
Enhanced oxygen evolution reaction of metallic nickel phosphide nanosheets by surface modification†
Zejun
Li
a,
Xinyu
Dou
b,
Yingcheng
Zhao
a and
Changzheng
Wu
*a
aHefei National Laboratory for Physical Sciences at the Microscale, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Hefei Science Center (CAS), and CAS Key Laboratory of Mechanical Behavior and Design of Materials, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: czwu@ustc.edu.cn
bCollege of Chemistry, Jilin University, Changchun, 130012, China
First published on 31st May 2016
Abstract
Employing water splitting as a practicable technique for alternative clean energy requires the development of efficient oxygen evolution electrocatalysts based on inexpensive and earth-abundant materials. Transition metal phosphides have been regarded as promising candidates due to their high electrochemical activity, high conductivity and widespread abundance. However, currently, the oxygen evolution capability of the transition metal phosphides is still limited. It can be noted that the surface state of the electrocatalysts also plays a crucial role in determining the electrocatalytic reaction. In this study, we demonstrate a surface modification strategy using oxygen incorporation in the metallic Ni2P nanosheets catalyst for the first time, leading to dramatically enhanced oxygen evolution activity. The oxygen-incorporated Ni2P nanosheets were achieved through a simple low-temperature phosphidation from the Ni(OH)2 nanosheets precursor. We found that the oxygen incorporation in the Ni2P nanosheets could actively promote the electrochemical performance and presented stable current density of 10 mA cm−2 at a small overpotential of 347 mV with a Tafel slope as low as 63 mV dec−1. Furthermore, the large electrocatalytic current is about 25 times enhanced compared with the pure Ni2P counterpart. Moreover, the high OER performance is superior to most of the reported pure metal phosphide electrocatalysts. These findings point to new opportunities for surface modification to manipulate and improve the reaction of electrocatalysts for designing high performance electrocatalysts and for the investigation of the underlying mechanism of the electrochemical reaction.
Introduction
Electrochemical water splitting into hydrogen and oxygen has been regarded as a promising approach for energy storage and conversion to address the energy crisis and environmental issues.1–7 However, the electrochemical water splitting process is greatly limited by the oxygen evolution reaction (OER) owing to the requirement of high overpotential and the complex four-electron redox process,8,9 thus a high-performance electrocatalyst is required to decrease the overpotential and promote the sluggish reaction process. Noble metal oxides, such as RuO2 and IrO2, have been identified as the most efficient OER electrocatalysts,10,11 but their high cost and low abundance have greatly hindered their large-scale applications. Numerous efforts have therefore been made to develop new and efficient noble metal-free OER electrocatalysts relying on their electrical conductivity and electrochemical activity. Over the past few years, many potential alternatives for precious metal-based OER electrocatalysts have been investigated, including abundant 3d transition metal (Fe, Co, Ni, Mn) compounds,12–18 perovskites19,20 and metal-free catalysts.9,21,22 Among these non-precious metal electrocatalysts, Ni-based compounds23–31 have shown high electrochemical activity and excellent OER performance.Recently, transition metal phosphides (TMP) have attracted intensive attention as low cost candidates in many electrochemistry fields, including electrocatalysis such as the hydrogen evolution reaction (HER).32–39 However, the investigation of transition metal phosphides as OER catalysts is still relatively rare. Similar to transition metal carbides and nitrides, transition metal phosphides possess high conductivity, high stability, corrosion resistance and good mechanical properties. Nevertheless, the transition metal phosphides exhibit a characteristic structure that is different from the carbides and nitrides. The carbides and nitrides generally form interstitial compounds owing to the small radius of the carbon and nitrogen ions, whereas the phosphides are prone to produce filling compounds in which the phosphorus atoms occupy the lattice sites because of the larger ionic radius.40 Moreover, the strength of the M–P bond is stronger than that in the carbides and nitrides, as well as the presence of the P–P bond in the transition metal phosphides.41 These special atomic arrangements and bonding types in the phosphides make them rich and tunable components; special lattice structures and electronic states enable the phosphides to expose more unsaturated surface atoms and obtain a large surface electrochemical activity density.42
However, although the transition metal phosphides show good conductivity, high chemical stability and activity density, the OER performance of phosphides is still unsatisfactory and more efforts are needed to optimize their OER activity. Of note, the surface properties of the catalysts are also a critical factor in determining the electrocatalytic performance and governing the electrochemical reactivity.43–46 As an example, an efficient route to enhance the HER performance of noble Pt in basic media is combination with metal hydroxides.47,48 This is due to the fact that Pt is effective for the adsorption and recombination of the reactive intermediates but generally inefficient in the prior step of water dissociation, whereas metal hydroxides are effective for cleaving the HO–H bond. Thus, the combination of Pt and metal hydroxides could facilitate the multistep OER process. Moreover, the surface oxygen vacancies favour the adsorption of H2O molecules on the surface of the catalyst during the OER process.16 Therefore, the surface state of the electrocatalyst plays an important role in electrochemical performance.
In this study, we successfully fabricated a new type of metallic Ni2P nanosheet through a thermal conversion reaction from the Ni(OH)2 nanosheets precursor but with a surface modification stemming from oxygen incorporation. As a proof of concept, the prepared oxygen-incorporated Ni2P nanosheets exhibit much enhancement of OER performance compared with the pure Ni2P counterpart and the pristine Ni(OH)2 precursor. Our oxygen-incorporated Ni2P nanosheets exhibit a large electrocatalytic current, about 25 times larger than that of pure Ni2P counterpart without oxygen incorporation. The highly improved OER performance of our metallic Ni2P nanosheets was attributed to the surface existence of Ni–O components that favours the water oxidation process, thus improving the intrinsic OER performance of the Ni2P sample. We anticipate that surface modification for electrocatalysts will be a powerful means to design high performance electrocatalysts and investigate the underlying mechanism of electrochemical reaction.
Experimental section
Materials
NiCl2·6H2O, hexamethylenetetramine, NaH2PO2, Ni powder, KOH and ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd. PPh3 was purchased from Alfa Aesar. All reagents were used as received without further purification.Synthesis of Ni(OH)2 nanosheets
In a typical synthesis, 0.5 mmol NiCl2·6H2O (0.12 g) and 168 mg hexamethylenetetramine were dissolved in 27 mL distilled water and 3 mL anhydrous ethanol under vigorous stirring for 30 min. Then, the solution was transferred into a Teflon-lined stainless steel autoclave (40 mL). The autoclave was sealed and maintained at 120 °C for 12 h in an electric oven. After the autoclave cooled slowly to room temperature, the product was taken out and washed with water thoroughly before being vacuum dried.Synthesis of pure Ni2P sample
The pure Ni2P samples were prepared according to literature.49 In brief, a mixture of the NaBH4-treated Ni powder and PPh3 with a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 was put into a quartz tube. The use of NaBH4 is to remove the surface oxides. The tube was then evacuated and sealed. The sealed tube was loaded into the furnace by a tilt angle of 5°, heated from room temperature to 380 °C in 100 min and maintained at this temperature for 15 h. Then, the final products were collected and washed for further characterization.
:1.2 was put into a quartz tube. The use of NaBH4 is to remove the surface oxides. The tube was then evacuated and sealed. The sealed tube was loaded into the furnace by a tilt angle of 5°, heated from room temperature to 380 °C in 100 min and maintained at this temperature for 15 h. Then, the final products were collected and washed for further characterization.
Electrochemical measurements
All the electrochemical measurements were performed using a three-electrode system on an electrochemical workstation (CHI660E). A Pt wire electrode was used as counter electrode and a KCl-saturated Ag/AgCl electrode was used as the reference electrode. Typically, 4 mg of catalyst and 30 μL of Nafion solution (Sigma Aldrich, 5 wt%) were dispersed in 1.0 mL water–ethanol solution with a volume ratio of 4:1 by sonicating for 1 h to form a homogeneous ink. Then, 5 μL of the catalyst ink (containing 20 μg of catalyst) was loaded onto a glassy carbon electrode with 3 mm diameter (loading = 0.285 mg cm−2). The potentials reported in our study were referenced to the reversible hydrogen electrode (RHE): ERHE = EAg/AgCl + 0.059 × pH + 0.1988. Linear sweep voltammetry with a scan rate of 5 mV s−1 was conducted in 0.1 M KOH solution. The electrochemical impedance spectroscopy (EIS) measurements were performed in the same configuration at an overpotential η = 400 mV over a frequency range from 100 kHz to 0.01 Hz at an amplitude of 5 mV. Cyclic voltammetry (CV) was conducted between 0.96 and 1.76 V vs. RHE at 50 mV s−1 to investigate the cycling stability. The current density (J) was based on the geometric area of the electrode (0.07 cm2) and all the polarization curves were corrected with iR-compensation that arose from the solution resistance.
The effective active surface area of the samples was evaluated approximately by using the electrochemical double-layer capacitance (Cdl). ECSA was determined by measuring the capacitive current associated with double-layer charging from the scan rate dependence of the CVs. The scan rates were 20, 40, 60, 80 and 100 mV s−1. The double-layer capacitance (Cdl) was estimated by plotting the (ΔJ = Ja − Jc) at 1.18 V vs. RHE against the scan rate (Fig. 5). The slope is twice the double-layer capacitance Cdl.
Characterization
The products were characterized using X-ray powder diffraction (XRD, performed on a Philips X'pert PRO X-ray diffractometer, Cu–Kα, λ = 1.54182 Å) and a scanning electron microscope (SEM, JSM-6700F). Transmission electron microscopy (TEM) images were obtained using an H-7650 (Hitachi, Japan) operated at an acceleration voltage of 100 kV. X-ray photoelectron spectra (XPS) were obtained on an ESCALAB MK II with Mg-Kα as the excitation source.Results and discussion
The oxygen-incorporated Ni2P nanosheets were synthesized by low-temperature phosphidation of the Ni(OH)2 nanosheets precursor, as shown in Fig. 1. In a typical procedure, the Ni(OH)2 nanosheets were first obtained (for details see Experimental section), then 10 mg Ni(OH)2 samples and 100 mg NaH2PO2 were placed at two separate positions in a quartz boat with the NaH2PO2 at the upstream side of the furnace. Subsequently, the samples were heated at 330 °C for 30 min in a static Ar atmosphere and then naturally cooled to ambient temperature under Ar. At 330 °C, NaH2PO2 decomposes into PH3 gas, which would react with the pristine Ni(OH)2 nanosheets to produce Ni2P nanosheets. Finally, the product was collected for further characterization and electrochemical property measurements.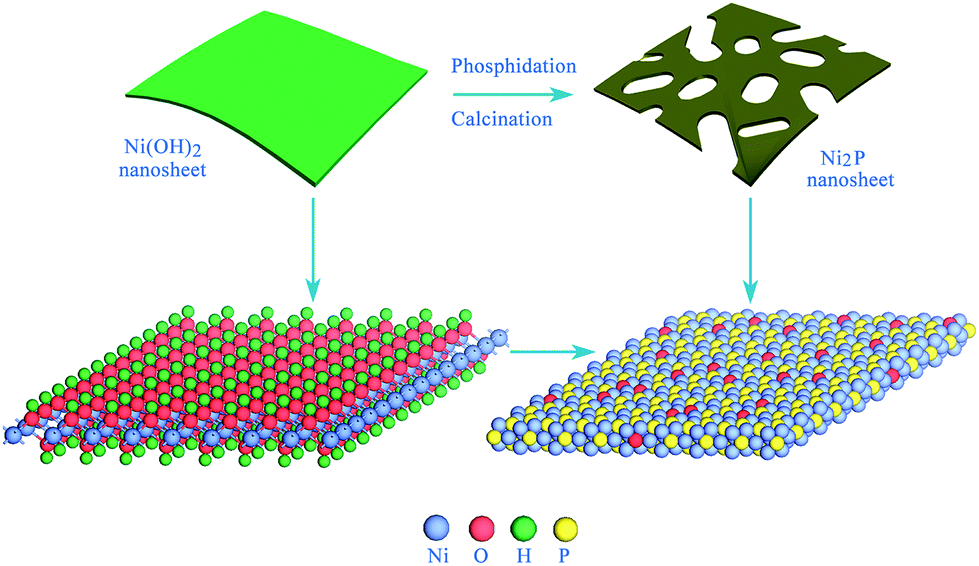 | ||
| Fig. 1 Schematic for the formation of the oxygen-incorporated Ni2P nanosheets using low-temperature phosphidation from the Ni(OH)2 nanosheets precursor. | ||
The oxygen-incorporated Ni2P nanosheets were successfully produced by the thermal conversion reaction, which were verified by a series of characterizations. Fig. 2a and b show the X-ray diffraction (XRD) pattern and transmission electron microscopy (TEM) image for the hydrothermally obtained Ni(OH)2 nanosheets precursor. The diffraction peaks for the Ni precursor can be well identified as the hexagonal Ni(OH)2 phase (JCPDS no. 74-2075). The TEM image reveals that the Ni(OH)2 precursor consists of ultrathin nanosheets with a smooth surface. Of note, the hexagonal phase Ni2P was obtained after the thermal conversion reaction. As shown in Fig. 2c, the phosphidated product presents sharp diffraction peaks corresponding to hexagonal Ni2P (JCPDS no. 74-1385), indicating successful phase conversion of Ni(OH)2 into highly crystalline Ni2P after the direct low-temperature calcination. Fig. 2d depicts the TEM image of the as-synthesized Ni2P, showing rough nanosheet morphology and porosity due to the gas release and dehydration of the Ni(OH)2 nanosheets precursor during the annealing process.50,51 The corresponding SEM images are shown in Fig. S1.† To investigate the electrical conductivity of our synthesized Ni2P nanosheets, temperature-dependent resistance measurement was carried out. As shown in Fig. 2e, the corresponding resistance of the obtained Ni2P increases with increasing temperature from 50 K to 300 K, indicating a typical metal character. All these results demonstrate that a simple phosphidation reaction provides an ideal platform and prerequisite for investigating the relationship between the structure and its intrinsic electrochemical properties.
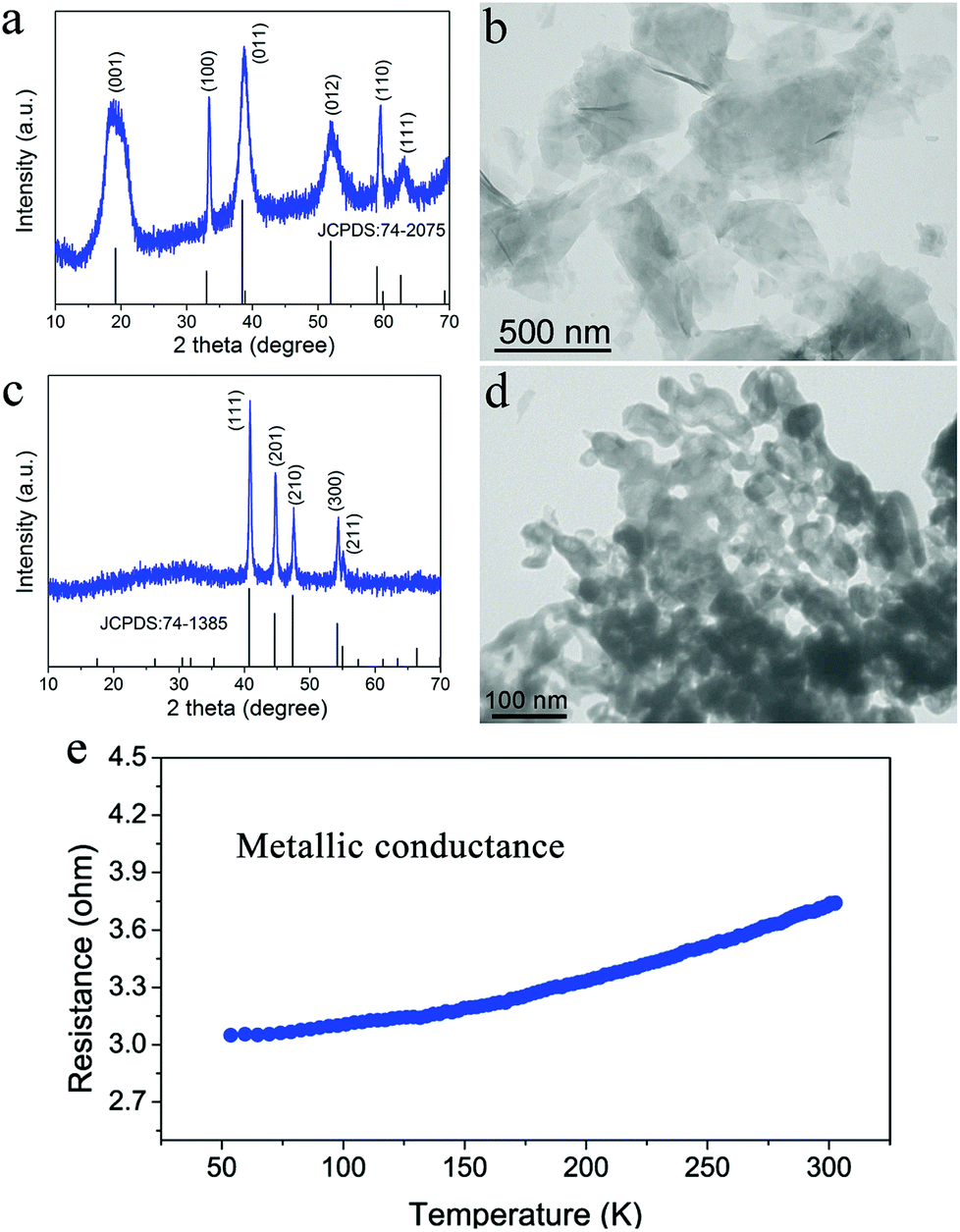 | ||
| Fig. 2 Characterization of the Ni(OH)2 nanosheets precursor and the obtained Ni2P nanosheets. (a) XRD pattern of the Ni(OH)2 nanosheets, (b) TEM image of the Ni(OH)2 nanosheets, (c) XRD pattern of the as-prepared oxygen-incorporated Ni2P nanosheets, (d) TEM image of the as-prepared oxygen-incorporated Ni2P nanosheets and (e) temperature-dependent resistance of the as-synthesized Ni2P nanosheets. | ||
The oxygen incorporation in the surface of the as-synthesized Ni2P nanosheets was verified from the X-ray photoelectron spectra (XPS). As is known, XPS is a powerful tool to determine the surface chemical composition of the materials. Thus, the XPS analysis was carried out to detect the surface state of the obtained Ni2P nanosheets. In Fig. 3a, the high resolution XPS of the Ni 2p core level for our as-synthesized Ni2P nanosheets (red curve) shows two characteristic peaks at 853.4 eV and 870.0 eV arising from Ni 2p3/2 and Ni 2p1/2, respectively, and two peaks located at 129.65 eV and 130.25 eV shown in Fig. S3† corresponding to P 2p3/2 and P 2p1/2, respectively. All peaks are consistent with the reported literature and belong to the Ni2P sample.52 Of note, four distinct peaks in the XPS spectrum of Ni 2p were found corresponding to the binding energy of Ni 2p in NiO53 and suggesting the existence of Ni–O bonds in our Ni2P nanosheets. This verifies successful oxygen inclusion in the Ni2P nanosheets’ surface and is defined as oxygen-incorporated Ni2P. The oxygen incorporation in our fabricated Ni2P nanosheets was attributed to the low-temperature phosphidation process. The conversion reaction process is not accomplished completely leading to remaining Ni–O bonds inherited from the initial Ni(OH)2 precursor, thus realizing oxygen incorporation in the as-obtained Ni2P product.
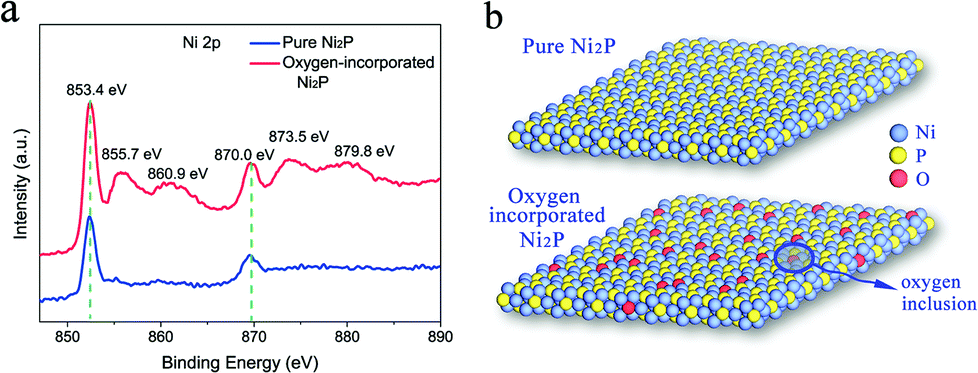 | ||
| Fig. 3 Chemical composition characterization of the as-synthesized Ni2P nanosheets and pure Ni2P samples (a) high resolution XPS spectra of Ni 2p core level of the obtained oxygen-incorporated Ni2P nanosheets and the pure Ni2P. (b) Atomic structure model of the pure Ni2P and obtained oxygen-incorporated Ni2P nanosheets. | ||
In contrast, the normal Ni2P sample was also synthesized using a previously reported method (see the Experimental section) and the XPS was also measured. As shown in the blue curve of Fig. 3a, only two characteristic peaks at 853.4 eV and 870.0 eV were obtained, which arise from Ni 2p3/2 and Ni 2p1/2, respectively, in Ni2P, suggesting the pure Ni–P composition without oxygen incorporation in the Ni2P sample, thus defined as a pure Ni2P sample. The temperature-dependent resistance measurement of the pure Ni2P sample was also performed and exhibited metallic behaviour (Fig. S4†). In this regard, to evaluate the role of the surface state of the catalysts in determining the catalytic reactions, two types of Ni2P samples were fabricated: a pure Ni2P sample and an oxygen-incorporated Ni2P sample with surface modification, as illustrated in Fig. 3b.
As expected, the OER performance of the Ni2P sample with surface oxygen incorporation showed a large enhancement as compared with the pure Ni2P sample. To characterize the performance of the obtained materials as OER electrocatalysts, they were loaded onto a glassy carbon electrode that served as the working electrode, which formed a uniform catalyst film with a catalyst loading of 0.285 mg cm−2.
Linear sweep voltammetry using the prepared electrode was carried out in O2-saturated 0.1 M KOH alkaline solution with a three-electrode set-up (see details in Experimental section). The oxygen-incorporated Ni2P, pure Ni2P and pristine Ni(OH)2 samples were tested using the same method. In fact, the ohmic potential drop (iR) losses caused by electrolyte resistance were compensated before comparison. As shown in Fig. 4a, the pure Ni2P and Ni(OH)2 precursor exhibit poor OER performance. Intriguingly, the oxygen-incorporated Ni2P nanosheets present remarkable enhancement of OER activity, its polarization curve (iR corrected) exhibits much higher current density and smaller onset potential in comparison with those of the pure Ni2P and pristine Ni(OH)2 samples. The oxygen-incorporated Ni2P nanosheets showed stable current density of 10 mA cm−2 at a small overpotential of 347 mV with a Tafel slope as low as 63 mV dec−1 (Table S1†). For example, at 1.6 V vs. RHE, the current density of the oxygen-incorporated Ni2P nanosheets is about 25 times larger than that of the pure Ni2P sample.
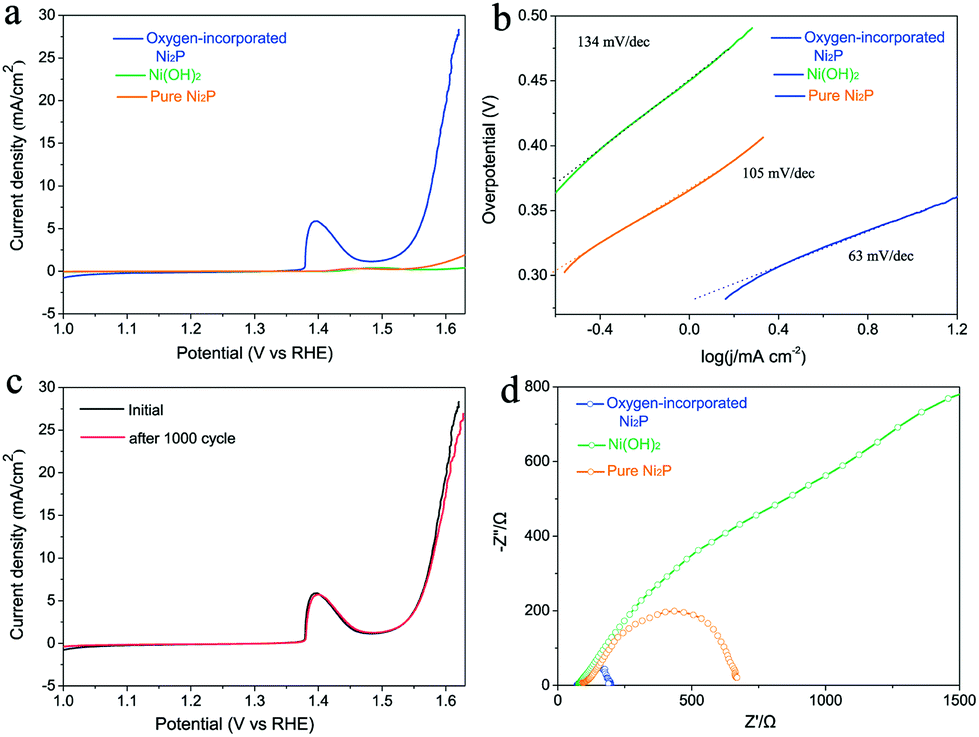 | ||
| Fig. 4 Water oxidation properties: (a) polarization curves and (b) corresponding Tafel plots of oxygen-incorporated Ni2P nanosheets, Ni(OH)2 precursor and pure Ni2P samples as the electrocatalyst. Catalyst loading is about 0.285 mg cm−2 for all samples, sweep rate 5 mV s−1, (c) polarization curves of the oxygen-incorporated Ni2P nanosheets before and after 1000 cycles of an accelerated stability test. Note: All the measurements were performed in O2-saturated 0.1 M KOH medium. (d) Nyquist plots of various catalysts at an overpotential of 400 mV. Z′ is the real impedance and Z′′ is the imaginary impedance. | ||
To gain further insights into the oxygen evolution activity, Tafel plots of the three samples derived from the polarization curves were constructed (Fig. 4b). The Tafel slope of the oxygen-incorporated Ni2P nanosheets is 63 mV dec−1, which is smaller than those of pure Ni2P (105 mV dec−1) and the Ni(OH)2 precursor (134 mV dec−1). Long durability is also one of the most important criteria for judging an effective electrochemical catalyst. To assess it, we cycle the catalyst continuously for 1000 cycles at a scan rate of 50 mV s−1. As shown in Fig. 4c, after long term cycling, the catalyst shows a similar polarization curve as before with negligible decay of the current density, which reveals the excellent stability of the oxygen-incorporated Ni2P nanosheets.
To further prove the better OER electrocatalytic efficiency of oxygen-incorporated Ni2P nanosheets, electrochemical impedance spectroscopy (EIS) was carried out to study the electrode kinetics under OER conditions. The Nyquist plots (Fig. 4d) reveal that the charge-transfer resistance of the oxygen-incorporated Ni2P nanosheets exhibits a significant decrease in comparison with pure the Ni2P and the Ni(OH)2 precursor. The finding suggests that oxygen-incorporated Ni2P nanosheets have the fastest charge transfer process among all three catalysts, which could be associated with surface oxygen incorporation and is in good agreement with the outstanding OER property.
Furthermore, electrochemical double-layer capacitances (Cdl) were measured to evaluate the effective surface area of various catalysts (Fig. 5). As shown in Fig. 5d, the oxygen-incorporated Ni2P nanosheets exhibit much larger Cdl of 5.61 μF than the Ni(OH)2 precursor (0.25 μF) and pure Ni2P (0.92 μF) samples, indicating the high exposure of effective active sites, which is responsible for the excellent electrocatalytic activity. Thus, the higher effective surface area contributes to the excellent electrocatalytic activity of the oxygen-incorporated Ni2P nanosheets.
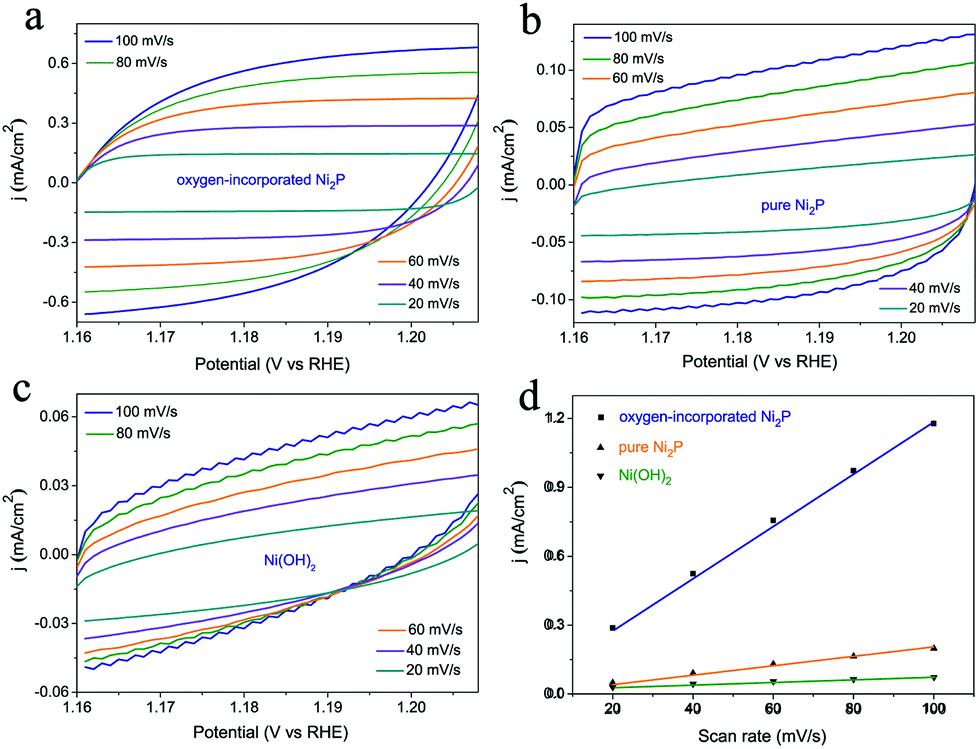 | ||
| Fig. 5 (a–c) Cyclic voltammetry curves of oxygen-incorporated Ni2P nanosheets, Ni(OH)2 precursor and pure Ni2P samples in the region of 1.16–1.21 V vs. RHE. (d) The differences in current density variation (ΔJ = Ja − Jc) at 1.18 V vs. RHE plotted against scan rates. The linear slope, equivalent to twice of the double-layer capacitance Cdl, was used to represent the ECSA. | ||
Finally, we deem that the remarkably enhanced activity of the oxygen-incorporated Ni2P nanosheets catalysts is due to the surface modification, as illustrated in Fig. 6, which has been well established.47,54 In our case, the Ni–O composition on the surface of our obtained Ni2P nanosheets might promote the OER reaction process, thus the OER performance could be highly enhanced compared with the pure Ni2P sample. Our findings demonstrate that the surface properties of catalysts play a critical role in electrochemical processes and offer an in-depth understanding of the electrocatalytic reaction.
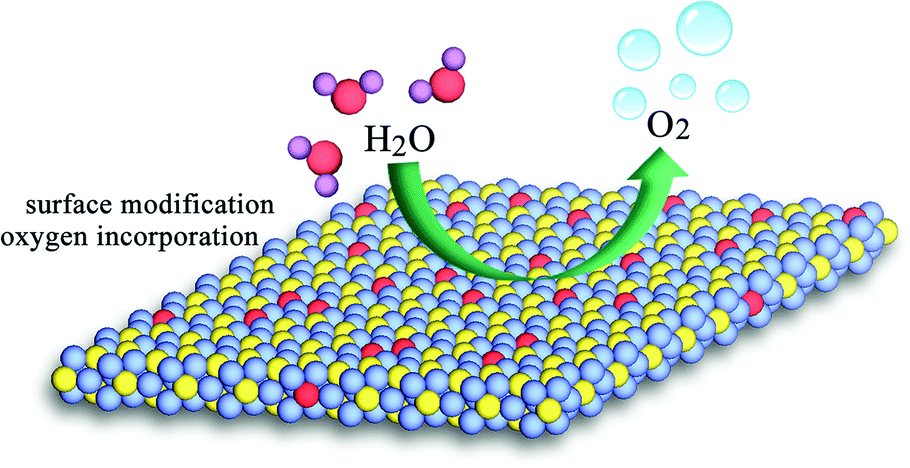 | ||
| Fig. 6 Schematic for the water oxidation on the modified surface of oxygen-incorporated Ni2P nanosheets. | ||
Conclusions
In summary, for the first time, we have successfully developed a surface modification strategy for oxygen incorporation in metallic Ni2P nanosheets through a thermal conversion from the Ni(OH)2 nanosheets precursor, providing a well controlled model system to explore the influence of surface states on the activity of electrocatalysts. As a proof of concept, the prepared oxygen-incorporated Ni2P nanosheets exhibited much higher OER activity compared with the pure Ni2P counterpart and the pristine Ni(OH)2 precursor. The oxygen incorporation in the Ni2P nanosheets presents a small overpotential of 347 mV at a current density of 10 mA cm−2 with a low Tafel slope of 63 mV dec−1. The large electrocatalytic current at 1.6 V is about 25 times enhanced compared with the pure Ni2P counterpart. The highly improved OER performance was attributed to the surface existence of Ni–O components on the Ni2P nanosheets, which promote the water oxidation reaction. Our results show that appropriate surface modification can improve the electrochemical activity of electrocatalysts and calls for the design of high performance electrocatalysts.Acknowledgements
This study was financially supported by the National Basic Research Program of China (2015CB932302), the National Natural Science Foundation of China (21222101, 21501164, U1432133, 11132009, 21331005, 11321503, J1030412), the National Young Top-Notch Talent Support Program, the Chinese Academy of Sciences (XDB01020300), the Fok Ying-Tong Education Foundation, China (Grant No. 141042), and the Fundamental Research Funds for the Central Universities (WK2060190027, WK2340000065).Notes and references
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- K. Yin, Z. D. Cui, X. R. Zheng, X. J. Yang, S. L. Zhu, Z. Y. Li and Y. Q. Liang, J. Mater. Chem. A, 2015, 3, 22770 CAS.
- K. Z. Y. Zeng, C. L. Tan, X. Huang, S. Y. Bao and H. Zhang, Energy Environ. Sci., 2014, 7, 797–803 Search PubMed.
- L. Yu, B. Y. Xia, X. Wang and X. W. Lou, Adv. Mater., 2016, 28, 92–97 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. X. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Q. P. Lu, Y. F. Yu, Q. L. Ma, B. Chen and H. Zhang, Adv. Mater., 2016, 28, 1917–1933 CrossRef CAS PubMed.
- X. Y. Yu, L. Yu, B. H. Wu and X. W. Lou, Angew. Chem., Int. Ed., 2015, 54, 5331–5335 CrossRef CAS PubMed.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 Search PubMed.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-Horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- V. Petrykin, K. Macounova, O. A. Shlyakhtin and P. Krtil, Angew. Chem., Int. Ed., 2010, 49, 4813–4815 CrossRef CAS PubMed.
- M. Gong, Y. G. Li, H. L. Wang, Y. Y. Liang, J. Z. Wu, J. G. Zhou, J. Wang, T. Regier, F. Wei and H. J. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- L. Kuai, J. Geng, C. Y. Chen, E. J. Kan, Y. D. Liu, Q. Wang and B. Y. Geng, Angew. Chem., Int. Ed., 2014, 53, 7547–7551 CrossRef CAS PubMed.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- S. Chen and S. Z. Qiao, ACS Nano, 2013, 7, 10190–10196 CrossRef CAS PubMed.
- J. Bao, X. D. Zhang, B. Fan, J. J. Zhang, M. Zhou, W. L. Yang, X. Hu, H. Wang, B. C. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- H. Hu, B. Y. Guan, B. Y. Xia and X. W. Lou, J. Am. Chem. Soc., 2015, 137, 5590–5595 CrossRef CAS PubMed.
- Z. Jiang, Z. Jiang, M. Thandavarayan and A. Manthiram, J. Mater. Chem. A, 2016, 4, 5877–5889 CAS.
- Y. Q. Guo, Y. Tong, P. Z. Chen, K. Xu, J. Y. Zhao, Y. Lin, W. S. Chu, Z. M. Peng, C. Z. Wu and Y. Xie, Adv. Mater., 2015, 27, 5989–5994 CrossRef CAS PubMed.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281–7285 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, Z. P. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- K. Fominykh, J. M. Feckl, J. Sicklinger, M. Doblinger, S. Bocklein, J. Ziegler, L. Peter, J. Rathousky, E. W. Scheidt, T. Bein and D. Fattakhova-Rohlfing, Adv. Funct. Mater., 2014, 24, 3123–3129 CrossRef CAS.
- M. R. Gao, W. C. Sheng, Z. B. Zhuang, Q. R. Fang, S. Gu, J. Jiang and Y. S. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed.
- X. Long, J. K. Li, S. Xiao, K. Y. Yan, Z. L. Wang, H. N. Chen and S. H. Yang, Angew. Chem., Int. Ed., 2014, 53, 7584–7588 CrossRef CAS PubMed.
- W. J. Zhou, X. J. Wu, X. H. Cao, X. Huang, C. L. Tan, J. Tian, H. Liu, J. Y. Wang and H. Zhang, Energy Environ. Sci., 2013, 6, 2921–2924 CAS.
- K. Xu, P. Z. Chen, X. L. Li, Y. Tong, H. Ding, X. J. Wu, W. S. Chu, Z. M. Peng, C. Z. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- L. Han, X. Y. Yu and X. W. Lou, Adv. Mater., 2016, 28, 4601–4605 CrossRef CAS PubMed.
- X. Y. Yu, Y. Feng, B. Guan and X. W. Lou, Energy Environ. Sci., 2016, 9, 1246–1250 CAS.
- M. Jiang, Y. Li, Z. Lu, X. Sun and X. Duan, Inorg. Chem. Front., 2016, 3, 630–634 RSC.
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015 DOI:10.1002/aenm.201500985.
- N. Jiang, B. You, M. Sheng and Y. Sun, ChemCatChem, 2016, 8, 106–112 CrossRef CAS.
- Z. Huang, Z. Chen, Z. Chen, C. Lv, M. G. Humphrey and C. Zhang, Nano Energy, 2014, 9, 373–382 CrossRef CAS.
- H. Li, W. Wang, Z. Gong, Y. Yu, L. Piao, H. Chen and J. Xia, J. Phys. Chem. Solids, 2015, 80, 22–25 CrossRef CAS.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- Y. M. Shi, Y. Xu, S. F. Zhuo, J. F. Zhang and B. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 2376–2384 CAS.
- J. Q. Tian, Q. Liu, A. M. Asiri and X. P. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- C. D. Wang, T. Ding, Y. Sun, X. L. Zhou, Y. Liu and Q. Yang, Nanoscale, 2015, 7, 19241–19249 RSC.
- S. T. Oyama, T. Gott, H. Y. Zhao and Y. K. Lee, Catal. Today, 2009, 143, 94–107 CrossRef CAS.
- D. Fuks, D. Vingurt, M. V. Landau and M. Herskowitz, J. Phys. Chem. C, 2010, 114, 13313–13321 CAS.
- D. Kanama, S. T. Oyama, S. Otani and D. F. Cox, Surf. Sci., 2004, 552, 8–16 CrossRef CAS.
- R. Parsons, Trans. Faraday Soc., 1958, 54, 1053 RSC.
- J. Greeley, J. K. Norskov, L. A. Kibler, A. M. El-Aziz and D. M. Kolb, ChemPhysChem, 2006, 7, 1032–1035 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, V. Van der, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef PubMed.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- H. Yin, S. Zhao, K. Zhao, A. Muqsit, H. Tang, L. Chang, H. Zhao, Y. Gao and Z. Tang, Nat. Commun., 2015, 6, 6430 CrossRef CAS PubMed.
- J. Wang, Q. Yang, Z. Zhang and S. Sun, Chem. – Eur. J., 2010, 16, 7916–7924 CrossRef CAS PubMed.
- L. Li, K. H. Seng, Z. Chen, Z. Guo and H. K. Liu, Nanoscale, 2013, 5, 1922–1928 RSC.
- J. Tian, Q. Liu, A. M. Asiri and X. P. Sun, J. Am. Chem. Soc., 2014, 136, 7587–7590 CrossRef CAS PubMed.
- A. Han, H. L. Chen, Z. J. Sun, J. Xu and P. W. Du, Chem. Commun., 2015, 51, 11626–11629 RSC.
- M. A. Peck and M. A. Langell, Chem. Mater., 2012, 24, 4483–4490 CrossRef CAS.
- P. A. Thiel and T. E. Madey, Surf. Sci. Rep., 1987, 7, 211–385 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6qi00078a |
| This journal is © the Partner Organisations 2016 |