
Harnessing uranyl oxo atoms via halogen bonding interactions in molecular uranyl materials featuring 2,5-diiodobenzoic acid and N-donor capping ligands†
Korey P.
Carter
,
Mark
Kalaj
and
Christopher L.
Cahill
*
Department of Chemistry, The George Washington University, 800 22nd Street, NW, Washington, D.C. 20052, USA. E-mail: cahill@gwu.edu; Tel: +1 (202) 994-6959
First published on 12th October 2016
Abstract
The syntheses and crystal structures of five new compounds containing the UO22+ cation, 2,5-diiodobenzoic acid, and a chelating N-donor (2,2′-bipyridine (bipy) (1), 1,10-phenanthroline (phen) (2 and 3), 2,2′:6′,2′′-terpyridine (terpy) (4), or 4′-chloro-2,2′:6′,2′′-terpyridine (Cl-terpy) (5)) are described and the spectroscopic properties (both vibrational and luminescent) and stretching and interaction force constants of complexes 2, 4, and 5 are reported. Single crystal X-ray diffraction analysis of these materials shows that variation of the chelating N-donor with the same benzoic acid featuring multiple, polarizable halogens at the periphery allows for the systematic accessing of uranyl oxo atoms for non-covalent assembly, which is notable as these atoms are generally terminal. Spectroscopic characterization of complexes 2, 4, and 5 indicate that oxo atom participation in halogen bonding interactions may complement the effects of the electron donating ability of the capping ligand on corresponding uranyl luminescence and vibrational spectra, each contributing to the observed bathochromic shifts.
Introduction
Supramolecular chemistry within hybrid materials incorporating hexavalent uranium is an area of recent interest as it presents a new route to yield potentially unique structure types and unexpected properties that are unobtainable via traditional metal-to-ligand coordination.1–3 Vital to this approach is the formation of predictable building units (tectons) and chemically robust, attractive motifs (synthons), which are capable of guiding molecular arrangement. This strategy, of directed assembly of molecular tectons into crystalline architectures via attractive, noncovalent synthons, provides an opportunity to circumvent synthetic challenges introduced by uranyl hydrolysis, and its continued development is contingent upon a comprehensive understanding of interaction and acceptor-donor pairing preferences. In order to fully utilize the potential of inorganic crystal engineering and design a material with a desired set of properties, the relationship between intermolecular interactions and the resulting structure must be fully understood,4 yet metrics for these remain underdeveloped in both lanthanide5–8 and actinide chemistry.9–11Adopting a directed approach to assembly in a uranyl system involves finding a way to restrict or “shut down” hydrolysis in order to end up with predictable molecular tectons. Our group has primarily focused on this problem via the synthesis of [UO2]2+ materials in highly acidic and high halide media,12–17 yet we have also been successful in generating discrete, reproducible tectons via the use of a chelating ligand in the first coordination sphere to force promotion of a single species with an inherent affinity for forming a specific coordination geometry about the [UO2]2+ cation.18–20
The synthesis of molecular uranyl materials based on a combination of coordination and supramolecular chemistry principles is a topic of continued interest in our lab, and whereas the literature is indeed rich with a wide variety of molecular uranyl species,2,3,21–25 efforts to promote extended structures via the use of non-covalent interactions remains underexplored. An early foray into this area utilized the composite synthesis methodology with a series of halogen functionalized benzoic acids and yielded uranyl coordination polymers, which employed halogen–halogen interactions for assembly.26 More recently we have adopted a dual-ligand approach, where a polypyridyl chelating N-donor ‘capping’ ligand thwarts hydrolysis and then halogen functionalized benzoic acids assemble molecular tectons via halogen or hydrogen bonding interactions, π–π stacking, or a combination thereof.19,20 Herein we continue this synthetic strategy and now involve the uranyl oxo atoms as participants in supramolecular assembly rather than limit uranyl cation connectivity to the equatorial plane. The uranyl oxo groups are typically terminal (hence the prevalence of bipyramidal building units),2,27,28 yet we will demonstrate these atoms can be utilized for assembly via the judicious selection of an electron-donating N-donor in the equatorial plane paired with benzoic acid linkers featuring polarizable halogen atoms at the periphery to promote halogen bonding.
Building on our previous work focusing on uranyl and lanthanide supramolecular assembly,19,20,29–32 we set out to explore a system that features the 2,5-diiodobenzoic acid ligand and chelating N-donor ligands of varying size (bipy, phen, terpy, and Cl-terpy) used for the purpose of controlling hydrolysis and tailoring assembly of the uranyl tectons. Changes in 2,5-diiodobenzoic acid coordination modes and chelating N-donor size yield two distinct molecular tectons containing a range of supramolecular synthons sites (i.e. halogen bonding interactions, I–I halogen–halogen interactions, I–Cl halogen–halogen interactions, and hydrogen bonding interactions) that lie at the edge of the immediate coordination sphere. Herein we report the synthesis, crystal structures and modes of supramolecular assembly for a family of five new uranyl-2,5-diiodobenzoic acid-N-donor materials. Additionally, the vibrational and luminescence spectra of complexes 2, 4, and 5 were collected and these results allow for a deeper discussion of uranyl structure–property relationships as we examine the spectroscopic manifestations of oxo participation in non-covalent assembly via the calculation of stretching force (k1) and interaction force (k12) constants using a valence bond potential. Whereas weak hydrogen-bonding with the uranyl oxo atoms is relatively common, there are limited examples of the uranyl oxo groups participating in ‘stronger’ non-covalent interactions (i.e. halogen bonding),10,16,19 thus finding new routes to systematically harness the oxo atoms of the uranyl for non-covalent assembly indeed represent a “frontier” of uranyl crystal chemistry, and have significant potential to contribute to our understanding of separations/fuel reprocessing methods, impact environmental transport of actinide (An) species, and enhance uranyl structure–property relationship delineations.
Experimental section
Materials and methods
Caution: Whereas the uranium oxyacetate dihydrate [UO2(CH3COO)2]·2H2O and uranyl nitrate hexahydrate [UO2(NO3)2]·6H2O used in this study contained depleted U, standard precautions for handling radioactive and toxic substances should be followed.All organic materials, 2,5-diiodobenzoic acid (Sigma Aldrich, 97%), and the chelating N-donors 2,2′-bipyridine (Sigma Aldrich, 99+%), 1,10-phenanthroline (Alfa Aesar, 99%), 2,2′:6′,2′′-terpyridine (Alfa Aesar, 97%), and 4′-chloro-2,2′:6′,2′′-terpyridine (Alfa Aesar, 97%), were purchased and used as received.
Synthesis
All complexes discussed herein were synthesized via hydrothermal methods at autogeneous pressure in a 23 mL Teflon-lined Parr bomb at an oven temperature of 120 °C for either two (complex 5) or three days (complexes 1–4). A molar ratio of (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:667—UO22+:2,5-diiodobenzoic acid:N-Donor:water—where N-Donors are bipy and phen) was used for complexes 1–3, whereas for complexes 4 and 5 a molar ratio of (1:2:1:667—UO22+:2,5-diiodobenzoic acid:N-Donor:water—where N-Donors are terpy and Cl-terpy) was optimal for single crystal growth. Complexes were synthesized at unadjusted pH (ca. 2.7), except for 3 which only appears at pH values >5.
:2:2:667—UO22+:2,5-diiodobenzoic acid:N-Donor:water—where N-Donors are bipy and phen) was used for complexes 1–3, whereas for complexes 4 and 5 a molar ratio of (1:2:1:667—UO22+:2,5-diiodobenzoic acid:N-Donor:water—where N-Donors are terpy and Cl-terpy) was optimal for single crystal growth. Complexes were synthesized at unadjusted pH (ca. 2.7), except for 3 which only appears at pH values >5.
Characterization
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Chemformula | C24H14N2I4O6U | C38H24N4I4O10U2 | C26H14N2I4 O6U | C29H17N3I4O6U | C29H16N3I4ClO6U |
| Formula weight | 1172.00 | 1680.27 | 1196.02 | 1249.08 | 1283.53 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/n | P21/c | P21/c |
| a (Å) | 7.6634(2) | 9.2236(8) | 19.2039(10) | 14.7607(9) | 15.720(7) |
| b (Å) | 25.2373(7) | 28.080(3) | 10.5773(5) | 16.8925(10) | 16.555(8) |
| c (Å) | 15.2304(4) | 8.6636(8) | 28.8136(14) | 13.3636(8) | 13.408(7) |
| α (°) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 101.744(4) | 104.853(11) | 96.772(7) | 104.521(11) | 106.011(6) |
| γ (°) | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 2883.95(14) | 2168.9(4) | 5811.9(5) | 3225.7(4) | 3354.0(3) |
| Z | 4 | 2 | 8 | 4 | 4 |
| T (K) | 293(2) | 293(2) | 293(2) | 293(2) | 293(2) |
| λ (Mo Kα) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| D calc (g cm−3) | 2.699 | 2.573 | 2.734 | 2.572 | 2.542 |
| μ (mm−1) | 9.950 | 10.362 | 9.878 | 8.906 | 8.646 |
| R int | 0.0470 | 0.0548 | 0.0467 | 0.0661 | 0.0465 |
| R 1 [I > 2σ(I)] | 0.0399 | 0.0362 | 0.0344 | 0.0396 | 0.0544 |
| wR2 [I > 2σ(I)] | 0.0987 | 0.0839 | 0.0709 | 0.0934 | 0.1436 |
Powder X-ray diffraction
Powder X-ray diffraction (PXRD) data on the bulk reaction product of complexes 1–5 (Fig. S5–S9, ESI†) were used to examine the purity of each sample. All data were collected on a Rigaku Miniflex (Cu Kα, 2θ = 3–60°) and were analyzed using the JADE software program.41 Initially, the bulk products of complexes 1 and 3 were found to contain multiple solid-state phases. Complex 3 was found to co-form with 2 (Fig. S7, ESI†) and efforts to isolate 3 as a single phase or increase the yield of 3 with respect to 2 were unsuccessful over a range of conditions. Attempts were made to identify and/or remove the impurities from 1 using a range of organic solvents and reaction conditions, yet they persisted and thus prevented further spectroscopic characterization of this material.Spectroscopic characterization
Infrared spectra of isolated single crystals of 2, 4, and 5 were collected from 400 to 4000 cm−1 using a Bruker Tensor 27 FT-IR microspectrometer. Crystals were placed on glass microscope slides and crushed using a diamond attenuated total reflectance (ATR) microscope objective.Raman spectra for single crystals of 2, 4, and 5 were collected using a Bruker Sentinel system linked via fiber optics to a video assisted Raman probe equipped with a 785 nm 400 mW and a high sensitivity TE-cooled, 1024 × 255 CCD array. The spectra were collected for fifteen seconds with four signal accumulations over the range of 80–3200 cm−1.
Low temperature (77 K) solid-state luminescence measurements were obtained for 2, 4, and 5 on a Horiba JobinYvon Fluorolog-3 spectrophotometer. Data were manipulated using the FluoroEssence software package and the final plots of the resulting solid-state spectra were made in Microsoft Excel. Samples were prepared by placing 10–15 single crystals (by visual inspection) into a quartz NMR tube, immersing the NMR tubes in liquid N2, and allowing the samples to equilibrate. Experimental parameters (slit width, integration time, etc.) were adjusted on a sample-by-sample basis to obtain suitable data with a sufficiently high signal-to-noise ratio.
Results
Description of structures
Single crystal X-ray crystallographic analyses revealed two unique building units in this series of molecular complexes: monomers (1, 3–5), and dimers (2). Local structures are described in detail for complexes 1–4, as they represent each of the unique coordination environments. Modes of supramolecular assembly are described for all complexes (1–5) as they are affected by changes in the coordination modes of the 2,5-diiodobenzoic acid ligands and the systematic change in the nature of the chelating N-donor ligands.Complex 1, [UO2(C10H8N2)(C7H3I2O2)2], crystallizes in the space group P21/c and features a local structure that closely resembles the nominally isostructural UO2-3,5-dichlorobenzoic acid-bipy analogue we reported recently.20 The single crystallographically unique [UO2]2+ cation adopts a distorted hexagonal bipyramidal geometry and features equatorial U1–O bonds to the two 2,5-diiodobenzoic acid ligands (O3–O6) at an average distance of 2.447 Å (Fig. 1). Additionally, U1–N distances to the bidentate bipy molecule (N1 and N2) are 2.638(6) Å (U1–N1) and 2.632(6) Å (U1–N2), respectively.
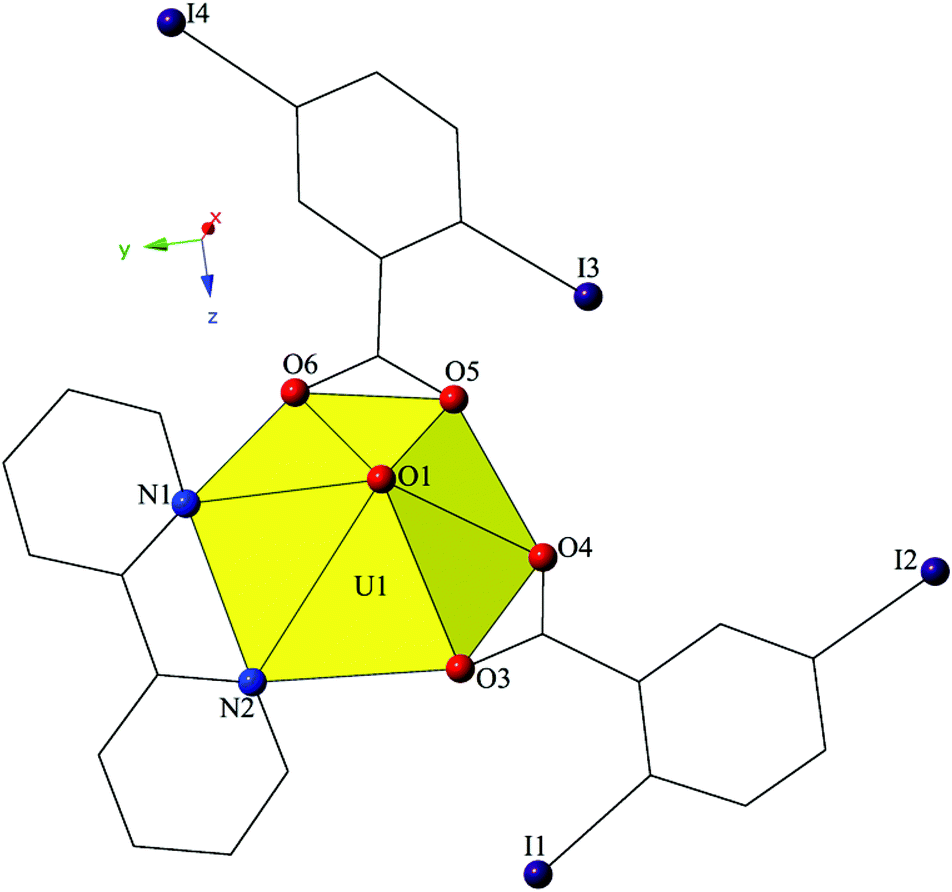 | ||
| Fig. 1 Polyhedral representation of local coordination geometry of 1. Yellow polyhedra represent uranium metal centers, whereas purple, red, and blue spheres represent iodine, oxygen, and nitrogen atoms, respectively. All H atoms have been omitted for clarity. | ||
Looking at the global structure of 1 (Fig. 2), the uranyl tectons are linked to form a 1D chain in [100] direction by a pair of alternating hydrogen-bonding interactions from two different hydrogen atoms on the chelating bipy ligand. The first interaction is between the uranyl oxo atom (O1) and the aromatic hydrogen atom (H4) at a distance of 2.65 Å, and the second interaction is from the other uranyl oxo atom (O2) to another aromatic H atom (H7) at a distance of 2.57 Å. Weak C–H⋯O hydrogen bonding interactions with the uranyl oxo atoms are relatively common,10,42–46 and occur more frequently than actually discussed, yet the occurrence in 1 is the only example of this synthon we observe in this family of materials.
 | ||
| Fig. 2 Complex 1 viewed along the [100] direction highlighting the hydrogen-bonding interactions that assemble the monomers of 1 into a 1D chain. | ||
The introduction of phen as a capping ligand yields complex 2, [UO2(OH)(C12H8N2)(C7H3I2O2)]2, which crystallizes in the space group P21/c. Complex 2 consists of a uranyl dimer where the single unique [UO2]2+ cation and its symmetry equivalent are bridged via point-sharing μ2-OH groups (O5, O5′) (confirmed via bond valence summations, Table S5, ESI†), and both feature coordination geometries that can be described as pentagonal bipyramidal (Fig. 3). U1–O bond distances to the point-sharing μ2-OH groups (O5, O5′) are 2.286(5) Å (U1–O5, U1′-O5′) and 2.341(5) Å (U1–O5′, U1′–O5), respectively. Completing the coordination sphere of each uranyl cation is a bidentate phen molecule with U1–N distances of 2.625(5) Å (U1–N1) and 2.587(6) Å (U1–N2), respectively, and a monodentate 2,5-diiodobenzoic acid ligand, coordinated via O3 at a distance of 2.346(5).
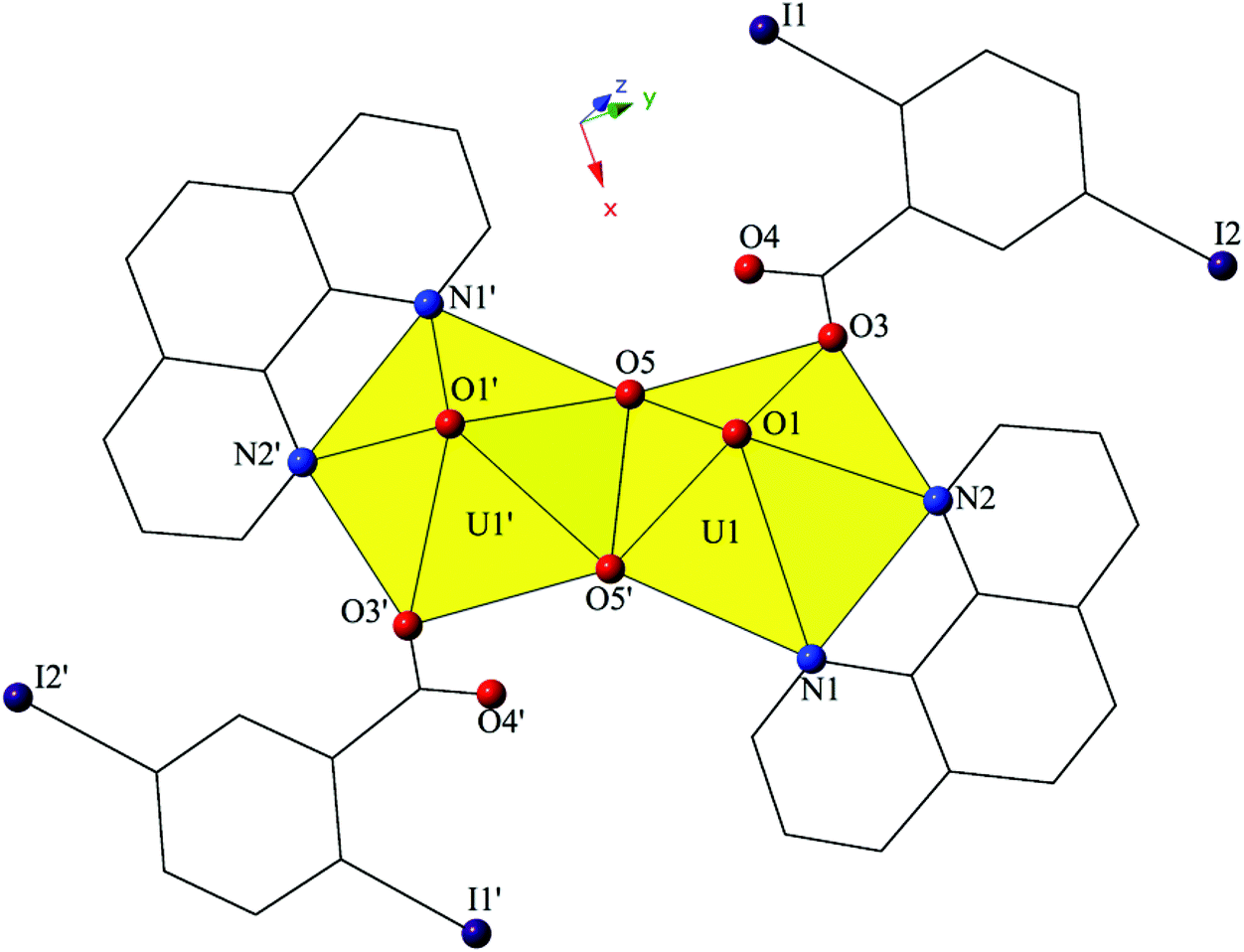 | ||
| Fig. 3 Polyhedral representation of local coordination geometry of 2. All H atoms have been omitted for clarity. | ||
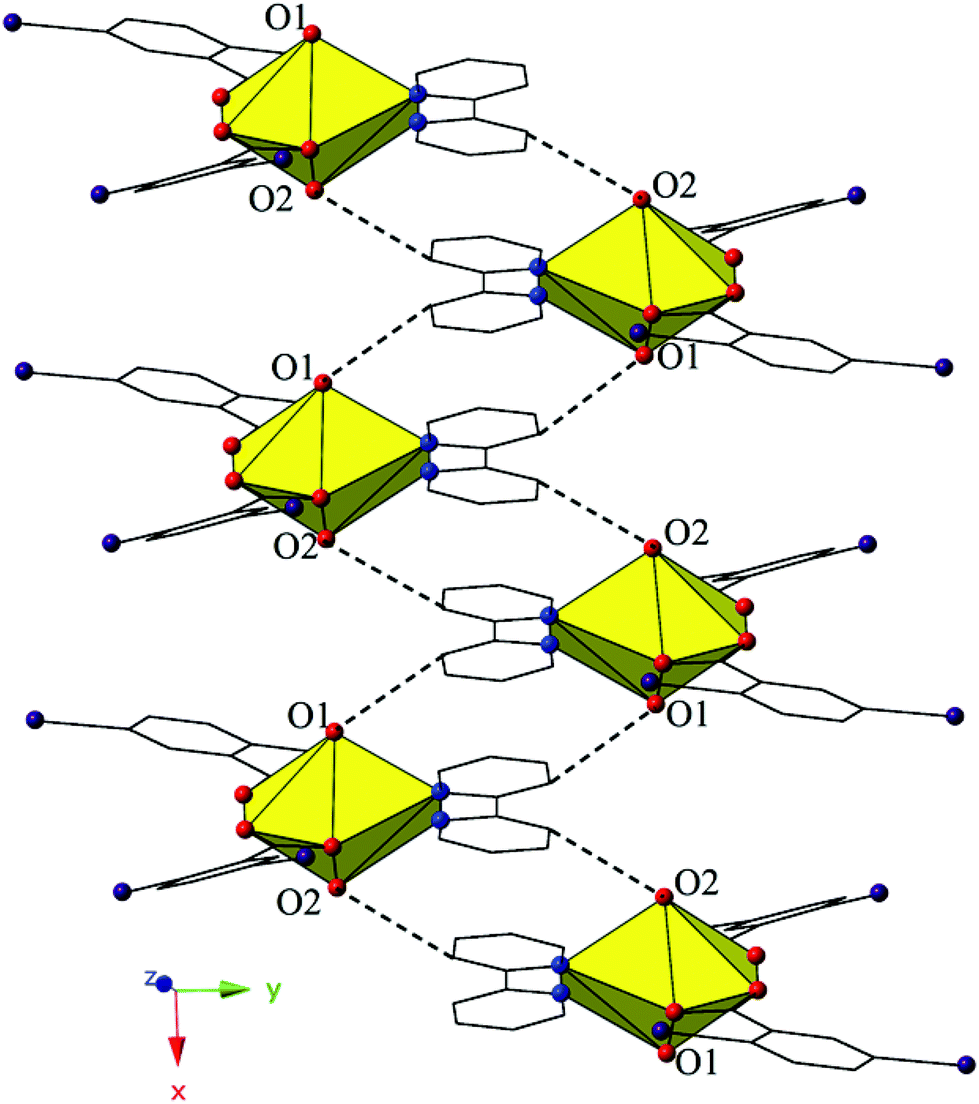
The dimers of 2 are linked to form a 1D chain that propagates in the [100] direction via halogen bonding interactions between iodine atoms from a 2,5-diiodobenzoic acid ligand on one unit with uranyl oxo atoms on the neighboring units (Fig. 4). The corresponding I–O interaction distance and angle are 3.356(5) Å (95.9% sum of the van der Waals radii) and ∠C–I⋯O 166.4(2)°, which is suggestive of a robust interaction between the iodine (I1) and the oxo atom (O1). This is notable as the uranyl oxo atoms are typically terminal and whereas explicit discussion of the uranyl oxo groups acting as hydrogen bond acceptors has been reported,10,47,48 examples of uranyl oxo atoms participating in halogen bonding remain rare.16,19 This occurrence in 2 is the first of four examples of the oxo groups adopting the same function we will describe herein.
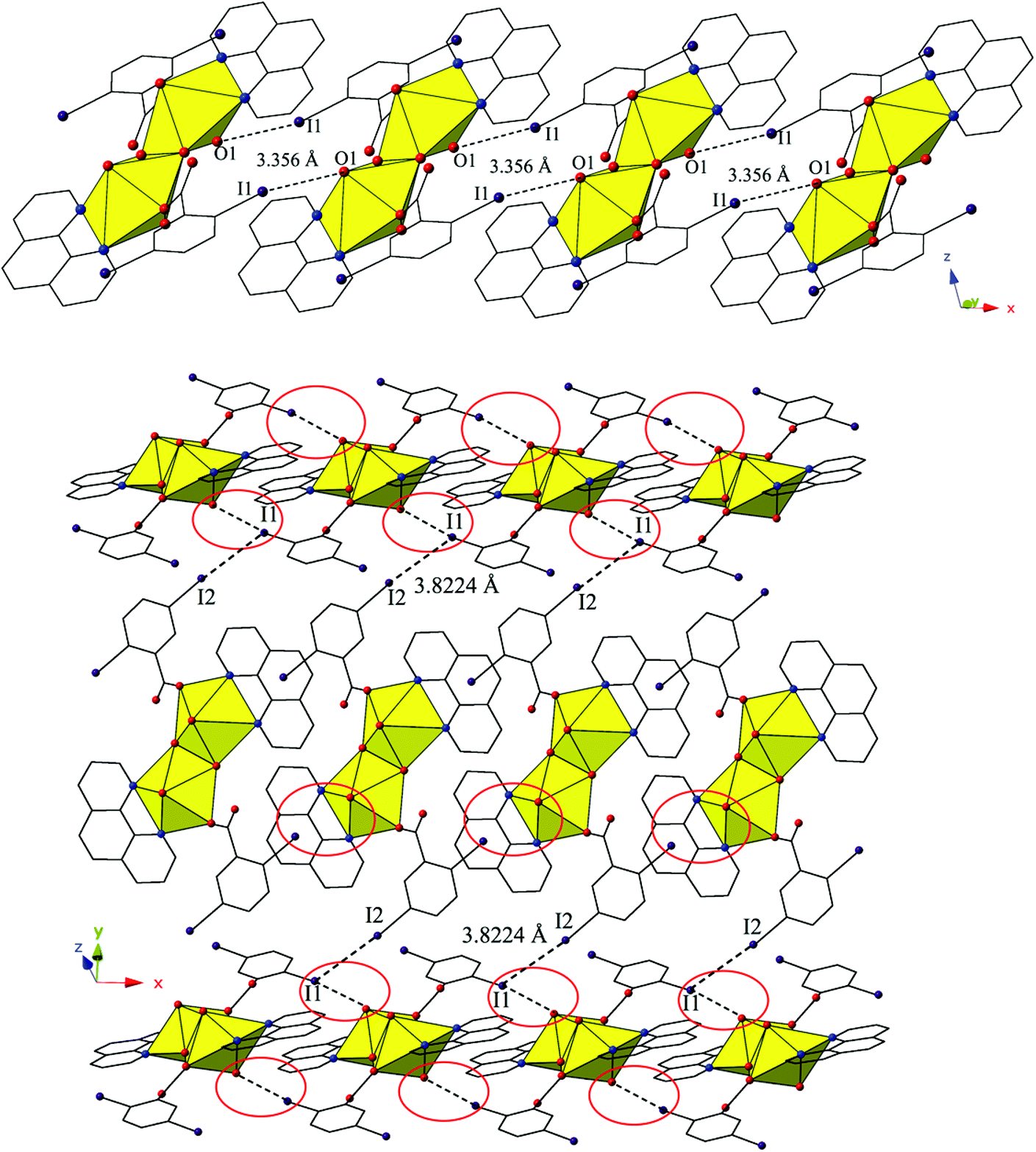 | ||
| Fig. 4 (Top) Complex 2 viewed along the [100] direction. Halogen bonding interactions with uranyl oxo atoms that link the dimers of 2 into 1D chain are shown. (Bottom) 2 in the (110) plane illustrating the Type II I–I interactions that further assemble UO22+ tectons into a supramolecular 2D sheet. Halogen bonding interactions with uranyl oxo groups that link tectons of 2 into 1D chain are highlighted by red circles. | ||
Further assembly of the monomers of 2 into a supramolecular 2D sheet is a result of Type II halogen–halogen interactions between iodine atoms from 2,5-diiodobenzoic acid ligands on neighboring 1D chains that align orthogonal with one another (Fig. 4). Halogen–halogen interactions generally adopt one of two geometries,49,50 and the interactions in 2 (I1–I2) are classified as Type II interactions based on the criteria delineated by Desiraju et al.51,52 The Type II interactions in 2 are between I1, which is participating in the oxo-halogen bonding interaction described above, and I2 on a neighboring orthogonal chain, and are at a distance of 3.8224(11) Å (96.5% sum of the van der Waals radii) with corresponding angles of 174.3(2)° (C18–I2–I1) and 119.48(17)° (C15–I1–I2), respectively.
Increasing the pH of the reaction that yielded 2 resulted in small amounts of complex 3, [UO2(C12H8N2)(C7H3I2O2)2]2, which crystallizes in the space group P21/n. The asymmetric unit of 3 consists of two crystallographically unique uranyl monomers, both of which have adopted hexagonal bipyramidal coordination geometries (Fig. 5). Both unique uranyl cations are capped by bidentate phen moieties and then chelated by two bidentate 2,5-diiodobenzoic acid ligands. U1–O bond distances to the 2,5-diiodobenzoic acid ligands (O3–O6) are at an average of 2.475 Å, whereas U2–O distances (O9–O12) are at an average of 2.470 Å. The hexagonal bipyramidal coordination spheres of both U1 and U2 are completed by bidentate phen moieties bound through their two nitrogen atoms (N1–N4) and are at average distances of 2.670 Å (U1–N) and 2.663 Å (U2–N), respectively.
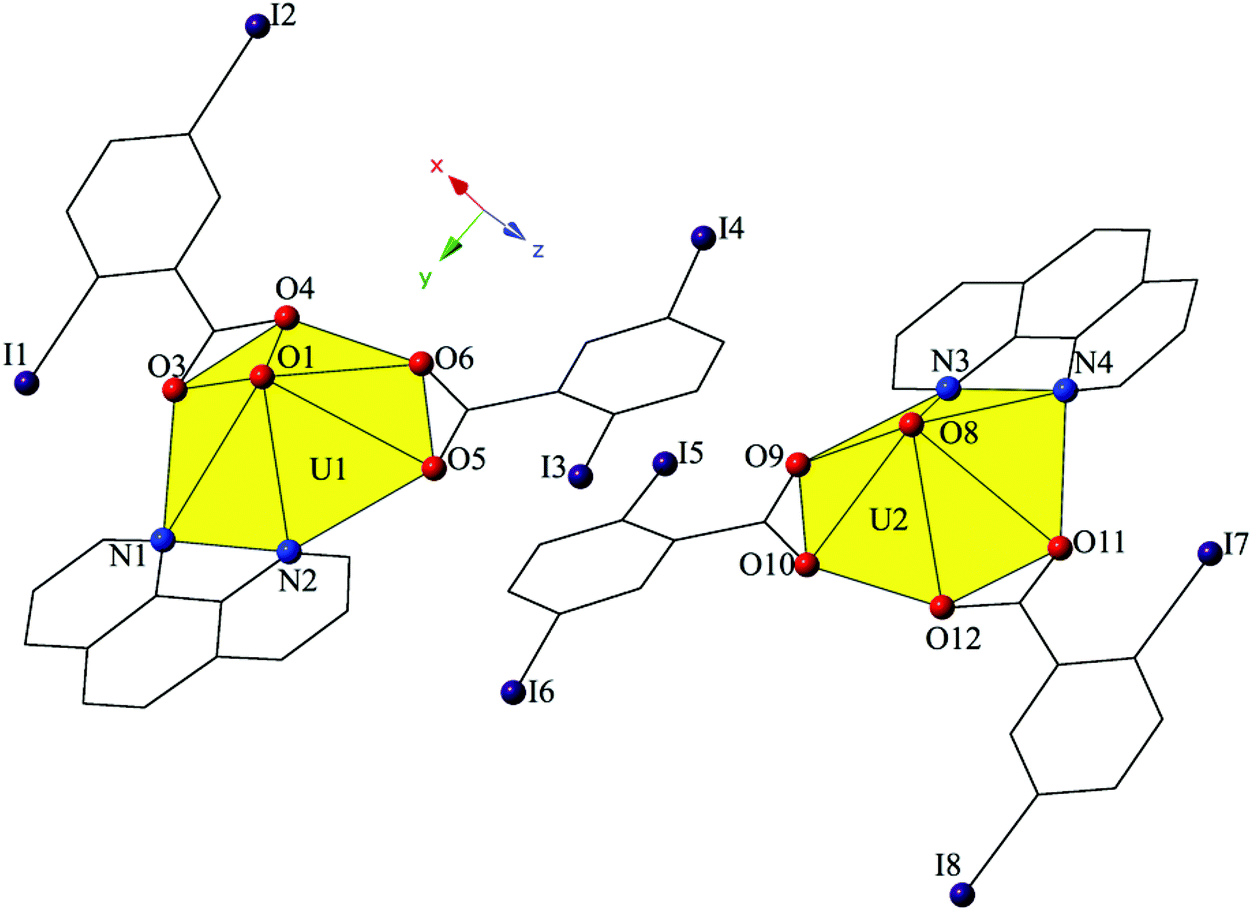 | ||
| Fig. 5 Polyhedral representation of local coordination geometry of 3. All H atoms have been omitted for clarity. | ||
The two unique monomers of 3 are linked into two unique, parallel 1D chains along the [010] direction by halogen–halogen interactions between iodine atoms on neighboring monomeric tectons (Fig. 6). The two interactions adopt geometries that can both be described as Type I,52 and are at distances of 3.8164(2) Å (I1–I2) and 3.7600 (2) Å (I7–I8), respectively. Corresponding angles (∠C–I–I) for the interactions are 158.27(1)° (C15–I1–I2) and 157.63(1)° (C18–I2–I1) (Type I interaction I1–I2) and 166.02(1)° (C48–I7–I8) and 161.08(1)° (C51–I8–I7) (Type I interaction I7–I8).
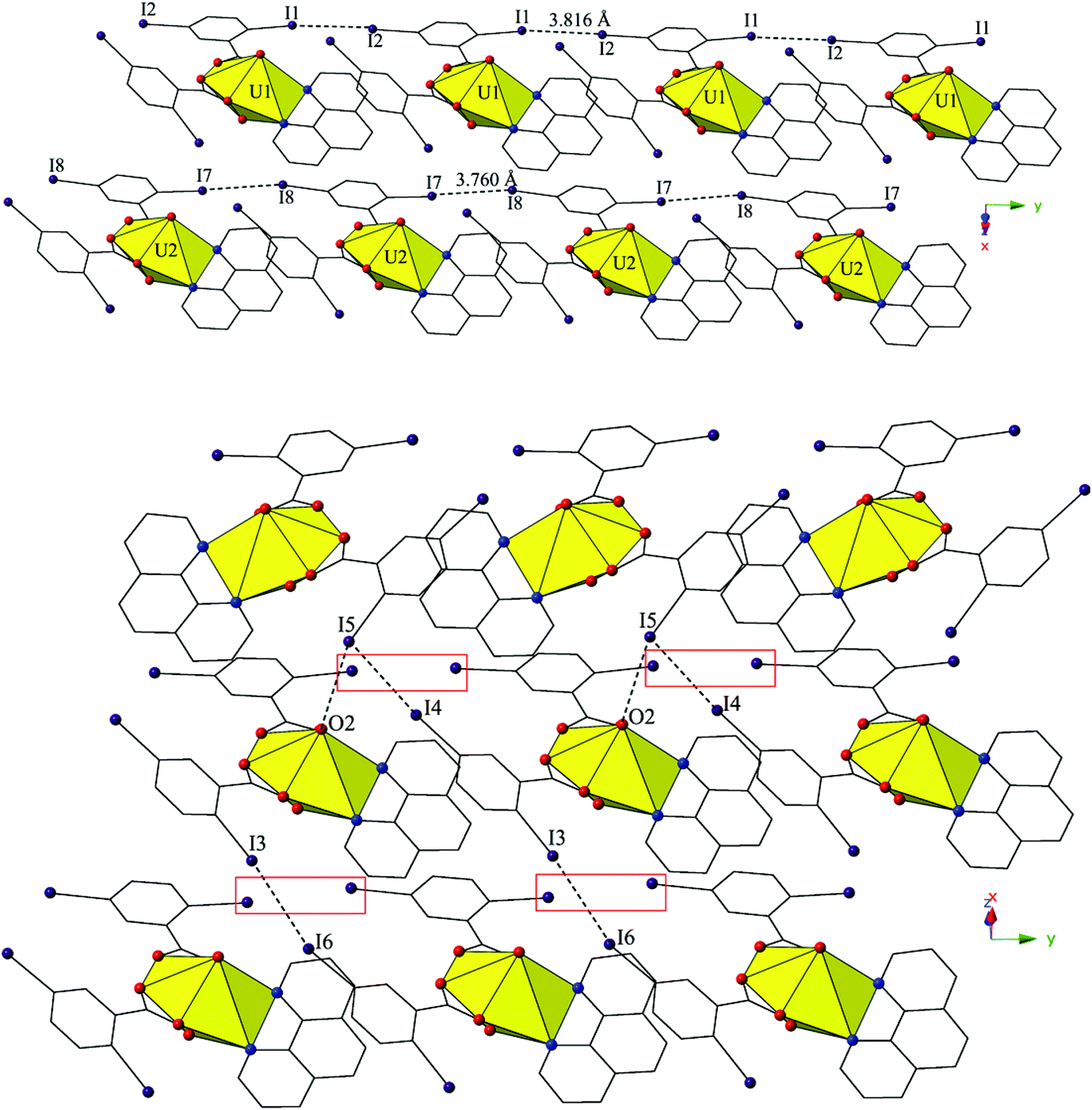 | ||
| Fig. 6 (Top) Complex 3 viewed along the [010] direction. Type I I–I interactions that link monomers of 3 into two parallel 1D chains are shown. (Bottom) 3 viewed in approximately the (110) plane illustrating halogen bonding interaction with the uranyl oxo group and Type I and II I–I interactions that further assemble the two unique chains of 3 into supramolecular 2D sheet. Type I I–I interactions that assemble tectons of 3 into parallel 1D chains are highlighted by red boxes. | ||
Further assembly of the unique supramolecular 1D chains of 3 into a 2D sheet is the result of an alternating combination of halogen bonding and halogen–halogen synthons (Fig. 6). The four iodine atoms not involved in the assembly of the monomers of 3 into 1D chains all participate in the interactions that link the two unique chains of 3 into a supramolecular 2D sheet. This additional connectivity is a result of alternating sets of synthons as chains of 3 are connected by bifurcated halogen bonding (I5–O2) and halogen–halogen (I4–I5) interactions and then by a second halogen–halogen (I3–I6) interaction. The bifurcated linkage features an iodine atom (I5) acting as both a halogen bond donor and acceptor, participating in both a Type II halogen–halogen interaction52 (I4–I5) and a halogen bonding interaction with one of the uranyl oxo atoms (O2). The Type II interaction52 is at a distance 3.7030(2) Å (93.5% sum of the van der Waals radii) with θ1 (C25–I4–I5) and θ2 (C41–I5–I4) values of 169.28(1)° and 102.29(1)°, respectively, and the halogen bonding (I5–O2) interaction with the uranyl oxo group is at a distance of 3.2588(2) Å with a corresponding angle of ∠C–I⋯O 157.21(1)°. The second interaction (I3–I6) adopts a Type I orientation,52 and is at a distance of 3.8888(2) Å (98.2% sum of the van der Waals radii) with θ1 (C22–I3–I6) and θ2 (C44–I6–I3) values of 166.87(1)° and 159.06(1)°, respectively.
Complex 4, [UO2(C15H11N3)(C7H3I2O2)2], crystallizes in the space group P21/c and features a local structure that closely resembles a UO2-3,5-dibromobenzoic acid-terpy material we reported previously.19 The asymmetric unit of 4 contains one pentagonal bipyramidal uranyl tecton where the crystallographically unique [UO2]2+ cation is chelated by a tridentate terpy molecule and further bound by two monodentate 2,5-diiodobenzoic acid ligands (Fig. 7). U1–O bond distances to the monodentate 2,5-diiodobenzoic acid ligands (O3 and O5) are 2.275(5) Å and 2.276(5) Å, respectively. The tridentate terpy molecule (N1, N2 and N3) caps the uranyl coordination sphere and the average U1–N distance is 2.581 Å.
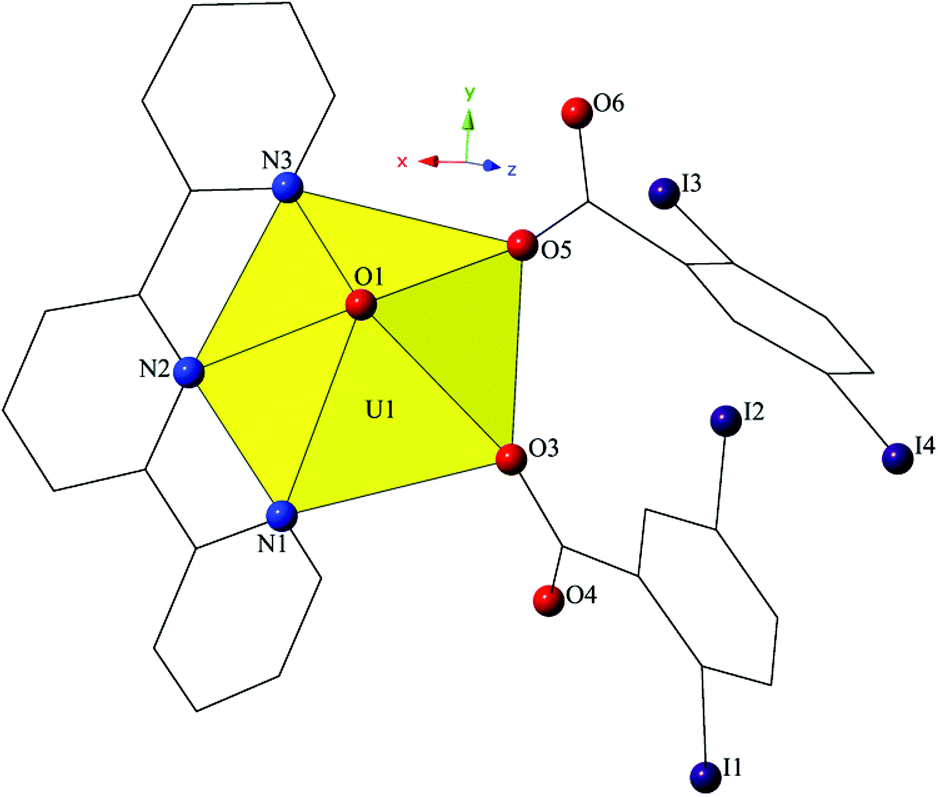 | ||
| Fig. 7 Polyhedral representation of asymmetric unit of 4. All H atoms have been omitted for clarity. | ||
The monomers of 4 are assembled into an undulating 2D sheet in the (011) plane by a pair of alternating halogen bonding interactions with both of the uranyl oxo atoms (Fig. 8). The halogen bonding interactions are between iodine atoms from 2,5-diiodobenzoic acid ligands and uranyl oxo atoms on the neighboring monomers and the corresponding I–O interaction distances and angles are 3.437(5) Å and ∠C–I⋯O 161.3(2)° (I1–O1) and 3.459(6) Å and ∠C–I⋯O 158.5(3)° (I3–O2), respectively. Both interactions are well within the sum of the van der Waals radii of iodine and oxygen (3.50 Å), and as far as we can surmise, this is the first reported instance of both uranyl oxo atoms simultaneously participating in halogen bonding interactions.
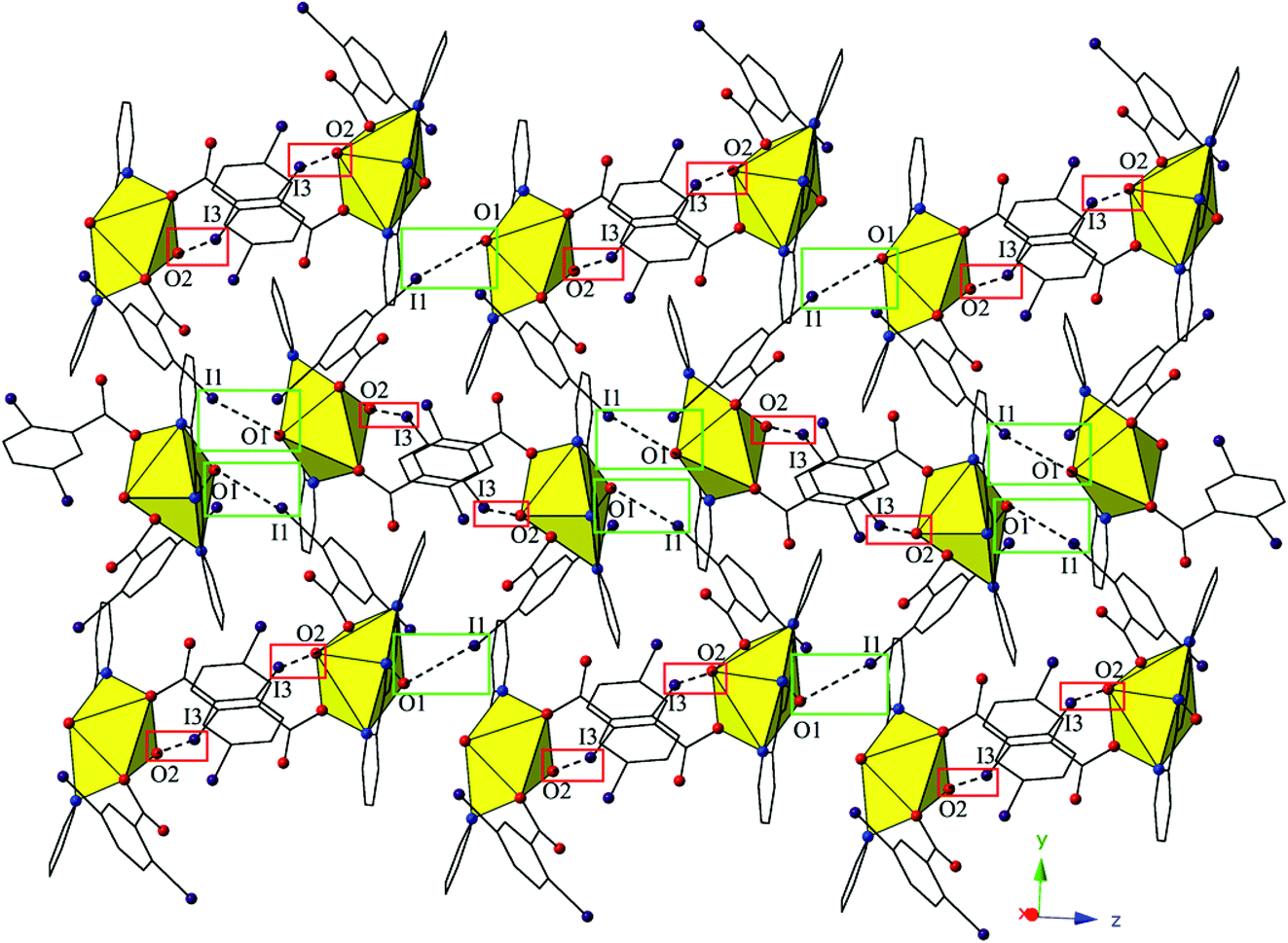 | ||
| Fig. 8 Complex 4 viewed in the (011) plane showcasing the two unique halogen-bonding interactions with the uranyl oxo atoms (O1 and O2) that assemble the monomers of 4 into a supramolecular 2D sheet. | ||
The 2D sheets of 4 are further linked into a supramolecular 3D network in the (110) plane via a third halogen bonding interaction between an iodine atom (I2) not participating in the formation of the 2D sheets and a carboxylate oxygen atom (O5) (Fig. S2, ESI†). The corresponding distance and angle for the halogen bonding interactions (I2–O5) are 3.363(5) Å (96.1% sum of the van der Waals radii) and ∠C–I⋯O 177.1(3)°.
Complex 5, [UO2(C15H10ClN3)(C7H3I2O2)2], crystallizes in the space group P21/c. Beyond the introduction of Cl-terpy as a capping ligand, 5 features a nearly identical local coordination geometry to 4 and will thus not be described in detail. The global structure of 5 is similar to 4, yet the undulating 2D sheet that forms in the (011) plane is the result of a halogen bonding interactions with a single uranyl oxo atom, rather than the two interactions we observed in 4 (Fig. 9). The halogen bonding interactions between the uranyl oxo atom (O2) and the iodine atom (I1) from one of the 2,5-diiodobenzoic ligands on a neighboring monomer is at a distance of 3.400(7) Å (97.1% sum of the van der Waals radii) and the corresponding interaction angle is ∠C–I⋯O 165.2(3)°.
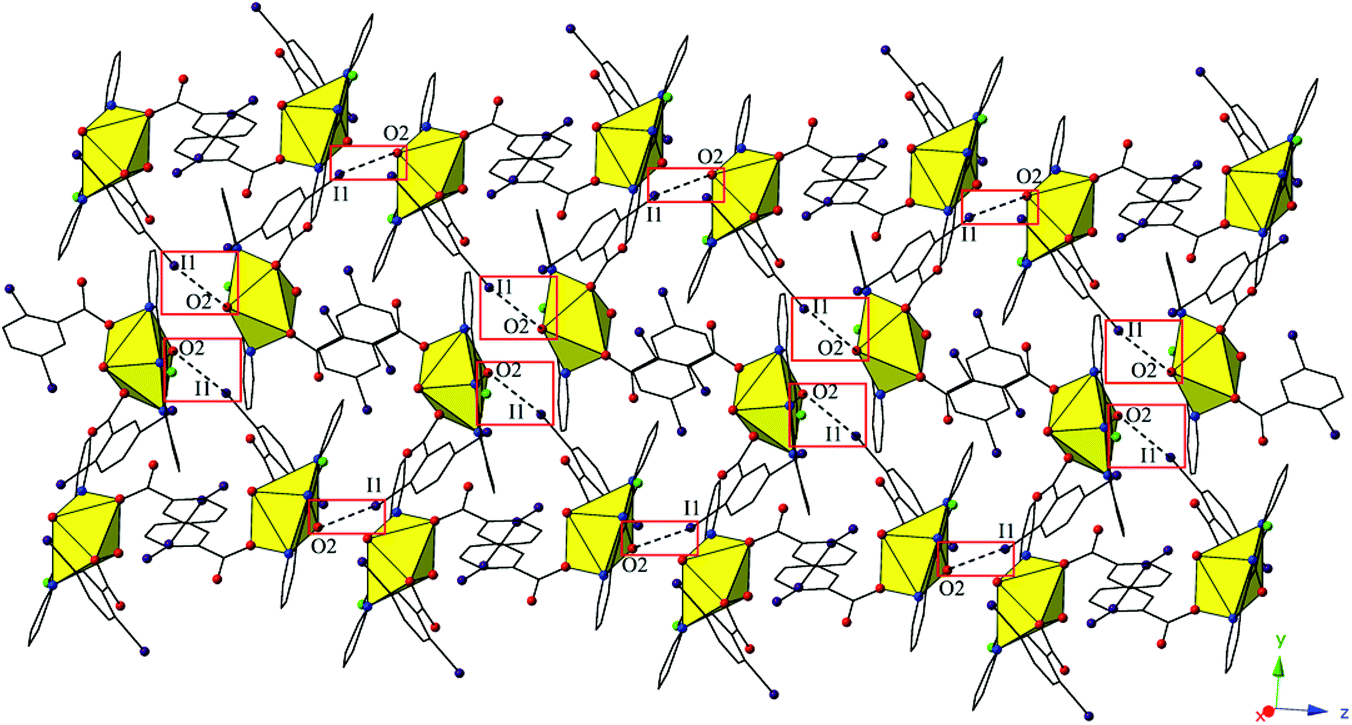 | ||
| Fig. 9 Complex 5 viewed in the (011) plane highlighting the halogen-bonding interactions with the uranyl oxo group that assemble monomers into a supramolecular 2D sheet. | ||
Similar to 4, the undulating 2D sheets of 5 are further assembled into a supramolecular 3D network, yet this is now a result of a halogen bonding interaction and an unsymmetrical halogen–halogen interaction,53 rather than a single halogen bonding interaction as observed in 4 (Fig. S3, ESI†). The halogen bonding interactions are highlighted in green boxes in Fig. S3† and are between an iodine atom (I2) not participating in the formation of the 2D sheets and a carboxylate oxygen atom (O5) with a corresponding I–O interaction distance and angle of 3.377(7) Å (96.5% sum of the van der Waals radii) and ∠C–I⋯O 165.3(4)°. The unsymmetrical halogen–halogen interactions are highlighted in red boxes in Fig. S3† and are between the chlorine atom (Cl1) at the 4′ position of the Cl-terpy capping ligand and an iodine atom (I4) from a 2,5-diiodobenzoic acid ligand on a different neighboring tecton. The I4–Cl1 halogen–halogen interaction meets the criteria for a Type I interaction52 and features an interaction distance of 3.539(5) Å (94.9% sum of the van deer Waals radii) and θ1 (C28–I4–Cl1) and θ2 (C8–Cl1–I4) values of 153.0(3)° and 139.5(5)°, respectively. This last interaction is notable as unsymmetrical halogen–halogen interactions (X1 ≠ X2) prefer type II orientation, yet type I I–Cl interactions are known in a few cases when the iodine acts as an electron donor and the chlorine atom as an electron acceptor as is likely the case in 5.52,53
Structural discussion
As complexes 1–5 were synthesized from similar reaction conditions, the resulting structure types and supramolecular synthons provide an opportunity to assess the influence of halogen polarizability and capping ligand size on supramolecular assembly (Table 2).| Complex | Observed synthons | Chelating ligand |
|---|---|---|
| 1 | H-bonding | Bipy |
| 2 | I–O (oxo), I–I | Phen |
| 3 | I–O (oxo), I–I (×4) | Phen |
| 4 | I–O (oxo) (×2), I–O (carboxylate) | Terpy |
| 5 | I–O (oxo), I–O (carboxylate), I–Cl | Cl-Terpy |
Complexes 1–5 all feature uranyl cations capped by chelating N-donors and further coordinated to 2,5-diiodobenzoic ligands adopting either monodentate or bidentate coordination modes. We primarily observe monomeric uranyl tectons in this family of materials, with the only exception being 2, which consists of hydroxide bridged dimers (suggestive of hydrolysis and subsequent metal-ion condensation), and note that herein metal-ion speciation seems to have less of an impact on local structures and modes of non-covalent assembly than it has had in other families of uranyl hybrid materials.19,54,55 Comparing 1–5 to our previous studies,19,20 which featured the capping ligands bipy, phen, terpy, and Cl-terpy with the similar 3,5-dichloro- and 3,5-dibromobenzoic acid ligands, we note variation in the observed building unit with phen and Cl-terpy (Chart 1). Whereas with 3,5-dichlorobenzoic acid and phen we observed a hydroxide bridged dimer, similar to 2, and with 3,5-dibromobenzoic acid we observed a pentagonal bipyramidal monomer, with 3 we now see two crystallographically unique monomers, both of which adopt hexagonal bipyramidal molecular geometry. Similarly with Cl-terpy, for the 3,5-dichloro- and 3,5-dibromobenzoic analogues we observed hydroxide bridged dimers, yet 5 is a monomer with local geometry that is very similar to a UO2-p-bromobenzoic acid-Cl-terpy material we have characterized in an earlier study.19 Please note that the UO2-3,5-dibromobenzoic acid-BPY complex included in Chart 1 was not prepared in our previous study19 (an oversight), and thus was synthesized and characterized as part of this work. We now refer to this material as complex 6 and its inclusion allows for a more comprehensive comparison of structural and supramolecular systematics across these three families of complexes featuring dichloro-, dibromo-, and diiodobenzoic acid ligands. As such see the ESI (Table S1, Fig. S1†) for complete crystallographic details and additional figures highlighting local structure and packing for complex 6.
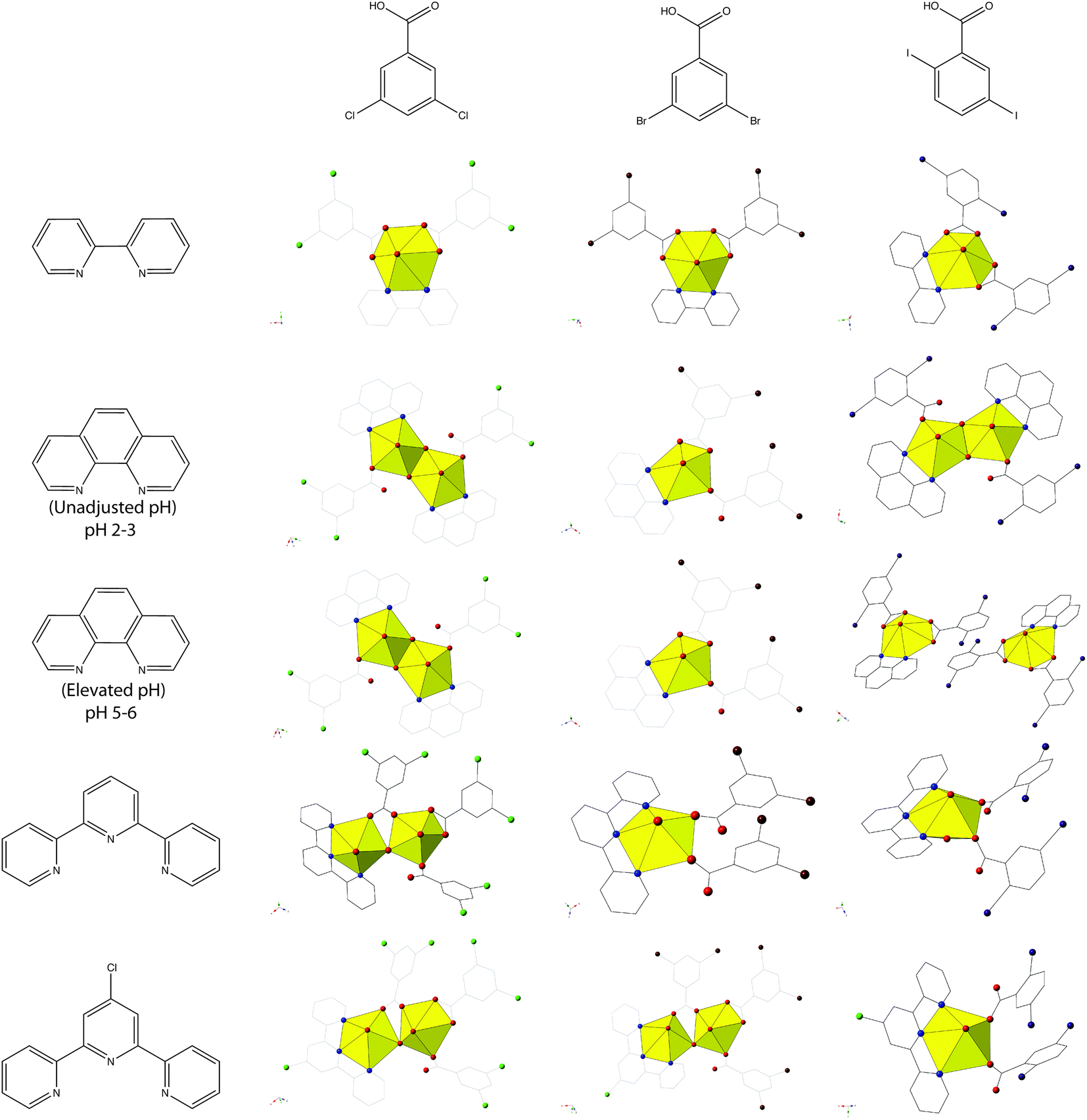 | ||
| Chart 1 Summary of observed local coordination environments for 1–5 along with 3,5-dichloro- and 3,5-dibromobenzoic acid analogues. Figures of 3,5-dichloro- and 3,5-dibromobenzoic acid structures re-made from ref. 19 and 20. Note the UO2-3,5-dibromobenzoic acid-BPY structure was not included in ref. 19 and was synthesized and characterized as a part of this work for completeness (Table S1, Fig. S1, ESI†). | ||
Looking at the modes of supramolecular assembly in 1–5 (Table 2, Chart 2), we note the prevalence of I–O (oxo) halogen bonding interactions in 2–5. Whereas occurrences of oxo interaction with 3,5-di![[c with combining low line]](https://www.rsc.org/images/entities/char_0063_0332.gif)
![[h with combining low line]](https://www.rsc.org/images/entities/char_0068_0332.gif)
![[l with combining low line]](https://www.rsc.org/images/entities/char_006c_0332.gif)
![[o with combining low line]](https://www.rsc.org/images/entities/char_006f_0332.gif)
![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) benzoic acid were absent20 and with 3,5-di
benzoic acid were absent20 and with 3,5-di![[b with combining low line]](https://www.rsc.org/images/entities/char_0062_0332.gif)
![[m with combining low line]](https://www.rsc.org/images/entities/char_006d_0332.gif) benzoic acid they were seemingly random,19 such interactions appear to be the norm with 2,5-di
benzoic acid they were seemingly random,19 such interactions appear to be the norm with 2,5-di![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif)
![[d with combining low line]](https://www.rsc.org/images/entities/char_0064_0332.gif) benzoic acid. As we have already introduced, additional bonding (or non-covalent interactions) at the nominally terminal uranyl oxo atoms in hybrid materials is not common. Moreover, specific ‘cation–cation’ interactions wherein the oxo group of one uranyl cation coordinates equatorially to another, while increasing in frequency,56–58 are also largely serendipitous. The work described herein builds on our recent results where we observed only four instances of the uranyl oxo atoms participating in halogen bonding interactions (out of twelve structures),19 and marks the first time where we explicitly aimed to promote the oxo atoms of the uranyl cation in non-covalent assembly via the judicious pairing of an electron N-donor ligand with a benzoic acid linker featuring polarizable halogen atoms at the periphery. This process of directing synthon(s) of choice for assembly of uranyl hybrid materials represents a step forward in the process of crystal engineering with the uranyl cation, and this process was recently defined by Bombicz et al. as ‘synthon engineering’.59
benzoic acid. As we have already introduced, additional bonding (or non-covalent interactions) at the nominally terminal uranyl oxo atoms in hybrid materials is not common. Moreover, specific ‘cation–cation’ interactions wherein the oxo group of one uranyl cation coordinates equatorially to another, while increasing in frequency,56–58 are also largely serendipitous. The work described herein builds on our recent results where we observed only four instances of the uranyl oxo atoms participating in halogen bonding interactions (out of twelve structures),19 and marks the first time where we explicitly aimed to promote the oxo atoms of the uranyl cation in non-covalent assembly via the judicious pairing of an electron N-donor ligand with a benzoic acid linker featuring polarizable halogen atoms at the periphery. This process of directing synthon(s) of choice for assembly of uranyl hybrid materials represents a step forward in the process of crystal engineering with the uranyl cation, and this process was recently defined by Bombicz et al. as ‘synthon engineering’.59
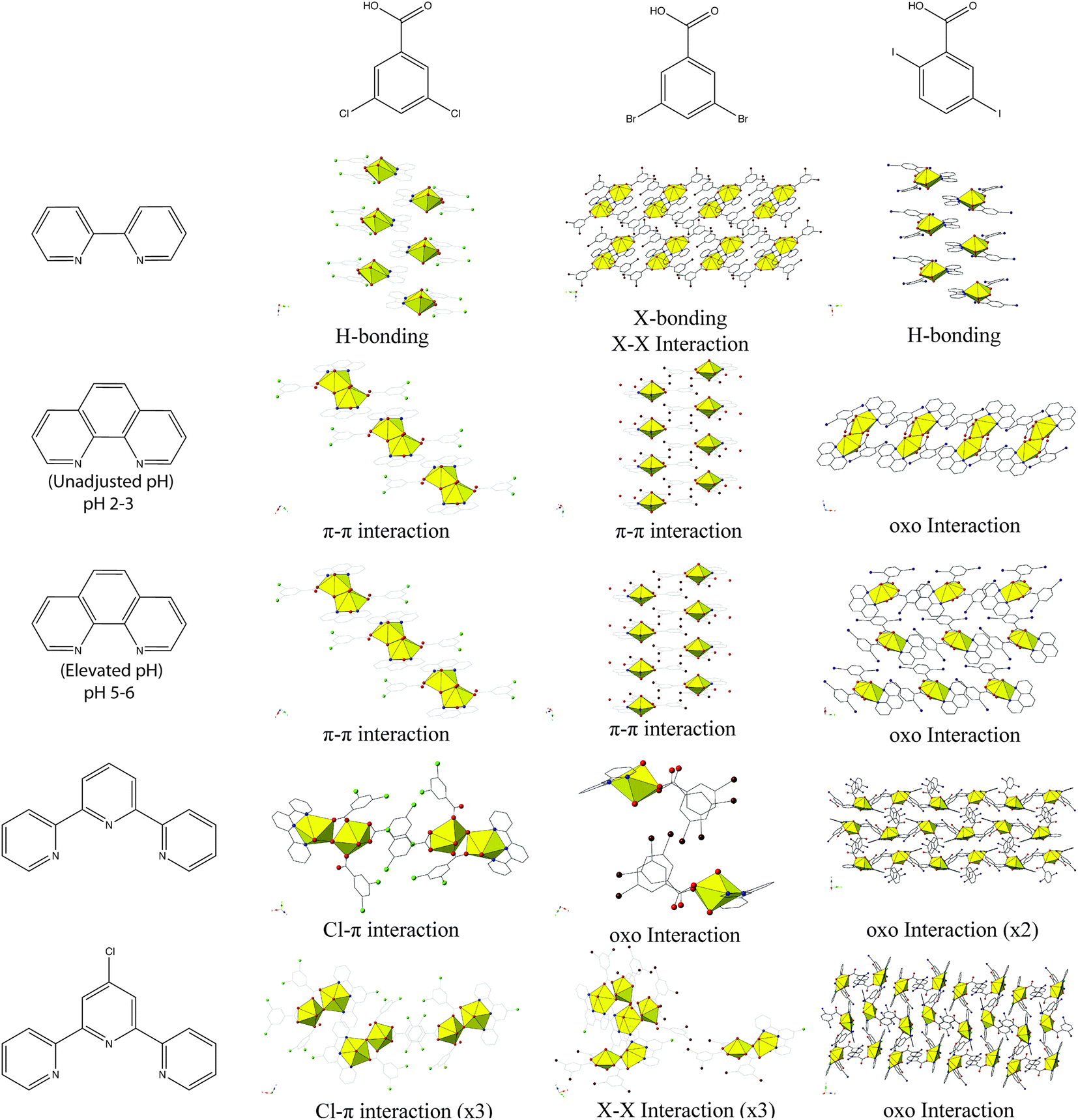 | ||
| Chart 2 Summary of observed modes of supramolecular assembly for 1–5 along with 3,5-dichloro- and 3,5-dibromobenzoic acid analogues. Figures of 3,5-dichloro- and 3,5-dibromobenzoic acid structures re-made from ref. 19 and 20. | ||
Vibrational spectroscopy
Triatomic molecules with a linear arrangement such as the uranyl moiety (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO) have three characteristic normal vibrational modes: a symmetric stretching mode (ν1, Raman active), a bending mode (ν2, infrared active), and an asymmetric stretching mode (ν3, infrared active).60,61 In aqueous solution, these three modes are often observed between 860 and 880 cm−1 (ν1), 200 and 210 cm−1 (ν2), and 930–960 (ν3).62 Previous studies have shown that when nucleophilic ligands are coordinated to the uranyl cation, electron density from the equatorial ligands can be transferred into the π*-antibonding orbitals of the uranyl ion leading to a concomitant redshift in both the associated ν1 and ν3 frequencies,20,63,64 and more recently this observation has been confirmed by quantum chemical calculations.65 Further, a decrease of between 30–60 cm−1 in ν1 and ν3 values, in comparison to the [UO2(H2O)5]2+, can be expected upon complexation of strongly electron donating ligands, such as the capping ligands used in 2, 4, and 5.46,66,67 Raman and infrared spectra have been collected for complexes 2, 4, and 5 these provide a platform for probing the effects of halogen bonding interactions with the oxo groups, as the effects of the electron-donating ability of the capping ligands on uranyl Raman spectra was described in our recent work.20
UO) have three characteristic normal vibrational modes: a symmetric stretching mode (ν1, Raman active), a bending mode (ν2, infrared active), and an asymmetric stretching mode (ν3, infrared active).60,61 In aqueous solution, these three modes are often observed between 860 and 880 cm−1 (ν1), 200 and 210 cm−1 (ν2), and 930–960 (ν3).62 Previous studies have shown that when nucleophilic ligands are coordinated to the uranyl cation, electron density from the equatorial ligands can be transferred into the π*-antibonding orbitals of the uranyl ion leading to a concomitant redshift in both the associated ν1 and ν3 frequencies,20,63,64 and more recently this observation has been confirmed by quantum chemical calculations.65 Further, a decrease of between 30–60 cm−1 in ν1 and ν3 values, in comparison to the [UO2(H2O)5]2+, can be expected upon complexation of strongly electron donating ligands, such as the capping ligands used in 2, 4, and 5.46,66,67 Raman and infrared spectra have been collected for complexes 2, 4, and 5 these provide a platform for probing the effects of halogen bonding interactions with the oxo groups, as the effects of the electron-donating ability of the capping ligands on uranyl Raman spectra was described in our recent work.20
Looking at the Raman spectrum of 2, the ν1 symmetric stretch is the most prominent signal and is observed as two sharp peaks at 831.5 cm−1 and 846 cm−1 (Fig. 10, Table 3). The ν1 frequency for 2 was assigned as the peak at 831.5 cm−1 based on the luminescence results shown in Fig. 11, yet the origin of the second peak at 846 cm−1 is the subject of ongoing investigations. The sloping baseline of the Raman spectrum of 4 is a result of interference stemming from concurrent uranyl fluorescence and these effects weaken the resulting Raman spectrum, although we were able to assign the ν1 frequency at 829 cm−1. Comparing this value to that of a UO2-3,5-dichlorobenzoic acid-TPY material without halogen bonding with the uranyl oxo atoms (complex 4),20 we note a redshift in the ν1 value of 7 cm−1. The ν1 frequency for 5 was observed at 826.5 cm−1 and compared to the UO2-3,5-dichlorobenzoic acid-Cl-TPY material without halogen bonding with the uranyl oxo atoms (complex 5),20 we note a redshift in the ν1 value of 14.5 cm−1.
 | ||
| Fig. 10 Raman spectra of complexes 2 (blue), 4 (red), and 5 (green) highlighting the symmetric stretch (ν1) of the uranyl cation in each material. | ||
 | ||
| Fig. 11 Low temperature, solid-state emission of 2 (blue), 4 (red), and 5 (green) upon excitation at 420 nm. | ||
AnO bond
| Complex | ν 1 (cm−1) | ν 3 (cm−1) | k 1 (mdyn Å−1) | k 12 (mdyn Å−1) | Supramolecular Int. | Sum of vdW (%) |
|---|---|---|---|---|---|---|
| UO22+ (aq) | 860–880 (ref. 62) | 930–960 (ref. 62) | ||||
| [UO2(OH)(C12H8N2)(C7H3I2O2)]2 (2) | 831.5 | 916 | 6.74 | −0.22 | I-oxo | 95.9 |
| [UO2(C15H11N3)(C7H3I2O2)2] (4) | 829 | 910 | 6.68 | −0.20 | I-oxo (×2) | 98.2, 98.8 |
| [UO2(C15H10ClN3)(C7H3I2O2)2] (5) | 826.5 | 910 | 6.66 | −0.22 | I-oxo | 97.1 |
The red shift in ν1 values from complexes 2 to 4 to 5 represents a small deviation from our previous work where we saw a direct correlation between the symmetric stretch value and the electron donating properties of capping ligand. The introduction of halogen bonding interactions at the uranyl oxo groups likely directs additional charge density towards the UO22+ center, increasing the ionic character of the U-oxo bond,65 further redshifting the ν1 values. Although we will further probe these results via the calculation of stretching (k1) and interaction (k12) force constants below, we acknowledge that these comments remain speculative absent rigorous theoretical study.
Looking at the ν3 asymmetric stretch values for 2, 4, and 5 in the infrared spectra we observe a series of strong signals at 916 cm−1, 910 cm−1, and 910 cm−1, respectively (Fig. S4, ESI,†Table 3). All three of these frequencies have shifted further in comparison to both the uranyl moiety in aqueous solution and those we observed previously,20 suggesting that while the capping ligands (phen, terpy, and Cl-terpy) do transfer electron density to the uranium metal center, the halogen bonding interactions may also influence the ν3 asymmetric stretch.
Actinyl force constants
The stretching force constant (k1) and interaction force constant (k12) for the AnO bond were calculated for complexes 2, 4, and 5 to explore the viability of quantifying halogen bonding with the uranyl oxo atoms via spectroscopic means as we have thus far only viewed these interactions through a crystallographic lens.16,19 Force constant values (k1, k12) can be calculated from ν1 and ν3 stretches of the actinyl compound using a valence potential model as first described by Herzberg60 and more recently outlined by Schnaars and Wilson.68,69 In a linear triatomic molecule like uranyl, the stretching force constant (k1) describes the uranyl UO bond, whereas the interaction force constant (k12) quantifies the interaction between the two uranyl oxygen atoms.
Utilizing our experimental values for ν1 and ν3 we have calculated the k1 values for 2, 4, and 5 to be 6.74, 6.68, and 6.66 mdyn Å−1, respectively. A decrease in the stretching force constant (k1) signifies a weakening of the AnO bond, which would be an expected manifestation of oxo participation in halogen bonding, and the halogen bonding interactions with the uranyl oxo atoms in 2, 4, and 5 were found to be at 95.9, 98.2 and 98.8, and 96.4% the sum of the van der Waals radii, respectively. Based on crystallographic metrics alone we would expect the weakest AnO bond in 2 as it features the oxo atom participating in the ‘strongest’ interaction, yet the vibrational spectroscopy and force constants highlighted in Table 3 paint a more complex picture. Stretching force constants indicate that there are subtle differences between the AnO bond strengths in 2, 4, and 5, and these changes mirror the redshifts observed in the Raman and IR spectra highlighted in Fig. 10 and S4.† This suggests the ligands in the equatorial plane also contribute to force constants of the AnO bond and the resulting ν1 and ν3 values,20,65,70 yet the relative contributions of equatorial ligands and the effects of halogen bonding with the actinyl unit, and the spectroscopic manifestations thereof, are still being explored.65,71
In contrast to the stretching force constants, the interaction force constant (k12) values for 2, 4, and 5 were found to be consistent, spanning only 0.02 mdyn Å−1, from −0.20 to −0.22 mdyn Å−1 (Table 3). The invariance in k12 values indicates that the interaction between oxo groups is relatively consistent for all three complexes, and suggests that oxo participation in non-covalent assembly does not impact the interaction force constant values, even in cases where both oxo atoms are involved in supramolecular interactions, as is the case in 4.
Luminescence
Low temperature solid-state luminescence studies were carried out on several single crystals from the bulk phases of 2, 4, and 5. Uranyl containing materials typically exhibit a characteristic green emission profile that results from ligand-to-metal charge transfer transitions between uranyl bonding (3σu, 3σg, 2πu, and 1πg) and non-bonding (5f δu and ϕu) molecular orbitals,63,72 and for 2, 4, and 5, characteristic emission (four to five major vibronic peaks) was observed upon excitation at 420 nm (Fig. 11). The spectrum of 5 also features four secondary vibronic shoulder peaks, which is notable and does require additional comment. Minor vibronic peaks in uranyl emission spectrum have been observed elsewhere and are generally attributed to additional coupling of the equatorial ligand vibrational modes and/or the uranyl bending mode with the uranyl excited state.72 For 5 we were not able to experimentally pinpoint the origin of the secondary peaks and the appearance of these additional peaks warrants additional theoretical treatment, which was not conducted as a part of this study.The average vibronic progression of the emission bands are coupled to the Raman active vibrational modes and were found to be at 828, 833, and 829 cm−1 for 2, 4, and 5, respectively, which is in excellent agreement with the measured Raman ν1 values for 2, 4, and 5 of 831.5, 829, and 826 cm−1 highlighted in Fig. 10. Additionally, red shifts of approximately 666 and 675 cm−1 are observed when comparing the luminescence spectra of 4 and 5, respectively, with that of 2. These results are consistent with our Raman and IR results described above, and mostly in agreement with the experimental findings of our group and Natrajan et al.,20,70 which qualitatively highlighted correlations between shifts in uranyl emission and the electron donating capabilities of equatorial ligands. Similar to the Raman results in Fig. 10, complex 5 features uranyl emission at the lowest energy values even though complex 4 features a capping ligand (terpy) with slightly greater electron donating ability. The energy differences between the major vibronic peaks of 4 and 5 are small (ca. 22 cm−1), yet notable, as previously we observed a much larger (ca. 200 cm−1) energy gap between similar UO2-3-5-dichlorobenzoic acid-TPY and UO2-3-5-dichlorobenzoic acid-Cl-TPY materials.20 We hesitate to comment definitively whether the red shift observed in Fig. 11 can be directly attributed to the change in capping ligands electron transfer characteristics, or that the additional vibrational and luminescent shifts we have observed herein, in comparison to the UO2-3-5-dichlorobenzoic acid-N-Donor materials synthesized previously,20 are a result of halogen bonding with the uranyl oxo groups, yet these initial results however, are indicative of possible correlations.65,73 Further, the effects of the strengths of supramolecular interactions in the equatorial plane, which also vary from 2 to 4 to 5, on uranyl luminescence have been recently been explored,17,74 yet remain relatively nascent and were not probed herein.
Conclusions
The syntheses and crystal structures of five uranyl complexes containing 2,5-diiodobenzoic acid and the chelating N-donors bipy, phen, terpy, and Cl-terpy are reported, their means of supramolecular assembly have been detailed, and their vibrational and luminescence spectra have been collected where possible. This family of materials is the first example of the uranyl oxo atoms being systematically involved in non-covalent assembly, and is complementary to the ongoing efforts in actinide organometallic chemistry exploring ‘oxo-functionalization’.46,75–77 Additionally, these materials are quite valuable for the ongoing development of uranyl supramolecular assembly criteria based on acceptor-donor pairings, as they indicate that one route to promote assembly via halogen bonding on a consistent basis is the combination of electron donating capping ligands paired with benzoic acid groups featuring multiple large, polarizable halogen atoms at the periphery. Follow up studies exploring the effects of the identity of the halogen, along with its position on the benzoic acid ligand, on oxo atom participation in supramolecular assembly are in progress and will be published in the near future. Further, computational efforts to improve understanding of halogen interaction strengths and their spectroscopic manifestations are ongoing.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This material was supported by the U. S. Department of Energy—Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Sciences, Office of Science, Heavy Elements Program, under grant number DE-FG02-05ER15736. K. P. C. would also like to acknowledge George Washington University for a Presidential Merit Fellowship award. The authors would also like to thank the ND Energy Center for Infrared and Raman microscope time.References
- R. J. Baker, Chem. – Eur. J., 2012, 18, 16258–16271 Search PubMed.
- M. B. Andrews and C. L. Cahill, Chem. Rev., 2013, 113, 1121–1136 Search PubMed.
- R. G. Surbella III and C. L. Cahill, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, Amsterdam, 2015, vol. 48, ch. 276, pp. 163–285 Search PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 Search PubMed.
- J.-C. G. Bünzli and C. Piguet, Chem. Rev., 2002, 102, 1897–1928 Search PubMed.
- P. Thuéry, CrystEngComm, 2012, 14, 8128–8136 Search PubMed.
- K. P. Carter and C. L. Cahill, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier, Amsterdam, 2015, vol. 47, ch. 271, pp. 147–208 Search PubMed.
- D. E. Barry, D. F. Caffrey and T. Gunnlaugsson, Chem. Soc. Rev., 2016, 45, 3244–3274 Search PubMed.
- M. Nyman and P. C. Burns, Chem. Soc. Rev., 2012, 41, 7354–7367 Search PubMed.
- J. de Groot, K. Gojdas, D. K. Unruh and T. Z. Forbes, Cryst. Growth Des., 2014, 14, 1357–1365 Search PubMed.
- S. Nuzzo, M. Browne, B. Twamley, M. Lyons and R. Baker, Inorganics, 2016, 4, 4 Search PubMed.
- N. P. Deifel and C. L. Cahill, CrystEngComm, 2009, 11, 2739–2744 Search PubMed.
- N. P. Deifel and C. L. Cahill, C. R. Chim., 2010, 13, 747–754 CrossRef CAS.
- M. B. Andrews and C. L. Cahill, Dalton Trans., 2012, 41, 3911–3914 Search PubMed.
- M. B. Andrews and C. L. Cahill, CrystEngComm, 2013, 15, 3082–3086 Search PubMed.
- R. G. Surbella III and C. L. Cahill, CrystEngComm, 2014, 16, 2352–2364 Search PubMed.
- R. G. Surbella III, M. B. Andrews and C. L. Cahill, J. Solid State Chem., 2016, 236, 257–271 Search PubMed.
- S. G. Thangavelu, M. B. Andrews, S. J. A. Pope and C. L. Cahill, Inorg. Chem., 2013, 52, 2060–2069 Search PubMed.
- K. P. Carter and C. L. Cahill, Inorg. Chem. Front., 2015, 2, 141–156 Search PubMed.
- K. P. Carter, M. Kalaj and C. L. Cahill, Eur. J. Inorg. Chem., 2016, 2016, 126–137 Search PubMed.
- N. W. Alcock, D. J. Flanders and D. Brown, J. Chem. Soc., Dalton Trans., 1985, 1001–1007 Search PubMed.
- J.-C. Berthet, M. Nierlich and M. Ephritikhine, Dalton Trans., 2004, 2814–2821 Search PubMed.
- P. Thuéry, Eur. J. Inorg. Chem., 2014, 2014, 58–68 Search PubMed.
- T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2014, 266–267, 69–109 Search PubMed.
- W. Yang, T. G. Parker and Z.-M. Sun, Coord. Chem. Rev., 2015, 303, 86–109 CrossRef CAS.
- N. P. Deifel and C. L. Cahill, Chem. Commun., 2011, 47, 6114–6116 Search PubMed.
- P. C. Burns, R. C. Ewing and F. C. Hawthorne, Can. Mineral., 1997, 35, 1551–1570 Search PubMed.
- P. C. Burns, Can. Mineral., 2005, 43, 1839–1894 Search PubMed.
- K. P. Carter, S. J. A. Pope and C. L. Cahill, CrystEngComm, 2014, 16, 1873–1884 Search PubMed.
- K. P. Carter, C. H. F. Zulato and C. L. Cahill, CrystEngComm, 2014, 16, 10189–10202 Search PubMed.
- K. P. Carter, C. H. F. Zulato, E. M. Rodrigues, S. J. A. Pope, F. A. Sigoli and C. L. Cahill, Dalton Trans., 2015, 44, 15843–15854 Search PubMed.
- K. P. Carter, K. E. Thomas, S. J. A. Pope, R. J. Holmberg, R. J. Butcher, M. Murugesu and C. L. Cahill, Inorg. Chem., 2016, 55, 6902–6915 Search PubMed.
- SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- APEXII, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 Search PubMed.
- TWINABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–435 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 Search PubMed.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 Search PubMed.
- Crystal Maker, Crystal Maker Software Limited, Bicester, England, 2009 Search PubMed.
- JADE, Materials Data Inc., Livermore, California, USA, 2003 Search PubMed.
- T. S. Franczyk, K. R. Czerwinski and K. N. Raymond, J. Am. Chem. Soc., 1992, 114, 8138–8146 Search PubMed.
- P. Thuéry and M. Nierlich, J. Chem. Soc., Dalton Trans., 1997, 1481–1482 RSC.
- B. Masci, M. Gabrielli, S. L. Mortera, M. Nierlich and P. Thuéry, Polyhedron, 2002, 21, 1125–1131 CrossRef CAS.
- P. L. Arnold, A. J. Blake, C. Wilson and J. B. Love, Inorg. Chem., 2004, 43, 8206–8208 Search PubMed.
- S. Fortier and T. W. Hayton, Coord. Chem. Rev., 2010, 254, 197–214 Search PubMed.
- D. L. Clark, S. D. Conradson, R. J. Donohoe, D. W. Keogh, D. E. Morris, P. D. Palmer, R. D. Rogers and C. D. Tait, Inorg. Chem., 1999, 38, 1456–1466 Search PubMed.
- L. A. Watson and B. P. Hay, Inorg. Chem., 2011, 50, 2599–2605 Search PubMed.
- F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, Chem. – Eur. J., 2006, 12, 8952–8960 Search PubMed.
- L. Brammer, G. Minguez Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 Search PubMed.
- G. R. Desiraju and R. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725–8726 Search PubMed.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 Search PubMed.
- S. Tothadi, S. Joseph and G. R. Desiraju, Cryst. Growth Des., 2013, 13, 3242–3254 Search PubMed.
- C. E. Rowland and C. L. Cahill, Inorg. Chem., 2010, 49, 6716–6724 Search PubMed.
- C. E. Rowland and C. L. Cahill, Inorg. Chem., 2010, 49, 8668–8673 Search PubMed.
- P. O. Adelani and P. C. Burns, Inorg. Chem., 2012, 51, 11177–11183 Search PubMed.
- J. Lhoste, N. Henry, P. Roussel, T. Loiseau and F. Abraham, Dalton Trans., 2011, 40, 2422–2424 Search PubMed.
- P. M. Cantos, L. J. Jouffret, R. E. Wilson, P. C. Burns and C. L. Cahill, Inorg. Chem., 2013, 52, 9487–9495 Search PubMed.
- P. Bombicz, T. Gruber, C. Fischer, E. Weber and A. Kalman, CrystEngComm, 2014, 16, 3646–3654 Search PubMed.
- G. Herzberg, Infrared and Raman Spectra of Polyatomic Molecules, D. Van Nostrand Company, Inc., New York, NY, 1946 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds Part A: Theory and Applications in Inorganic Chemistry, John Wiley & Sons, Inc., New York, NY, 5th edn, 1997 Search PubMed.
- L. H. Jones and R. A. Penneman, J. Chem. Phys., 1953, 21, 542–544 Search PubMed.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- G. S. Groenewold, A. K. Gianotto, M. E. McIlwain, M. J. Van Stipdonk, M. Kullman, D. T. Moore, N. Polfer, J. Oomens, I. Infante, L. Visscher, B. Siboulet and W. A. de Jong, J. Phys. Chem. A, 2008, 112, 508–521 CrossRef CAS PubMed.
- P. Di Pietro and A. Kerridge, Inorg. Chem., 2016, 55, 573–583 Search PubMed.
- C. Nguyen Trung, G. M. Begun and D. A. Palmer, Inorg. Chem., 1992, 31, 5280–5287 Search PubMed.
- F. Quilès, C. Nguyen-Trung, C. Carteret and B. Humbert, Inorg. Chem., 2011, 50, 2811–2823 Search PubMed.
- D. D. Schnaars and R. E. Wilson, Inorg. Chem., 2013, 52, 14138–14147 Search PubMed.
- D. D. Schnaars and R. E. Wilson, Inorg. Chem., 2014, 53, 11036–11045 Search PubMed.
- M. P. Redmond, S. M. Cornet, S. D. Woodall, D. Whittaker, D. Collison, M. Helliwell and L. S. Natrajan, Dalton Trans., 2011, 40, 3914–3926 Search PubMed.
- P. Di Pietro and A. Kerridge, Phys. Chem. Chem. Phys., 2016, 18, 16830–16839 Search PubMed.
- L. S. Natrajan, Coord. Chem. Rev., 2012, 256, 1583–1603 Search PubMed.
- S. Tsushima, Dalton Trans., 2011, 40, 6732–6737 Search PubMed.
- J. M. Harrowfield, N. Lugan, G. H. Shahverdizadeh, A. A. Soudi and P. Thuéry, Eur. J. Inorg. Chem., 2006, 2006, 389–396 Search PubMed.
- P. L. Arnold, A.-F. Pécharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056–1061 Search PubMed.
- A. J. Lewis, H. Yin, P. J. Carroll and E. J. Schelter, Dalton Trans., 2014, 43, 10844–10851 Search PubMed.
- P. L. Arnold, A.-F. Pécharman, R. M. Lord, G. M. Jones, E. Hollis, G. S. Nichol, L. Maron, J. Fang, T. Davin and J. B. Love, Inorg. Chem., 2015, 54, 3702–3710 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Single crystal XRD data for complex 6, X-ray crystallographic files in CIF format, ORTEP figures of all complexes, PXRD spectra of all complexes, IR spectra of complexes 2, 4, and 5, and tables of selected supramolecular interaction distances and bond lengths are all available. CCDC 1501011–1501016 for compounds 1–6. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00352d |
| This journal is © the Partner Organisations 2017 |