
Asymmetric total synthesis of Lycopodium alkaloids α-obscurine, N-desmethyl-α-obscurine, β-obscurine and N-desmethyl-β-obscurine†
Jian-Guo
Fu
,
Guang-Qiang
Xu
,
Rui
Ding
,
Guo-Qiang
Lin
and
Bing-Feng
Sun
*
CAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, 345 Lingling Road, Shanghai 200032, China. E-mail: bfsun@sioc.ac.cn
First published on 18th November 2015
Abstract
The asymmetric total synthesis of α-obscurine (1), β-obscurine (2), N-desmethyl-α-obscurine (3), and N-desmethyl-β-obscurine (4) was accomplished. Key reactions in the construction of the A/B/C-ring system include the Buchwald–Hartwig coupling reaction, the Heck cyclization, and the diastereoselective hydrogenation.
Among the Lycopodium alkaloids, lycodine-type alkaloids constitute a unique family to which the well-known memory-enhancing natural product huperzine A belongs.1 α-Obscurine (1), β-obscurine (2), N-desmethyl-α-obscurine (3), and N-desmethyl-β-obscurine (4) are lycodine-type alkaloids. Some of these historic molecules were identified as early as seven decades ago. Interestingly, these natural products may be biogenetically relevant to huperzine A. As proposed previously, compound 5, which in principle could be a general intermediate to all Lycopodium alkaloids, may engender deacetylflabellidine, a natural product, via a Mannich-type cyclization.2,7c,9b Deacetylflabellidine might undergo oxidation, dehydrogenation and methylation to produce 1–4, prior to further oxidative modifications leading to huperzine A (Scheme 1).
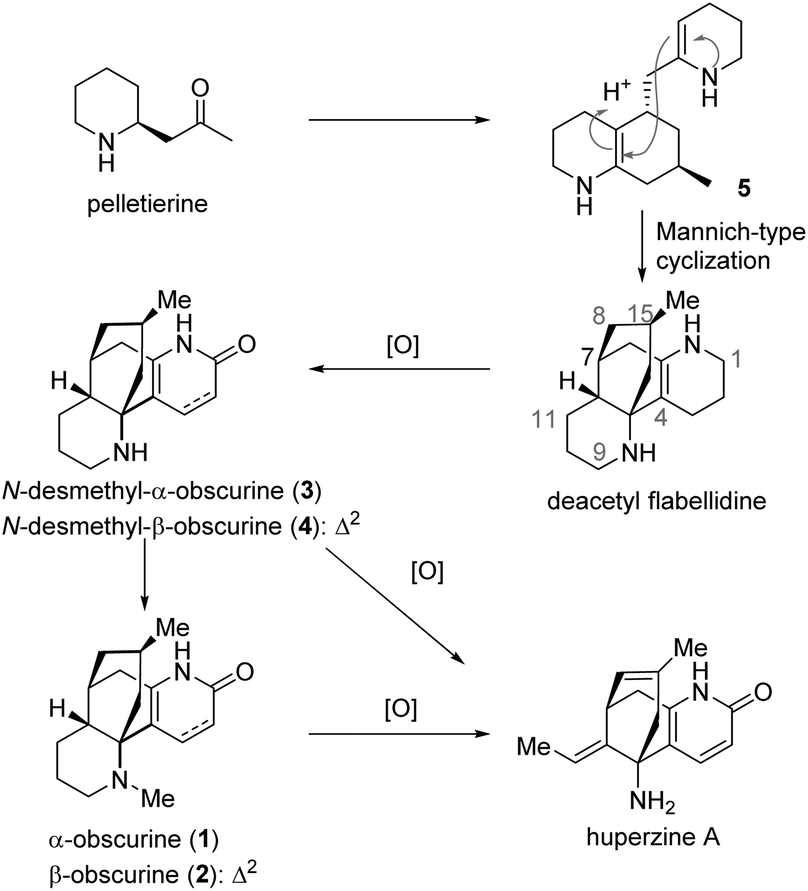 | ||
| Scheme 1 Obscurines 1–4 in the proposed biosynthetic pathway leading to huperzine A. | ||
Obscurine was first isolated in 1942 by Manske and Marion3 from Lycopodium obscurum and was shown by Moore and Marion4 in 1953 to be actually a mixture of α-obscurine (1) and β-obscurine (2). In 1962, Ayer and co-workers successfully established the structure of 1 and 2 with the relative as well as the absolute stereochemistry by using a chemical correlation strategy.5 Moreover, they isolated N-desmethyl-α-obscurine (3) as a natural product and demonstrated that it could be obtained by demethylation of 1.5 In the same paper, Ayer reported the preparation of N-desmethyl-β-obscurine (4) from β-obscurine (2).5 And in 1989 N-desmethyl-β-obscurine (4) was verified to be a natural product.6
After Ayer's chemical transformations of α-obscurine (1) and β-obscurine (2) to N-desmethyl-α-obscurine (3) and N-desmethyl-β-obscurine (4), respectively, Schumann accomplished the first total synthesis of α-obscurine (1) and N-desmethyl-α-obscurine (3) as racemic forms in 1983.7 Schumann's elegant synthesis featured a highly convergent construction of the tetracyclic skeleton, which assembled A/D- and C-ring segments by an endgame biomimetic Mannich cyclization forming a B-ring. In 2010, by harnessing Schumann's strategy, Sarpong and co-workers rendered an asymmetric synthesis of N-desmethyl-α-obscurine (3) en route to the total synthesis of (+)-complanadine A.8 To the best of our knowledge, these constitute the only synthetic endeavours toward these molecules.
In the past several years, we have been involved in the total syntheses of biologically significant natural products.9 Among these, a collective total synthesis of huperzine A, huperzine B and huperzine U has been accomplished efficiently by employing a unified synthetic strategy.9a,b In this paper, we report the asymmetric total synthesis of 1–4 by harnessing the same synthetic strategy. As depicted in Scheme 2, the tricyclic compound 6 containing the A/B/C-ring system was chosen as the key precursor to obscurines 1–4. The C12 chiral centre in the target molecules was anticipated to be established via a diastereoselective hydrogenation of the olefinic double bond after the D-ring was constructed. Compound 6 with its [3.3.1] bicyclic core would stem from 7 and may be further traced back to (R)-pulegone.9a The establishment of the C15 stereogenic centre was envisaged to be a critical undertaking.
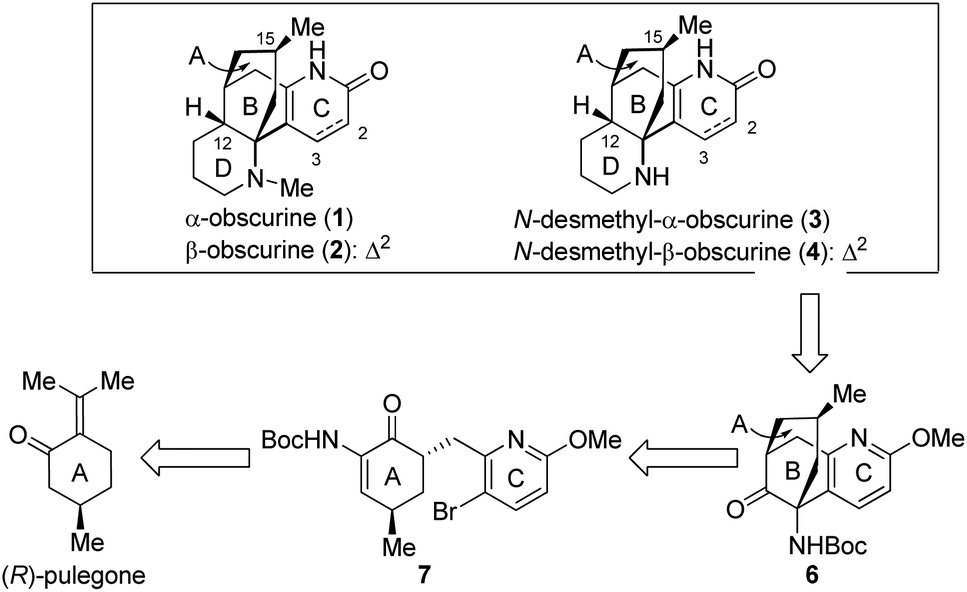 | ||
| Scheme 2 Retrosynthetic analysis for 1–4. | ||
Initially, a reductive Heck cyclization was envisioned to transform 7 to 6 with the stereocenter conserved. Thus, the synthetic journey commenced with the preparation of 7 and 8 from (R)-pulegone on a deca-gram scale.9a,b The reductive Heck cyclization of 8 was attempted. Unfortunately, under various tested conditions, the anticipated cyclization was not observed while compound 9 was isolated as a by-product. We reasoned that the reductive debromination might be significantly faster compared to the desired Heck cyclization, leading to the formation of 9 (Scheme 3).
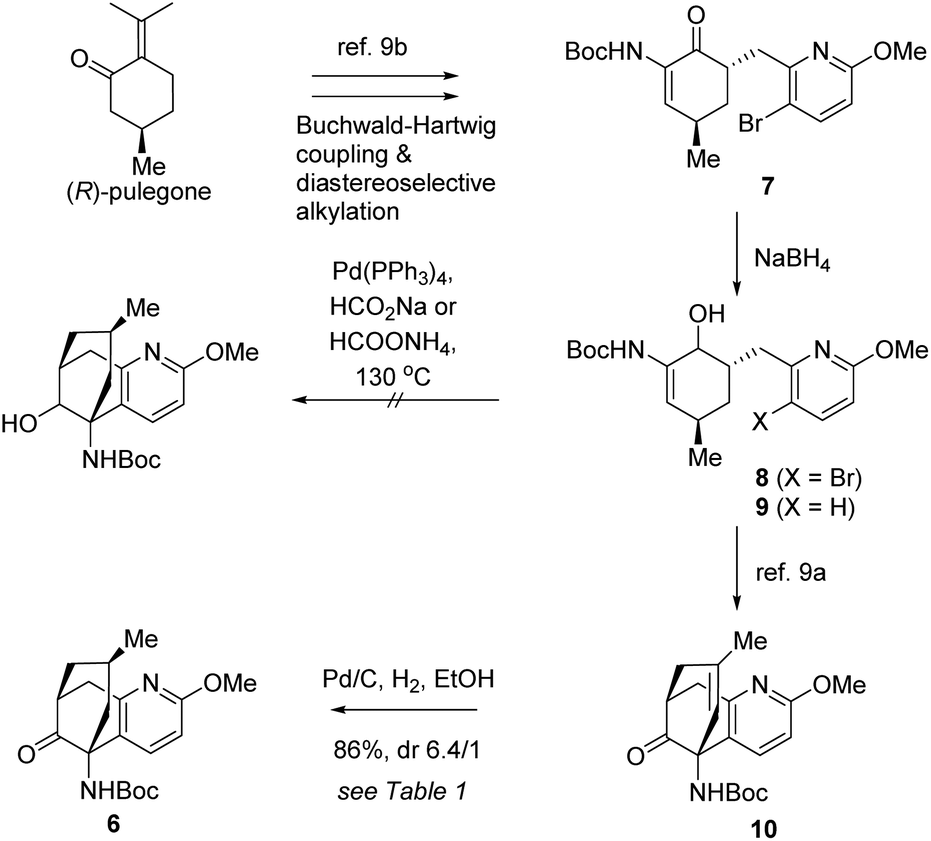 | ||
| Scheme 3 Synthesis of 6. | ||
We sought to attack this problem by resorting to a stepwise strategy involving Heck cyclization and the subsequent diastereoselective hydrogenation. The Heck cyclization had favourably been achieved before9a,b which, however, would eliminate the original chirality at the carbon to be C15 in the target molecules. As a consequence, it would become necessary to effect a diastereoselective hydrogenation to restore the chiral center after the Heck reaction.
In light of this strategy, 8 was first transformed to 10 according to our previous conditions.9a With 10 in hand, we investigated the crucial diastereoselective hydrogenation. Under a hydrogen atmosphere of normal pressure, several conventional catalysts were tested for the reaction and the results are summarized in Table 1. Crabtree's catalyst did not show any catalytic activity for this reaction (entry 1). The RANEY® nickel catalysed hydrogenation resulted in a 1/1 mixture of diastereoisomers (entry 2). Delightfully, hydrogenation catalysed by palladium on carbon furnished a favourable 6.4/1 diastereoselectivity (entry 3). The stereochemistry of the major product will be confirmed at a later stage.
| Entry | Conditions | Yield% | dr |
|---|---|---|---|
| 1 | Crabtree's cat. | — | — |
| 2 | RANEY® Ni | — | 1/1 |
| 3 | Pd/C, EtOH | 86 | 6.4/1 |
With the key intermediate 6 in hand, we focused ourselves on the construction of the last piperidine D-ring (Scheme 4). Allylation of 6 generated 11 and 12-epi-11 as a separable 2.7/1 mixture. The structure of the major isomer 11 was established by X-ray crystallographic analysis,10 thereby confirming the structure of 6. Hydroboration of 11 followed by oxidative workup produced diol 12, which was subjected to a one-pot protocol involving mesylation, deprotection, and basification, to furnish 13. Dehydration of 13 resulted in 14, which underwent diastereoselective hydrogenation on Pd/C to deliver 15 with all the stereochemistry properly loaded.
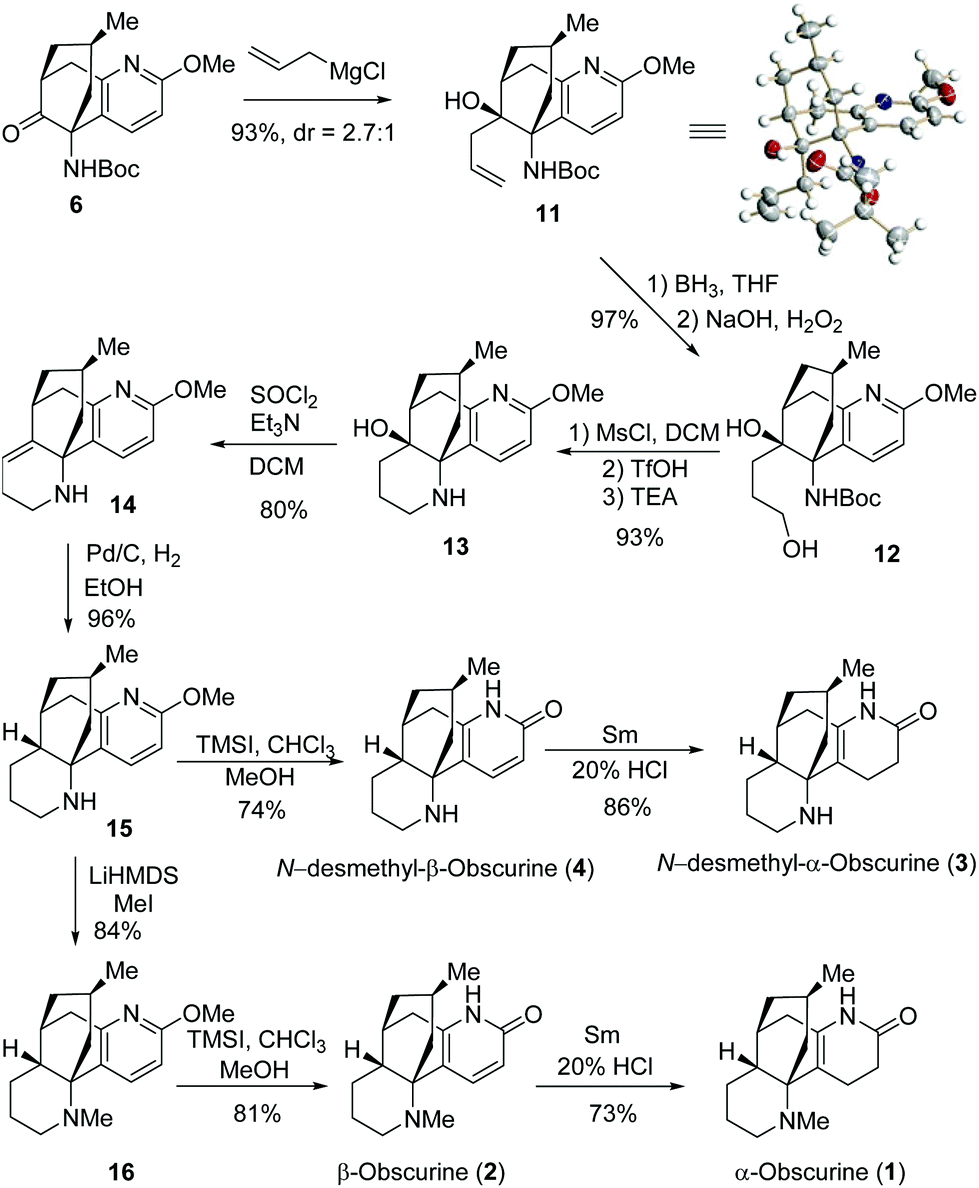 | ||
| Scheme 4 Total synthesis of obscurines 1–4. | ||
Eventually, compound 15 was converted to the target molecules (Scheme 4). Treatment of 15 with TMSI provided N-desmethyl-β-obscurine (4) in 74% yield.11 Reduction of 4 with Sm/HCl delivered N-desmethyl-α-obscurine (3) in 86% yield.12 On the other hand, 15 underwent N-methylation with LiHMDS/MeI to give 16. Under similar conditions, 16 was subjected to O-demethylation to furnish β-obscurine (2), which was reduced to afford 1 in good yields. The spectroscopic data of the synthetic samples of 1–4 matched those in the literature.
The facial selectivity in the hydrogenation of 10 is intriguing and deserves more comments. At a first glance, it seems that the undesired product stemming from the β-face approach would be more favoured. As depicted in Scheme 5A, the β face of the olefinic double bond corresponds to the convex side of the bicyclic framework. Therefore, the β face should have been more accessible for the hydrogenation, leading to the undesirable facial selectivity. However, the observed predominant product 6 resulted from the α face hydrogenation. This discrepancy possibly originated from the haptophilicity of the heteroatoms in the substrate.13 Moreover, the N-Boc group could also play a critical role by shielding the β face of the olefin, presumably as a result of minimizing its interaction with the pyridine moiety (Scheme 5B). However, the free ketoamine derived from 10 was not available for the hydrogenation reaction due to its lability.
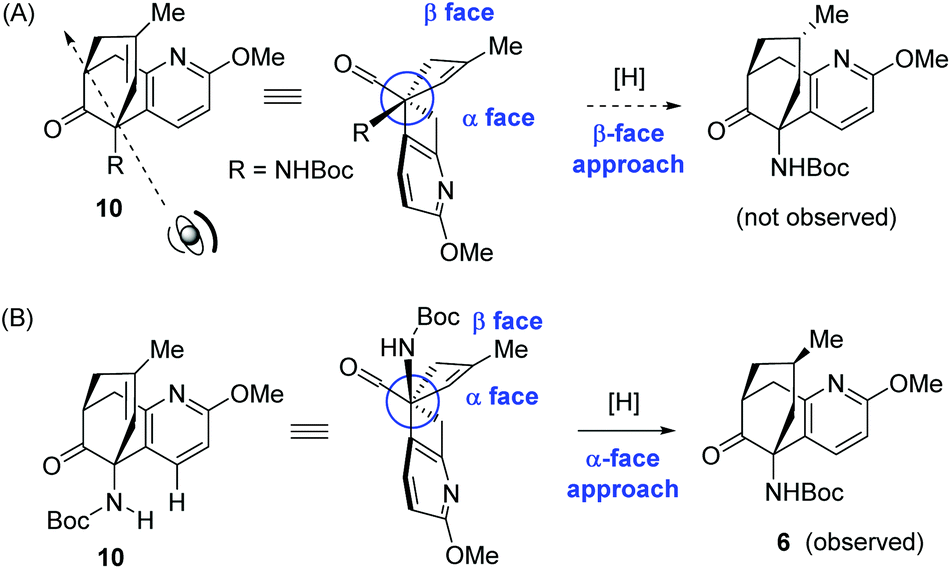 | ||
| Scheme 5 Conformational analysis for the hydrogenation of 10. | ||
To further gain an insight into the facial selectivity of the hydrogenation in this unique molecular framework, the catalytic hydrogenation reactions of more substrates containing the A/B/C ring system were investigated (Scheme 6). The hydrogenation of 17, catalysed either by Pd/C or RANEY® Ni, gave 18 essentially as a sole stereoisomer resulting from the β-face approach. In contrast, 19 underwent divergent hydrogenation reactions, furnishing predominantly 20 or 21 contingent on the catalytic conditions. The hydrogenation of 22 catalysed by RANEY® Ni furnished 23.10 Further, the parallel hydrogenation reactions of 24 and 26 provided preferably 25 and 27, respectively. These results clearly demonstrated that the Boc group benefited the hydrogenation reactions catalysed by Pd/C to deliver products with the desired facial selectivity while the OH group exerted a strong haptophilic effect in the RANEY® Ni-catalysed hydrogenation leading to the opposite facial selectivity. Importantly, these results can be of particular synthetic interest in view of the fact that both configurations at this stereocenter are present in natural products, such as acrifoline and annofoline.14
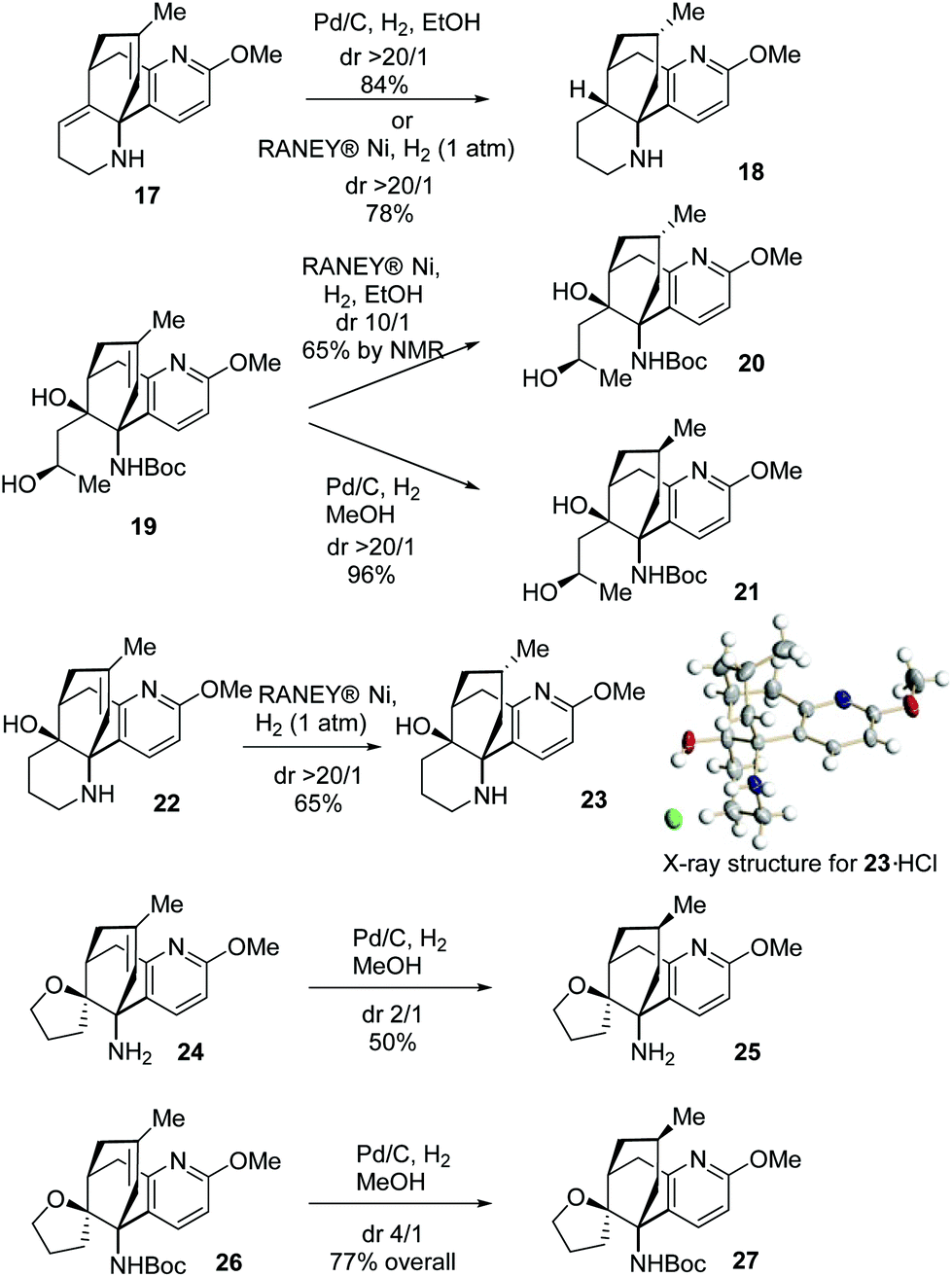 | ||
| Scheme 6 Catalytic hydrogenation of 17, 19, 22, 24 and 26. | ||
Conclusions
In summary, we have accomplished the asymmetric total synthesis of α-obscurine (1), β-obscurine (2), N-desmethyl-α-obscurine (3), and N-desmethyl-β-obscurine (4) with a new strategy, which features the approach to the A/B/C-ring system prior to the construction of the D-ring. Key reactions include the previously realized Buchwald–Hartwig coupling and the Heck cyclization reactions, and the newly developed diastereoselective hydrogenation, in a combined fashion to attain the A/B/C-ring system. In particular, the enabling hydrogenation reaction of 10 that fostered the critical C15 stereocenter, together with the hydrogenation reactions of 17, 19, 22, 24 and 26, constitute a collection of intriguing examples that can readily lend themselves to the total synthesis of relevant natural products. Endeavours along this line are currently underway and will be reported in due course.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (grant No. 21172246, 21290180, 21472210), and Youth Innovation Promotion Association CAS.Notes and references
- (a) Y. Hirasawa, J. Kobayashi and H. Morita, Heterocycles, 2009, 77, 679 CrossRef CAS; (b) G. T. Ha, R. K. Wong and Y. Zhang, Chem. Biodiversity, 2011, 8, 1189 CrossRef CAS PubMed; (c) A. R. Desilets, J. J. Gickas and K. C. Dunican, Ann. Pharmacother., 2009, 43, 514 CrossRef CAS PubMed; (d) A. P. Kozikowski and W. Tuckmantel, Acc. Chem. Res., 1999, 32, 641 CrossRef CAS.
- (a) X. Q. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752 RSC; (b) T. Hemscheidt, Top. Curr. Chem., 2000, 209, 175 CrossRef CAS; (c) T. Hemscheidt and I. D. Spenser, J. Am. Chem. Soc., 1996, 118, 1799 CrossRef CAS.
- R. H. F. Manske and L. Marion, Can. J. Res., 1942, 20, 87 CrossRef CAS.
- B. P. Moore and L. Marion, Can. J. Chem., 1953, 31, 952 CrossRef CAS.
- W. A. Ayer, J. A. Berezowsky and G. C. Iverach, Tetrahedron, 1962, 18, 567 CrossRef CAS.
- W. A. Ayer and G. C. Kasitu, Can. J. Chem., 1989, 67, 1077 CrossRef CAS.
- (a) D. Schumann, H. J. Müller and A. Naumann, Liebigs Ann. Chem., 1982, 1700 CrossRef CAS; (b) D. Schumann, H. J. Müller and A. Naumann, Liebigs Ann. Chem., 1982, 2057 CrossRef CAS; (c) D. Schumann and A. Naumann, Liebigs Ann. Chem., 1983, 220 CrossRef CAS.
- D. F. Fischer and R. Sarpong, J. Am. Chem. Soc., 2010, 132, 5926 CrossRef CAS PubMed.
- (a) R. Ding, B. F. Sun and G. Q. Lin, Org. Lett., 2012, 14, 4446 CrossRef CAS PubMed; (b) R. Ding, J. G. Fu, G. Q. Xu, B. F. Sun and G. Q. Lin, J. Org. Chem., 2014, 79, 240 CrossRef CAS PubMed; (c) J. Wang, B.-F. Sun, K. Cui and G.-Q. Lin, Org. Lett., 2012, 14, 6354 CrossRef CAS; (d) J. Wang, S.-G. Chen, B.-F. Sun, G.-Q. Lin and Y.-J. Shang, Eur. J. Chem., 2013, 19, 2539 CrossRef CAS PubMed; (e) X.-L. Wang, Y.-Y. Lu, J. Wang, X. Wang, H.-Q. Yao, G.-Q. Lin and B.-F. Sun, Org. Biomol. Chem., 2014, 12, 3562 RSC; (f) J. Wang, W.-B. Sun, Y.-Z. Li, X. Wang, B.-F. Sun, G.-Q. Lin and J.-P. Zou, Org. Chem. Front., 2015, 2, 674 RSC.
- CCDC 1412716 (11) and CCDC 1412717 (23) contain the supplementary crystallographic data for this paper.
- (a) L. Qian and R. Ji, Tetrahedron Lett., 1989, 30, 2089 CrossRef CAS; (b) Y. Xia and A. P. Kozikowski, J. Am. Chem. Soc., 1989, 111, 4116 CrossRef CAS.
- Y. Kamochi and T. Kudo, Chem. Pharm. Bull., 1995, 43, 1442 CrossRef.
- For selected examples of haptophilic hydrogenation, see: (a) H. W. Thompson and R. E. Naipawer, J. Am. Chem. Soc., 1973, 95, 6379 CrossRef CAS; (b) H. W. Thompson and J. K. Wong, J. Org. Chem., 1985, 50, 4270 CrossRef CAS; (c) L. E. Overman and A. L. Tomasi, J. Am. Chem. Soc., 1998, 120, 4039 CrossRef CAS; (d) J. Tamiya and E. J. Sorensen, J. Am. Chem. Soc., 2000, 122, 9556 CrossRef CAS; (e) J. Tamiya and E. J. Sorensen, Tetrahedron, 2003, 59, 6921 CrossRef CAS; (f) Q. Zhou, X. Chen and D. Ma, Angew. Chem., Int. Ed., 2010, 49, 3513 CrossRef CAS PubMed; (g) K. Molawi, N. Delpont and A. M. Echavarren, Angew. Chem., Int. Ed., 2010, 49, 3517 CrossRef CAS PubMed; (h) J. Wang, S.-G. Chen, B.-F. Sun, G.-Q. Lin and Y.-J. Shang, Chem. – Eur. J., 2013, 19, 2539 CrossRef CAS PubMed.
- (a) W. N. French and D. B. MacLean, Can. J. Chem., 1961, 39, 2100 CrossRef CAS; (b) R. H. Burnell and D. R. Taylor, Tetrahedron, 1961, 15, 173 CrossRef CAS; (c) W. A. Ayer, L. M. Browne, A. W. Elgersma and P. P. Singer, Can. J. Chem., 1990, 68, 1300 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, and copies of 1H, 13C and 2D NMR spectra. CCDC 1412716 and 1412717. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00355e |
| This journal is © the Partner Organisations 2016 |