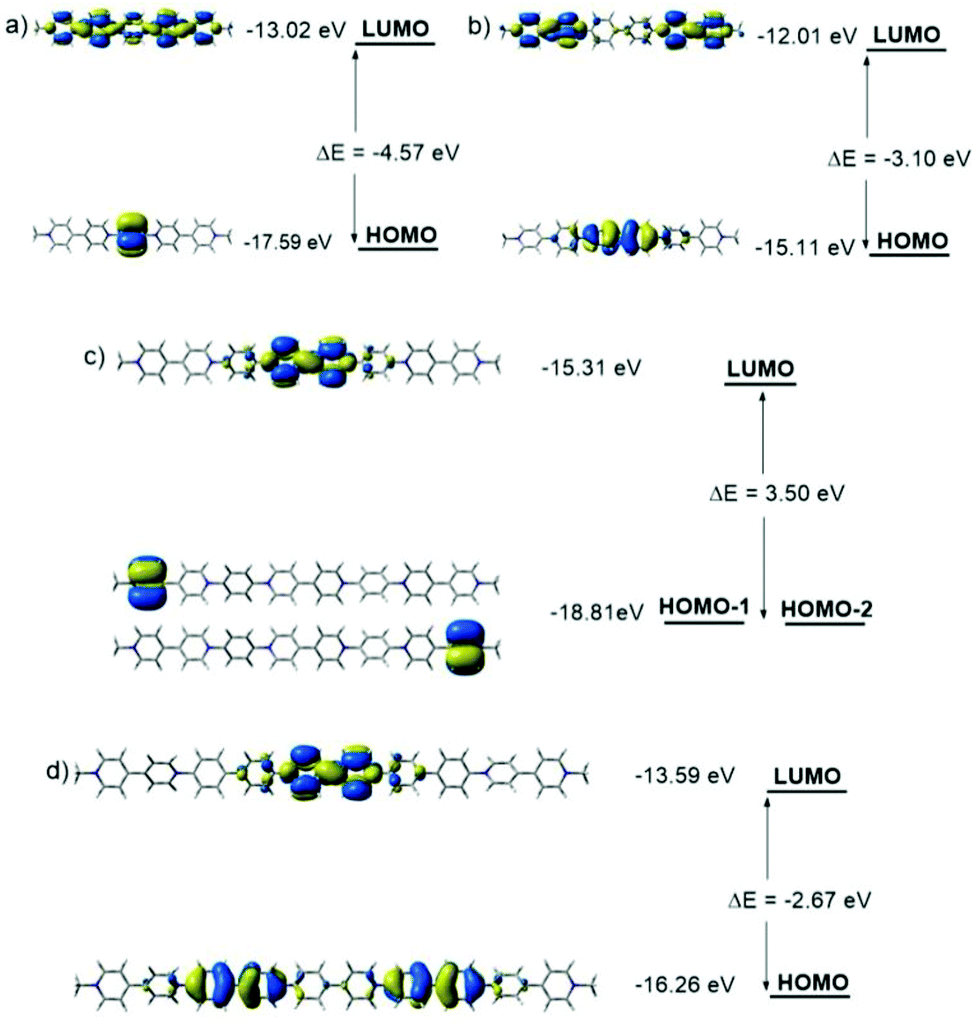
The FMOs of the radical cations (PV2)˙2+, (PV3)˙3+, (BPV2)˙2+ and (BPV3)˙3+ were also calculated by the DFT method at the UB3LYP/6-31G (d) level of theory. For the oligomers with different valence, two different spin states were considered respectively: spin1 (singlet) and spin3 (triplet) for the dicationic radicals (PV2)˙2+ and (BPV2)˙2+, and spin2 (doublet) and spin4 (quartet) for the triscationic radicals (PV3)˙3+ and (BPV3)˙3+ (Fig. S9†). For (PV2)˙2+, the MO energy of the spin3 state was 20.41 kJ mol−1 lower than that of the spin1 state; all the FMOs delocalized through the whole conjugated molecular backbone both in the spin1 and spin3 states to a great extent (Fig. 4a), and the band gap Eg of the spin1 state (0.67 eV) was much smaller than that of the spin3 state (2.83 and 2.47 eV). However, for the dicationic radicals (BPV2)˙2+, the MO energy of the spin3 state was 48.48 kJ mol−1 lower than that of the spin1 state, and the FMOs delocalized to a lower extent (Fig. 4b). The FMO energy level of spin1 was so close (Eg = 0.05 eV) and thus was nearly negligible compared to that of the spin3 state (2.74 and 2.61 eV). All the FMO energy levels of (BPV2)˙2+ were higher than the corresponding ones of (PV2)˙2+. This might be attributed to the twisted biphenyl linker that hindered the delocalization of the FMOs through the backbone of (BPV2)˙2+.
 | ||
| Fig. 4 FMOs energy diagrams in different spin states of (a) (PV2)˙2+, (b) (BPV2)˙2+, (c) (PV3)˙3+ and (d) (BPV3)˙3+ generated by DFT calculations at the UB3LYP/6-31G(d) level. | ||
In the case of the triscationic radicals (PV3)˙3+ (Fig. 4c) and (BPV3)˙3+ (Fig. 4d), the energy differences between their spin2 and spin4 states were only 5.51 kJ mol−1 and 0.41 kJ mol−1, respectively. For (PV3)˙3+, the band gap of the spin2 state (1.04 eV) was smaller than that of the spin4 state (2.80 and 2.22 eV). Similarly, the spin2 state band gap (1.09 eV) of (BPV3)˙3+ was smaller than that of its spin4 state (2.76 and 2.41 eV). It was clear that the FMOs of both (PV3)˙3+ and (BPV3)˙3+ delocalized to a much lower extent than that of the dicationic radicals (PV2)˙2+ and (BPV2)˙2+, and their highest occupied spin orbitals (HOSOs) were almost located on the BIPY˙+ unit. Similar to the above dicationic radicals, the FMO energy level of (BPV3)˙3+ was higher than the corresponding ones of (PV3)˙3+, although the Eg values between their LUMOs and HOSOs were quite close. Moreover, the energy-lowest HOSOs of the triscationic radicals (PV3)˙3+ and (BPV3)˙3+ are almost localized in the centre of their molecular backbones, indicating that the central BIPY unit would be reduced prior to the terminal ones.
Aggregation of radical cations
Although the first example of dimerization of a methyl viologen (MV) radical cation was reported more than 50 year ago,19c such a phenomenon did not attract much attention until it was found to be a powerful binding strategy in the construction of some exotic supramolecular architectures very recently.21a The aggregating properties of the radical cations generated from the above COVs were then investigated in aqueous media by various techniques.UV-vis-NIR spectroscopy was first used to study the aggregating behavior of these radical cations. The spectra were recorded in a sodium phosphate buffer (pH = 7.0) solution at 20 °C. As illustrated in Fig. 5a, dicationic radical (PV2)˙2+ exhibits three maximum absorption bands (385, 560 and 1080 nm). The new broad absorption signal centered at 1080 nm was regarded as the characteristic of radical–radical dimerization of the BIPY˙+units of (PV2)˙2+, which served as a valuable and discrete indicator for the presence of radical dimers in solution.19c Compared to that of a non-conjugated BIPY˙+ dimer whose absorption appears around 880 nm,30 this dramatically red-shift to the near infrared region phenomenon should be attributed to an extended conjugated backbone of (PV2)˙2+. UV-vis-NIR spectra for (BPV2)˙2+, (PV3)˙3+ and (BPV3)˙3+ were also recorded, and their corresponding radical dimer absorption bands appeared at 1085, 1095 and 1148 nm, respectively (Fig. S10†), which showed continuous red-shifts compared to that of (PV2)˙2+. What's more, the color of the aqueous solution (0.1 mM) changed immediately when excess reducing agent, Na2S2O3 (0.05 M), was added (Fig. S11a and b†). At a higher concentration, small particles that generated in situ from the radical cations could be observed, which could be attributed to strong stacking of the conjugated radical cations.31
 | ||
| Fig. 5 (a) UV spectra of PV2 (0.05 mM) before and after being reduced by Na2S2O3 (0.05 M) in H2O (pH = 7) at 20 °C; (b) UV titration spectra of PV2 (0.025 mM) with CB[8] (0–1.5 eq.) in Na2S2O3 (0.05 M) aqueous solution (pH = 7) at 20 °C; (c) Job's plot of PV2 with CB[8] in Na2S2O3 (0.05 M) aqueous solution (pH = 7) at 20 °C; (d) DLS plot of (0.05 mM PV2), (0.05 mM PV2 + 1.0 eq. CB[8]), (0.05 mM PV2 + 0.05 M Na2S2O3) and (0.05 mM PV2 + 1.0 eq. CB[8] + 0.05 M Na2S2O3) at 20 °C. | ||
It has been proved that the dimerization of BIPY˙+ unit could be dramatically weakened and even fully decomposed into monomers by introducing organic solvents or increasing temperature.26a The disassociating tendency of these di- or tris-cationic radical cations was then investigated by variable-temperature UV-vis-NIR spectroscopy and in a mixed aqueous media containing different ratios of organic solvents. The characteristic peaks of radical dimers around 560 and 1100 nm were absent at 60 °C, which means the aggregates formed by the dimerization of these radical cations decomposed completely at this temperature (Fig. S12†). On the other hand, partial disassociation of the aggregates was observed upon the addition of THF, CH3OH, CH3CN or DMSO into the aqueous solutions, as indicated by the decreasing absorption intensities of the absorptions of radical dimers (Fig. S13–16†).
It is well-known that the dimerization of a (MV)˙+ radical cation could be dramatically enhanced upon encapsulation of one radical dimer in the cavity of a cucurbit[8]uril (CB[8]) to form a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 host–guest complex.30 Therefore, CB[8] was introduced in the aqueous solutions of these COV radical cations. It was found that the color of the solutions changed to deep blue-green when 1.0 equivalent of CB[8] was added (Fig. S11d†), indicating that the dimerization of the COV radical cations was enhanced in the presence of CB[8]. In order to get deeper insight into the binding behaviour of these COV radical cations, UV-vis-NIR dilution experiments in the absence and presence of CB[8] were performed (Fig. S17 and 18†) and their apparent association constants (Ka) were estimated (Table S2†). It was revealed that the Ka values of COV radical cations were hundreds of folds larger than that of (MV)˙+, and increased as the conjugation backbone extended from (PV2)˙2+ to (BPV3)˙3+. Moreover, the Ka values further increased upon the addition of 1.0 equivalent of CB[8], again indicating that host-encapsulation enhanced the radicals’ dimerization.
:2 host–guest complex.30 Therefore, CB[8] was introduced in the aqueous solutions of these COV radical cations. It was found that the color of the solutions changed to deep blue-green when 1.0 equivalent of CB[8] was added (Fig. S11d†), indicating that the dimerization of the COV radical cations was enhanced in the presence of CB[8]. In order to get deeper insight into the binding behaviour of these COV radical cations, UV-vis-NIR dilution experiments in the absence and presence of CB[8] were performed (Fig. S17 and 18†) and their apparent association constants (Ka) were estimated (Table S2†). It was revealed that the Ka values of COV radical cations were hundreds of folds larger than that of (MV)˙+, and increased as the conjugation backbone extended from (PV2)˙2+ to (BPV3)˙3+. Moreover, the Ka values further increased upon the addition of 1.0 equivalent of CB[8], again indicating that host-encapsulation enhanced the radicals’ dimerization.
Moreover, the UV-vis-NIR titration spectra of the radical cations with CB[8] revealed that the absorbance around 400 and 560 nm increased significantly and 20–30 nm red-shifts occurred. In contrast, the absorption band around 1100 nm exhibited a blue-shift around 900 nm without an obvious enhancement of absorbance (Fig. 5b and S19†). Job's plots indicated a 1:1 stoichiometry for CB[8] and the radical cations (Fig. 5c and S20†). Dynamic light scattering (DLS) experiments were further carried out, which revealed that the conjugated cation radical (PV2)˙2+ formed large aggregates in solution with an average hydrodynamic diameter (Dh) of about 160 nm, increasing to about 210 nm in the presence of 1.0 equivalent CB[8] (Fig. 5d). On the basis of the above results, the formation of linear host–guest supramolecular polymers driven by the enhanced head-to-tail radical–radical dimerization was proposed. For (BPV2)˙2+, (PV3)˙3+ and (BPV3)˙3+, similar phenomena were observed (Fig. S21†), and aggregates with hundreds of nanometres formed from the triscationic radicals under the same concentration, which could be attributed to their more extended conjugated backbones.
Electron paramagnetic resonance (EPR) spectroscopic experiments in H2O were further carried out to gain more insight into these COV radical cations and their assembly behaviors. Unlike the methyl viologen radical cation (MV)˙+ which exhibited hyperfine splitting EPR signal in water,26a (PV2)˙2+ displayed a broad signal with a g-factor value of 2.0045 (Fig. 6a), which is close to that of free electron (g = 2.0023) and thus confirmed its organic radical feature.32 In the cases of the other three radical cations (PV3)˙3+, (BPV2)˙2+ and (BPV3)˙3+, their spectra showed two broad EPR signals (Fig. 6a). The additional EPR signals might be attributed to low lying triplet states resulted from radical–radical interactions which lead to small HOMO–LUMO gaps. For all the four radical cations, increasing the temperature from 25 to 60 °C did not result in the appearance of a hyperfine structure. However, the intensities of the signals were found to increase with the increasing temperature (Fig. S22†). When 1.0 equivalent of CB[8] was added into the in situ generated (PV2)˙2+ solution, its EPR signal was slightly shifted and became more intensive. However, (BPV2)˙2+, (PV3)˙3+ and (BPV3)˙3+ exhibited different behaviour: the EPR signals at a higher magnetic field disappeared after the addition of CB[8], while the signals at a lower magnetic field became more intensive and shifted slightly (Fig. 6b). These observations suggest that the introduction of CB[8] has a significant impact on the assembly of the COV radical cations. Furthermore, the variable-temperature EPR experiments revealed that the resulting radical cations involving host–guest complexes also displayed a good thermal stability (Fig. S23†). For the experimental results of the EPR studies, although some interesting phenomena were observed for the COV radical cations, it should also be pointed out that it was hard to give a definitive answer for their behaviour from the available EPR data.
 | ||
| Fig. 6 EPR spectra of (PV2)˙2+, (PV3)˙3+, (BPV2)˙2+ and (BPV3)˙3+ (a) in the absence and (b) in the presence of 1.0 eq. CB[8] at 25 °C. The concentration was 0.1 mM for all samples. | ||
In order to shed further light on the structural features of the supramolecular polymeric structures formed in water in the presence of CB[8], DFT calculations of the M06 flavor22a was used to provide a quantum mechanical description of the minimized superstructures. Firstly, the model superstructure for (MV˙+)2⊂CB[8] was carried out at the m06/6-31g (d, p) level. It was clearly viewed that two MV˙+ molecules stacked in a face-to-face manner with a distance of 0.35 nm (Fig. 7a) and their HOSO orbitals were partially overlapped, indicating that host enhanced dimerization interactions occurred between the BIPY˙+ units in the cavity of CB[8] (Fig. 7b). Thus, in the presence of 1.0 equivalent of CB[8], the encapsulation of two stacked BIPY˙+ units of the conjugated viologen oligomers in the cavity of a CB[8] molecule could be expected, and it would lead to the formation of rigid linear supramolecular radical polymers (Fig. 7c). The formation of such supramolecular polymers could also be expected in the absence of CB[8], which is driven by the strong dimerization of the rigid rod-like radical cations.
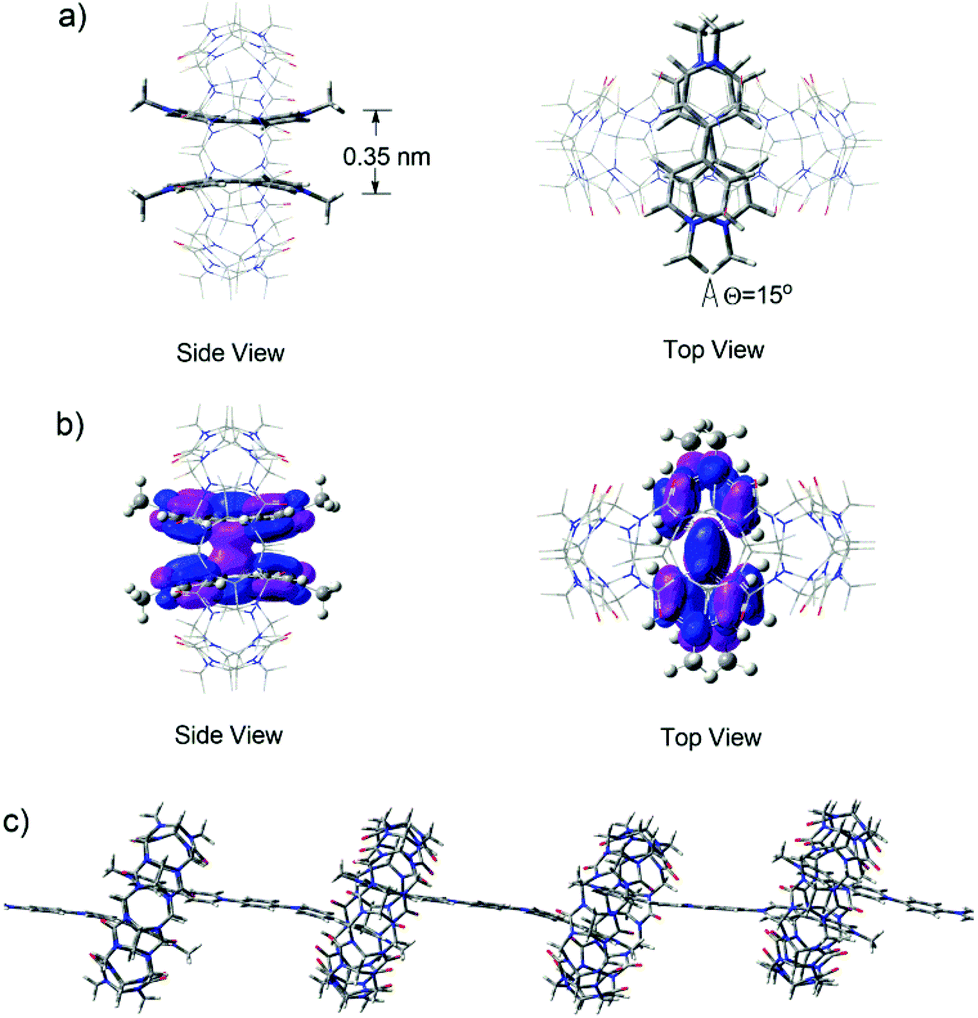 | ||
| Fig. 7 Different views of (a) optimized structure and (b) molecular orbital of (2MV˙+)⊂CB[8] model obtained by DFT calculation at the m06/6-31g (d, p) level. (c) Illustration of the formation of rigid linear supramolecular polymers from (PV2)˙2+ after the encapsulation of the BIPY˙+ units in the cavity of CB[8] generated by semi-empirical/upm6 calculations. | ||
The formation of supramolecular radical polymers was visualized by cryo-transmission electron microscopy (cryo-TEM). As revealed in Fig. 8a, straight stick-like objects formed from (PV2)˙2+ in water were observed, which is consistent with the expectation for rigid linear polymeric chains. The width of the polymer is significantly larger than the width of (PV2)˙2+, which might be attributed to the further stacking of the as-formed individual linear supramolecular polymers driven by the interaction between BIPY˙+ radicals. When 1.0 equivalent of CB[8] was added, similar linear structures were also observed (Fig. 8b), which could also be attributed to the formation of linear supramolecular polymers driven by CB[8]-encapsulated radical dimerization. The width of the rigid linear polymeric chain observed was estimated to be about tens of nanometres, also suggesting further aggregation of the individual polymer chains. The aggregation of polymeric chains in the presence of CB[8] could be attributed to the outer-surface interactions between the convex face of CB[8].33
 | ||
| Fig. 8 Cryo-TEM image of (a) (PV2)˙2+ and (b) (PV2)˙2+ with 1.0 eq. CB[8] in water, the concentration was 0.025 mM. | ||
Similar linear morphologies were also observed by cryo-TEM for the other three COV radical cations (Fig. S24†), also suggesting the formation of linear supramolecular polymers through the dimerization of the radical cations. However, the width of these supramolecular polymers were much smaller than that of (PV2)˙2+. It might be attributed to the more twisted backbones of (PV3)˙3+, (BPV2)˙2+ and (BPV3)˙3+ which resulted in weaker stacking interactions.
On the basis of the above results, a polymerization mechanism of the supramolecular radical polymers was proposed. As illustrated in Fig. 9, upon the reduction of the conjugated oligomeric viologens (PV2, PV3, BPV2 and BPV3) in water, the resulting radical cations tended to dimerize by taking advantage of strong radical–radical stacking between multiple BIPY˙+ units and hydrophobic interactions as a driving force, and further formed strip-like aggregates. What's more, the dimerization of BIPY˙+ units could be further enhanced by the encapsulation of CB[8].
 | ||
| Fig. 9 Cartoon representation of the formation of rigid linear supramolecular radical polymers and their further aggregation into bundles. PV2 and BPV2 were used as the representatives. | ||
Conclusions
In summary, a series of π-conjugated oligomeric viologens (COVs) linked by phenyl or biphenyl units have been designed and synthesized. Compared to their unconjugated counterparts previously reported, these oligomers exhibit interesting electrochemical and photophysical features, which originated from the conjugate-bridged multiple viologen units. Upon the treatment of the aqueous solutions of these COVs with sodium dithionite, they were reduced to radical cations and the resulting radicals spontaneously dimerized in water to generate linear supramolecular radical polymers. Furthermore, the dimerization of the radical cations could be significantly enhanced by the introduction of CB[8], and similar rigid linearly supramolecular radical polymers were also constructed. The comprehensive DFT calculation studies provide an in-depth insight into the structure–property relationships for the COVs, their radical cations and the self-assembly behavior of the radicals. This work demonstrates some interesting properties of π-conjugated oligomeric viologens, which endow them with great potential in developing novel supramolecular systems and functional materials in future.Acknowledgements
We thank the National Natural Science Foundation of China (91127007, 21502216) and Technology Commission of Shanghai Municipality (15ZR1449500) for financial support. We also thank Prof. Yu-Xue Li for the helpful discussion and suggestions.Notes and references
- (a) F. M. Raymo and J. F. Stoddart, Chem. Rev., 1999, 99, 1643 CrossRef CAS PubMed; (b) L. Fang, M. A. Olson, D. Benitez, E. Tkatchouk, W. A. Goddard III and J. F. Stoddart, Chem. Soc. Rev., 2010, 39, 17 RSC.
- (a) V. Balzani, A. Credi and M. Venturi, Molecular Devices and Machines- A Journey into the Nano World, Wiley-VCH, Weinheim, 2003 CrossRef; (b) E. R. Kay and D. A. Leigh, Top. Curr. Chem., 2005, 262, 133 CrossRef CAS.
- (a) Y. H. Ko, E. Kim, I. Hwang and K. Kim, Chem. Commun., 2007, 1305 RSC; (b) Y. Zhang, T.-Y. Zhou, K.-D. Zhang, J.-L. Dai, Y.-Y. Zhu and X. Zhao, Chem. – Asian J., 2014, 9, 1530 CrossRef CAS PubMed; (c) H. Yang, B. Yuan, X. Zhang and O. A. Scherman, Acc. Chem. Res., 2014, 47, 2106 CrossRef CAS PubMed.
- (a) H. Li, D. X. Chen, Y.-L. Sun, Y. B. Zheng, L.-L. Tan, P. S. Weiss and Y.-W. Yang, J. Am. Chem. Soc., 2013, 135, 1570 CrossRef CAS PubMed; (b) M. Zayats, S. P. Pogorelova, A. B. Kharitonov, O. Lioubashevski, E. Katz and I. Willner, Chem. – Eur. J., 2003, 9, 6108 CrossRef CAS PubMed; (c) B. Strehlitz, B. Gründig, W. Schumacher, P. M. H. Kroneck, K.-D. Vorlop and H. Kotte, Anal. Chem., 1996, 68, 807 CrossRef CAS PubMed.
- (a) S. Zahavy, M. Seiler, S. Marx-Tibbon, E. Joselevich, I. Willner, H. Dürr, D. O'Connor and A. Harriman, Angew. Chem., Int. Ed. Engl., 1995, 34, 1005 CrossRef; (b) Y. Nishikitani, S. Uchida, T. Asano, M. Minami, S. Oshima, K. Ikai and T. Kubo, J. Phys. Chem. C, 2008, 112, 4372 CrossRef CAS; (c) A. S. Perera, N. K. Subbaiyan, M. Kalita, S. O. Wendel, T. N. Samarakoon, F. D'Souza and S. H. Bossmann, J. Am. Chem. Soc., 2013, 135, 6842 CrossRef CAS PubMed.
- (a) N. Leventis, M. Chen, A. Liapis, J. W. Johnson and A. Jain, J. Electrochem. Soc., 1998, 145, L55 CrossRef CAS; (b) K.-C. Ho, Y.-W. Fang, Y.-C. Hsu and L.-C. Chen, Solid State Ionics, 2003, 165, 279 CrossRef CAS; (c) S.-h. Kim, N. Shim, H. Lee and B. Moon, J. Mater. Chem., 2012, 22, 13558 RSC; (d) L.-c. Cao, M. Mou and Y. Wang, J. Mater. Chem., 2009, 19, 3412 RSC; (e) G. Wang, X. Fu, J. Huang, C. Wu, L. Wu and Q. Du, Org. Electron., 2011, 12, 1216 CrossRef CAS.
- (a) Y. Liu, K. Liu, Z. Wang and X. Zhang, Chem. – Eur. J., 2011, 17, 9930 CrossRef CAS PubMed; (b) Y. Liu, Y. Yu, J. Gao, Z. Wang and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 6576 CrossRef CAS PubMed; (c) J. del Barrio, P. N. Horton, D. Lairez, G. O. Lloyd, C. Toprakcioglu and O. A. Scherman, J. Am. Chem. Soc., 2013, 135, 11760 CrossRef CAS PubMed; (d) F. Lin, T.-G. Zhan, T.-Y. Zhou, K.-D. Zhang, G.-Y. Li, J. Wu and X. Zhao, Chem. Commun., 2014, 50, 7982 RSC.
- (a) R. J. Coulston, S. T. Jones, T.-C. Lee, E. A. Appel and O. A. Scherman, Chem. Commun., 2011, 47, 164 RSC; (b) E. A. Appel, F. Biedermann, U. Rauwald, S. T. Jones, J. M. Zayed and O. A. Scherman, J. Am. Chem. Soc., 2010, 132, 14251 CrossRef CAS PubMed; (c) E. A. Appel, X. J. Loh, S. T. Jones, F. Biedermann, C. A. Dreiss and O. A. Scherman, J. Am. Chem. Soc., 2012, 134, 11767 CrossRef CAS PubMed; (d) E. A. Appel, R. A. Forster, A. Koutsioubas, C. Toprakcioglu and O. A. Scherman, Angew. Chem., Int. Ed., 2014, 53, 10038 CrossRef CAS PubMed.
- (a) K.-D. Zhang, J. Tian, D. Hanifi, Y. Zhang, A. C.-H. Sue, T.-Y. Zhou, L. Zhang, X. Zhao, Y. Liu and Z.-T. Li, J. Am. Chem. Soc., 2013, 135, 17913 CrossRef CAS PubMed; (b) J. Tian, T.-Y. Zhou, S.-C. Zhang, S. Aloni, M. V. Altoe, S.-H. Xie, H. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Nat. Commun., 2014, 5, 5574 CrossRef CAS PubMed; (c) X. Zhang, C.-B. Nie, T.-Y. Zhou, Q.-Y. Qi, J. Fu, X.-Z. Wang, L. Dai, Y. Chen and X. Zhao, Polym. Chem., 2015, 6, 1926 Search PubMed; (d) T.-Y. Zhou, Q.-Y. Qi, Q.-L. Zhao, J. Fu, Y. Liu, Z. Ma and X. Zhao, Polym. Chem., 2015, 6, 3018 RSC.
- (a) C. M. Gothard, C. J. Bruns, N. A. Gothard, B. A. Grzybowski and J. F. Stoddart, Org. Lett., 2012, 14, 5066 CrossRef CAS PubMed; (b) Z. Zhu, H. Li, Z. Liu, J. Lei, H. Zhang, Y. Y. Botros, C. L. Stern, A. A. Sarjeant, J. F. Stoddart and H. M. Colquhoun, Angew. Chem., Int. Ed., 2012, 51, 7231 CrossRef CAS PubMed.
- (a) S. Heinen and L. Walder, Angew. Chem., Int. Ed., 2000, 39, 806 CrossRef CAS; (b) F. Marchioni, M. Venturi, A. Credi, V. Balzani, M. Belohradsky, A. M. Elizarov, H.-R. Tseng and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 568 CrossRef CAS PubMed; (c) C. M. Ronconi, J. F. Stoddart, V. Balzani, M. Baroncini, P. Ceroni, C. Giansante and M. Venturi, Chem. – Eur. J., 2008, 14, 8365 CrossRef CAS PubMed; (d) S. Asaftei and E. D. Clercq, J. Med. Chem., 2010, 53, 3480 CrossRef CAS PubMed; (e) M. Kathiresan and L. Walder, Macromolecules, 2011, 44, 8563 CrossRef CAS; (f) N. Katir, J. P. Majoral, A. E. Kadib, A.-M. Caminade and M. Bousmina, Eur. J. Org. Chem., 2012, 269 CrossRef CAS; (g) K. Ciepluch, N. Katir, A. E. Kadib, A. Felczak, K. Zawadzka, M. Weber, B. Klajnert, K. Lisowska, A.-M. Caminade, M. Bousmina, M. Bryszewska and J. P. Majoral, Mol. Pharm., 2012, 9, 448 CrossRef CAS PubMed.
- (a) A. Nelson, J. M. Belitsky, S. Vidal, C. S. Joiner, L. G. Baum and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 11914 CrossRef CAS PubMed; (b) J. M. Belitsky, A. Nelson and J. F. Stoddart, Org. Biomol. Chem., 2006, 4, 250 RSC; (c) J. M. Belitsky, A. Nelson, J. D. Hernandez, L. G. Baum and J. F. Stoddart, Chem. Biol., 2007, 14, 1140 CrossRef CAS PubMed.
- T.-G. Zhan, B.-Y. Lu, F. Lin, T.-Y. Zhou, X. Zhao and Z.-T. Li, Org. Chem. Front., 2015, 2, 1578 RSC.
- (a) J. C. Barnes, M. Juríček, N. L. Strutt, M. Frasconi, S. Sampath, M. A. Giesener, P. L. McGrier, C. J. Bruns, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 183 CrossRef CAS PubMed; (b) M. Juríček, J. C. Barnes, N. L. Strutt, N. A. Vermeulen, K. C. Ghooray, E. J. Dale, P. R. McGonigal, A. K. Blackburn, A.-J. Avestro and J. F. Stodddart, Chem. Sci., 2014, 5, 2724 RSC; (c) J. C. Barnes, M. Juríček, N. A. Vermeulen, E. J. Dale and J. F. Stoddart, J. Org. Chem., 2013, 78, 11962 CrossRef CAS PubMed; (d) M. Juríček, J. C. Barnes, E. J. Dale, W.-G. Liu, N. L. Strutt, C. J. Bruns, N. A. Vermeulen, K. Ghooray, A. A. Sarjeant, C. L. Stern, Y. Y. Botros, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 12736 CrossRef PubMed.
- J. Sun, M. Frasconi, Z. Liu, J. C. Barnes, Y. Wang, D. Chen, C. L. Stern and J. F. Stoddart, Chem. Commun., 2015, 51, 1432 RSC.
- (a) E. J. Dale, N. A. Vermeulen, A. A. Thomas, J. C. Barnes, M. Juríček, A. K. Blackburn, N. L. Strutt, A. A. Sarjeant, C. L. Stern, S. E. Denmark and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 10669 CrossRef CAS PubMed; (b) N. Hafezi, J. M. Holcroft, K. J. Hartlieb, E. J. Dale, N. A. Vermeulen, C. L. Stern, A. A. Sarjeant and J. F. Stoddart, Angew. Chem., Int. Ed., 2015, 54, 456 CAS.
- (a) R. M. Young, S. M. Dyar, J. C. Barnes, M. Juríček, J. F. Stoddart, D. T. Co and M. R. Wasielewski, J. Phys. Chem. A, 2013, 117, 12438 CrossRef CAS PubMed; (b) S. M. Dyar, J. C. Barnes, M. Juríček, J. F. Stoddart, D. T. Co, R. M. Young and M. R. Wasielewski, Angew. Chem., Int. Ed., 2014, 53, 5371 CrossRef CAS PubMed.
- M. Juríček, N. L. Strutt, J. C. Barnes, A. M. Butterfield, E. J. Dale, K. K. Baldridge, J. F. Stoddart and J. S. Siegel, Nat. Chem., 2014, 6, 222 CrossRef PubMed.
- (a) L. Michaelis and E. S. Hill, J. Gen. Physiol., 1933, 16, 859 CrossRef CAS PubMed; (b) L. Michaelis, Chem. Rev., 1935, 16, 243 CrossRef CAS; (c) E. M. Kosower and J. L. Cotter, J. Am. Chem. Soc., 1964, 86, 5524 CrossRef CAS; (d) C. L. Bird and A. T. Kuhn, Chem. Soc. Rev., 1981, 10, 49 RSC; (e) P. M. S. Monk, The Radical Cation: Dimer Formation, in The Viologens: Physicochemical Properties, Synthesis, and Applications of the Salts of 4, 4′-Bipyridine, Wiley, New York, 1998, p. 115 Search PubMed.
- (a) E. M. Kosower and J. Hajdu, J. Am. Chem. Soc., 1971, 93, 2534 CrossRef CAS; (b) W. Geuder, S. Hünig and A. Suchy, Tetrahedron, 1986, 42, 1665 CrossRef CAS.
- (a) D.-W. Zhang, J. Tian, L. Chen, L. Zhang and Z.-T. Li, Chem. – Asian J., 2015, 10, 56 CrossRef CAS PubMed; (b) K. Wadhwa, S. Nuryyeva, A. C. Fahrenbach, M. Elhabiri, C. Platas-Iglesias and A. Trabolsi, J. Mater. Chem. C, 2013, 1, 2302 RSC; (c) K. Nchimi-Nono, P. Dalvand, K. Wadhwa, S. Nuryyeva, S. Alneyadi, T. Prakasam, A. C. Fahrenbach, J.-C. Olsen, Z. Asfari, C. Platas-lglesias, M. Elhabiri and A. Trabolsi, Chem. – Eur. J., 2014, 20, 7334 CrossRef CAS PubMed.
- (a) A. Trabolsi, N. Khashab, A. C. Fahrenbach, D. C. Friedman, M. T. Colvin, K. K. Cot, D. Benítez, E. Tkatchouk, J.-C. Olsen, M. E. Belowich, R. Carmielli, H. A. Khatib, W. A. Goddard III, M. R. Wasielewski and J. F. Stoddart, Nat. Chem., 2010, 2, 42 CrossRef CAS PubMed; (b) A. C. Fahrenbach, J. C. Barnes, D. A. Lanfranchi, H. Li, A. Coskun, J. J. Gassensmith, Z. Liu, D. Benítez, A. Trabolsi, W. A. Goddard, M. Elhabiri and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 3061 CrossRef CAS PubMed; (c) J. C. Barnes, A. C. Fahrenbach, D. Cao, S. M. Dyar, M. Frasconi, M. A. Giesener, D. Benítez, E. Tkatchouk, O. Chernyashevskyy, W. H. Shin, H. Li, S. Sampath, C. L. Stern, A. A. Sarjeant, K. J. Hartlieb, Z. Liu, R. Carmieli, Y. Y. Botros, J. W. Choi, A. M. Z. Slawin, J. B. Ketterson, M. R. Wasielewski, W. A. Goddard III and J. F. Stoddart, Science, 2013, 339, 429 CrossRef CAS PubMed.
- (a) H. Li, A. C. Fahrenbach, S. V. Dey, S. Basu, A. Trabolsi, Z. Zhu, Y. Y. Botros and J. F. Stoddart, Angew. Chem., Int. Ed., 2010, 49, 8260 CrossRef CAS PubMed; (b) H. Li, A. C. Fahrenbach, A. Coskun, Z. Zhu, G. Barin, Y. Zhao, Y. Y. Botros, J.-P. Sauvage and J. F. Stoddart, Angew. Chem., Int. Ed., 2011, 50, 6782 CrossRef CAS PubMed; (c) A. C. Fahrenbach, Z. Zhu, D. Cao, W.-G. Liu, H. Li, S. K. Dey, S. Basu, A. Trabolsi, Y. Y. Botros, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 16275 CrossRef CAS PubMed; (d) Z. Zhu, A. C. Fahrenbach, H. Li, J. C. Barnes, Z. Liu, S. M. Dyar, H. Zhang, J. Lei, R. Carmieli, A. A. Sarjeant, C. L. Stern, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2012, 134, 11709 CrossRef CAS PubMed; (e) H. Li, Z. Zhu, A. C. Fahrenbach, B. M. Savoie, C. Ke, J. C. Barnes, J. Lei, Y.-L. Zhao, L. M. Lilley, T. J. Marks, M. A. Ratner and J. F. Stoddart, J. Am. Chem. Soc., 2013, 135, 456 CrossRef CAS PubMed; (f) L. S. Witus, K. J. Hartlieb, Y. Wang, A. Prokofjevs, M. Frasconi, J. C. Barnes, E. J. Dale, A. C. Fahrenbach and J. F. Stoddart, Org. Biomol. Chem., 2014, 12, 6089 RSC; (g) C. J. Bruns, M. Frasconi, J. Iehl, K. J. Hartlieb, S. T. Schneebeli, C. Cheng, S. I. Stupp and J. F. Stoddart, J. Am. Chem. Soc., 2014, 136, 4714 CrossRef CAS PubMed.
- Y. Wang, M. Frasconi, W.-G. Liu, Z. Liu, A. A. Sarjeant, M. S. Nassar, Y. Y. Botros, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 876 CrossRef CAS PubMed.
- A. C. Fahrenbach, S. Sampath, D. J. Late, J. C. Barnes, S. L. Kleinman, N. Valley, K. J. Hartlieb, Z. Liu, V. P. Dravid, G. C. Schatz, R. P. Van Duyne and J. F. Stoddart, ACS Nano, 2012, 6, 9964 CrossRef CAS PubMed.
- (a) C. Zhou, J. Tian, J.-L. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Polym. Chem., 2014, 5, 341 RSC; (b) L. Zhang, T.-Y. Zhou, J. Tian, H. Wang, D.-W. Zhang, X. Zhao, Y. Liu and Z.-T. Li, Polym. Chem., 2014, 5, 4715 RSC; (c) D.-W. Zhang, J. Tian, L. Chen, L. Zhang and Z.-T. Li, Chem. – Asian J., 2015, 10, 56 CrossRef CAS PubMed; (d) L. Chen, H. Wang, D.-W. Zhang, Y. Zhou and Z.-T. Li, Angew. Chem., Int. Ed., 2015, 54, 4028 CrossRef CAS PubMed.
- L. Chen, H. Willcock, C. J. Wedge, F. Hartl, H. M. Colquhoun and B. W. Greenland, Org. Biomol. Chem., 2016, 14, 980 CAS.
- M. Nanasawa, M. Miwa, M. Hirai and T. Kuwabara, J. Org. Chem., 2000, 65, 593 CrossRef CAS PubMed.
- I. Azumaya, H. Kagechika, Y. Fujiwara, M. Itoh, K. Yamaguchi and K. Shudo, J. Am. Chem. Soc., 1991, 113, 2833 CrossRef CAS.
- W. S. Jeon, H.-J. Kim, C. Lee and K. Kim, Chem. Commun., 2002, 1828 RSC.
- (a) C. R. Martinez and B. L. Iverson, Chem. Sci., 2012, 3, 2191 RSC; (b) M. T. Stone, J. M. Heemstra and J. S. Moore, Acc. Chem. Res., 2006, 39, 11 CrossRef CAS PubMed; (c) M. L. Waters, Acc. Chem. Res., 2013, 46, 873 CrossRef CAS PubMed.
- D. Schweinfurth, M. Zalibera, M. Kathan, C. Shen, M. Mazzolini, N. Trapp, J. Crassous, G. Gescheidt and F. Diederich, J. Am. Chem. Soc., 2014, 136, 13045 CrossRef CAS PubMed.
- X.-L. Ni, X. Xiao, H. Cong, Q.-J. Zhu, S.-F. Xue and Z. Tao, Acc. Chem. Res., 2014, 47, 1386 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional UV-vis, fluorescence and EPR spectra, CV profiles, Job's plots, DLS profiles and NMR spectra of compounds. See DOI: 10.1039/C6QO00298F |
| This journal is © the Partner Organisations 2016 |