
Six- and seven-coordinate Fe(II) and Zn(II) compounds ligated by unsymmetric xanthene-based ligands: characterization and magnetic properties†
Ruth M.
Haas
a,
Muhammad
Arshad‡
b,
James
Anthony
b,
Philipp J.
Altmann
a,
Alexander
Pöthig
a,
Frank H.
Köhler
a and
Corinna R.
Hess
*a
aTechnische Universität München, Department of Chemistry and Catalysis Research Center, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: corinna.hess@ch.tum.de
bDurham University, Department of Chemistry, South Road, Durham, DH1 3LE, UK
First published on 4th February 2016
Abstract
The synthesis of a set of novel unsymmetric ligands, iXa and iXa-2, and of their corresponding FeII and ZnII complexes are described. The ligands incorporate a redox-active α-diimine moiety alongside (2-pyridylmethyl)amino groups. Electrochemical data highlight ligand-centered reduction processes available in these systems. The molecular structures of the series of six- and seven-coordinate metal complexes reveal a pentagonal bipyramidal geometry for all compounds. The ligands and complexes were further characterized in the solution state by NMR spectroscopy, with assignments supported by DFT calculations. The ferrous-iXa and -iXa-2 complexes have a high spin, S = 2 configuration. Magnetic susceptibility studies reveal the unusual low-temperature ferromagnetism of these complexes. Intermolecular interactions between the iron centers are mediated by the PF6− counterions, as evidenced by 31P-NMR studies.
 Corinna R. Hess | Corinna Hess received a B.S. in Chemistry from the University of Chicago, where she carried out undergraduate research in the group of Prof. Gregory Hillhouse. She completed her PhD studies in the area of bioinorganic chemistry at the California Institute of Technology under the supervision of Prof. Harry Gray. Corinna further developed her interests in catalysis and inorganic spectroscopy during postdoctoral studies at the University of California, Berkeley (with Prof. Judith Klinman) and the Max Planck Institute for Bioinorganic Chemistry (with Prof. Karl Wieghardt). The research projects focused on the mechanisms of copper oxygenases, and the study of metal complexes coordinated by redox-active ligands. She began her independent career as a lecturer at Durham University, and was appointed as Assistant Professor of Bioinorganic Chemistry at the Technische Universität München in 2014. Her research centers on the design and electronic structure studies of new transition metal complexes, and the development of catalysts for small molecule activation. |
Introduction
Seven coordinate metal centres represent a minor subset of transition metal compounds (<2% of structures in the Cambridge Structural Database (CSD); 1% of Fe-containing structures, 1% of Zn-containing structures1) and are even rarer in nature, limited to a handful of enzymes containing Mo, Mn or Cd.2 Reviews of heptacoordinate transition metal complexes have tended to focus on the structural aspects, rather than on their reactivity or applications.2,3 However, seven coordinate iron complexes – of relevance to this work – have exhibited interesting magnetic properties and reactivities that merit further examination of such higher coordinate systems. The superoxide dismutase activities of the pentagonal bipyramidal Fe(dapsox) and pentaaza macrocyclic iron compounds provide noteworthy examples of biological relevance.4–6 The reports of atom transfer reactions by pentagonal bipyramidal (PBP) Fe-oxido and amido complexes based on the qpy (2,2′:6′,2′′:6′′,2′′′:6′′′,2′′′′-quinquepyridine) and tris(2-picolyl)amine ligands complement the well-established atom transfer chemistry of lower-coordinate iron compounds.7,8 Reports of seven coordinate iron complexes that exhibit spin crossover,9–12 single-molecule magnet behaviour,13,14 or Light-Induced Excited Spin State Trapping (LIESST) phenomena,15,16 also are found in the literature.We herein present pentagonal bipyramidal Fe- and Zn-containing complexes ligated by a set of novel unsymmetric xanthene-based molecules, as shown in Scheme 1. The ligands were generated as part of ongoing work in our group pertaining to the development of unsymmetric coordination scaffolds containing redox-active functionalities. The molecules, iXa and iXa-2, contain a redox-active α-iminopyridine along with the innocent (2-pyridylmethyl)amino group(s) in the opposing position. The arrangement resembles that of the phenylenediamine based ligand, L1, reported previously, which supported bimetallic complexes.17 We examined the binding capabilities of the more structured xanthene framework. In this case, the multidentate N4O or N5O framework of iXa and iXa-2 encapsulates a single metal ion, yielding mononuclear complexes. The M-(iXa-2) complexes are six-coordinate, while heptacoordinate compounds result from metal-ligation to iXa. NMR spectroscopy, electrochemistry, magnetic susceptibility studies and DFT computational methods have been used to characterize the complexes in the solution and solid state; the results are described in this report. The data highlight intriguing magnetic properties of the iron-containing complexes.
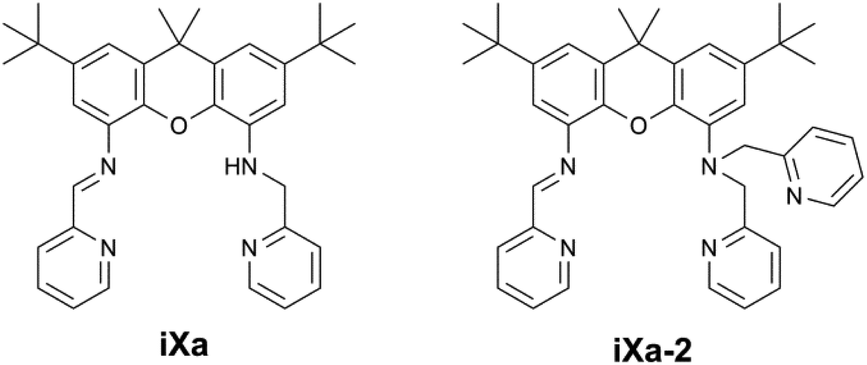 | ||
| Scheme 1 Ligands described in the present work. | ||
Results and discussion
Synthesis of ligands and metal-containing compounds
The newly reported ligands, iXa and iXa-2, were synthesized from 2,7-di-tert-butyl-9,9-dimethyl-4,5-xanthene diamine according to the route detailed in Scheme 2. The bis(2-pyridylmethyl)amino groups were attached to the BOC-protected 1 upon reaction with either 1 or 2 equiv. of chloromethylpyridine. Following removal of the BOC group, the redox-active α-iminopyridine moiety was added to the xanthene backbone via the reaction of 3a or 3b with pyridine carboxaldehyde. The corresponding ferrous complexes, [Fe(iXa)(CH3CN)2](PF6)2 (4) and [Fe(iXa-2)](PF6)2 (5), were subsequently obtained upon reaction of the ligands with [Fe(CH3CN)6](PF6)2, while the Zn-containing analogues [Zn(iXa)(OTF)2] (6) and [Zn(iXa-2)](OTF)2 (7) were obtained using Zn(OTf)2 as the metal ion source (Scheme 3).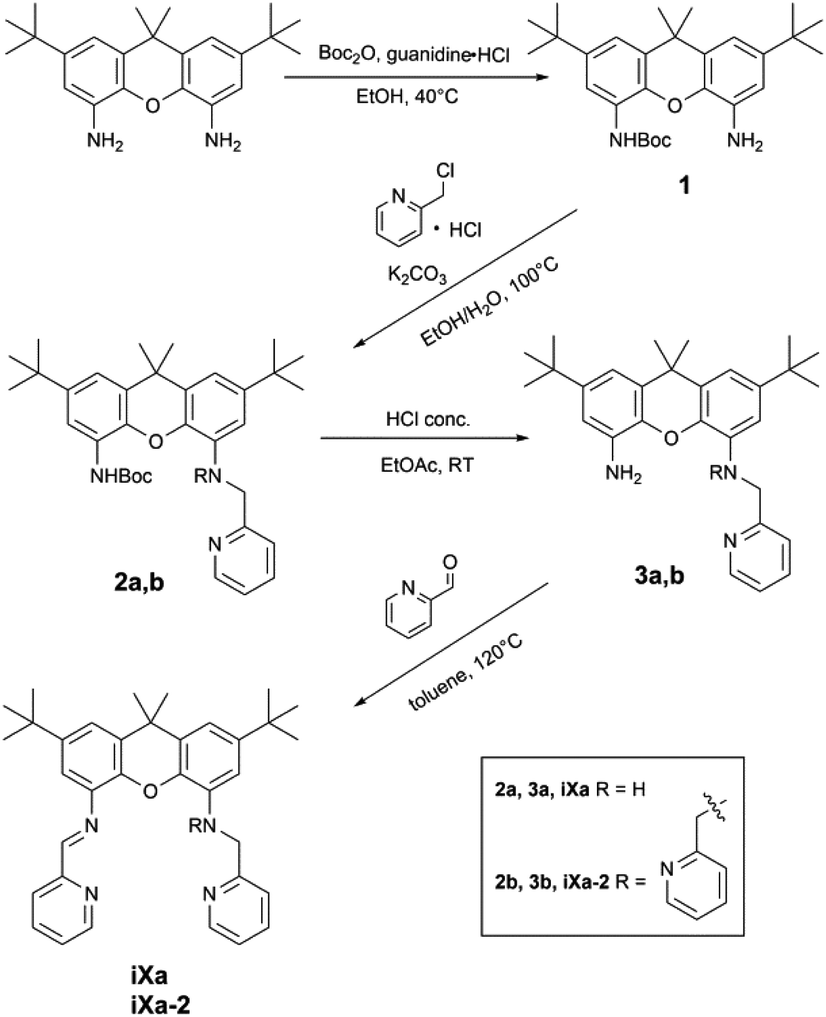 | ||
| Scheme 2 Synthesis of iXa and iXa-2. | ||
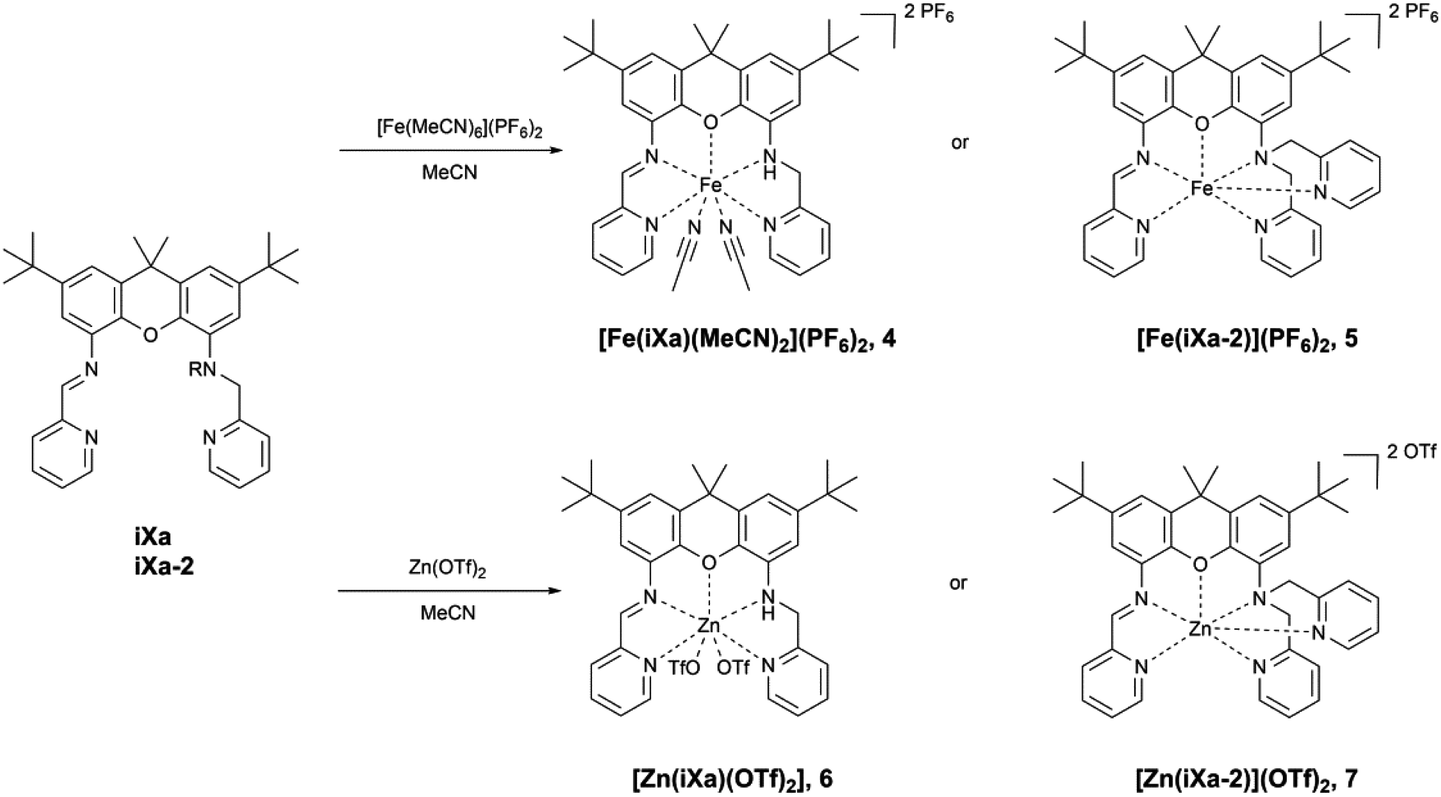 | ||
| Scheme 3 Synthesis of 4–7. | ||
Xanthene-based coordination complexes containing iminopyridine functionalities have been reported previously. The symmetric ligand frameworks in these related systems, which consist of identical iminopyridine groups at the 4- and 5-positions of the xanthene, can accommodate two metal ions yielding binuclear Pt and Cu complexes.18–20 In contrast, the unsymmetric iXa and iXa-2 act as multidentate chelators and sequester a single metal ion within the NxO (x = 4, 5) cavity. The solid state structures of the mononuclear compounds 4–7 are shown in Fig. 1 (crystallographic data are provided in Table 1). The molecular structures suggest a high spin configuration for the ferrous compounds, with average Fe–N bond distances of Fe–Navg = 2.24 Å and Fe–Navg = 2.17 Å for 4 and 5 respectively (Fig. 1). Compound 4, with one less pyridine moiety in comparison to 5, additionally ligates two acetonitrile molecules, adopting a seven-coordinate distorted pentagonal bipyramidal geometry. Among the reported examples of heptacoordinate complexes, the pentagonal bipyramidal geometry commonly arises from the use of pentadentate chelating ligands, such as depicted in Scheme 4.10,21–24
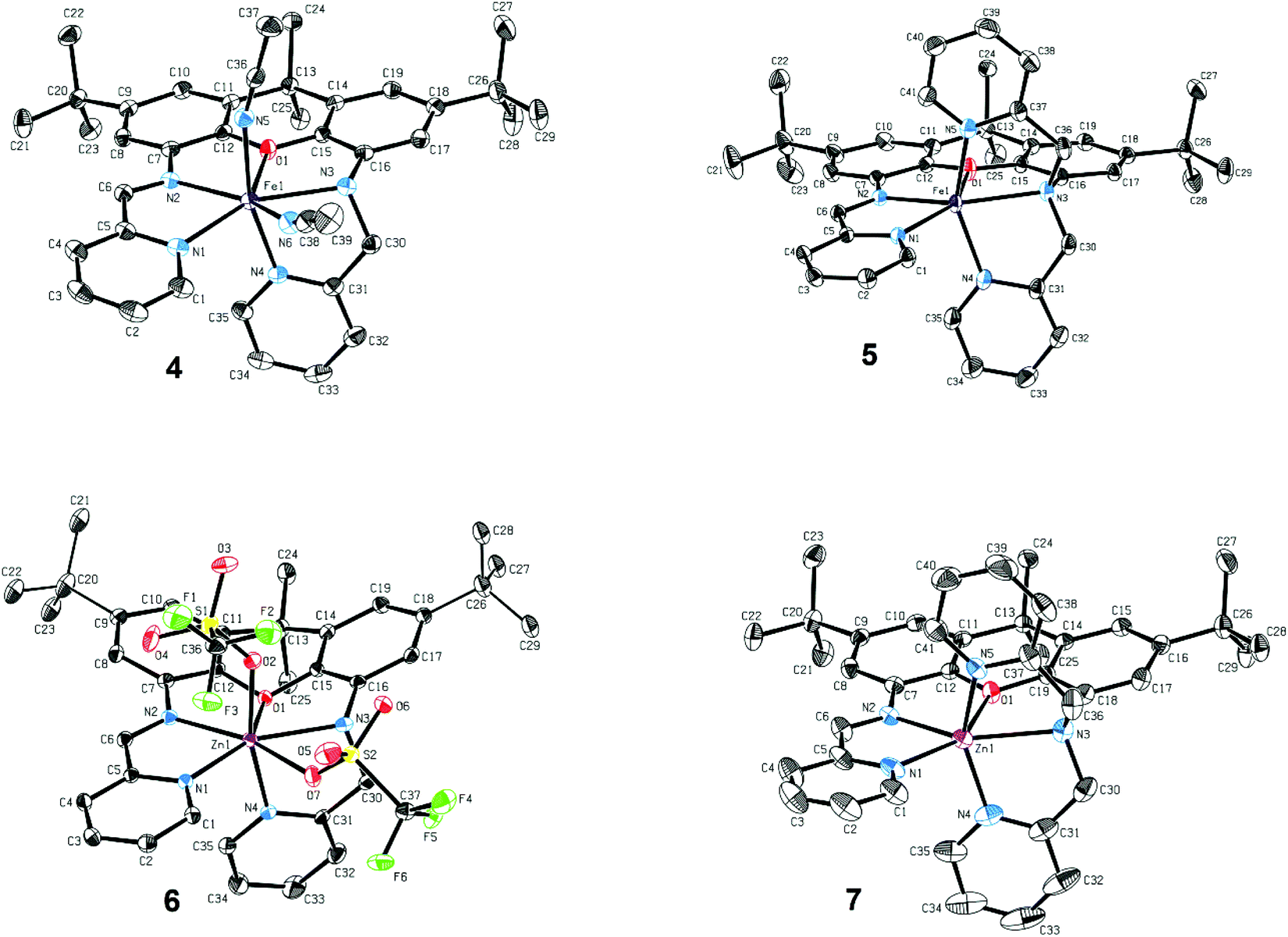 | ||
| Fig. 1 Molecular structures of 4–7 (50% probability ellipsoids). Hydrogen atoms, PF6− and OTf− counterions and solvent molecules omitted for clarity. Selected bond length (Å): 4: Fe1–N1 2.244(15), Fe1–N2 2.246(14), Fe1–N3 2.335(14), Fe1–N4 2.134(14), Fe1–N5 2.152(15), Fe1–N6 2.329(16), Fe1–O1 2.380(11); 5: Fe1–N1 2.185(11), Fe1–N2 2.152(11), Fe1–N3 2.308(11), Fe1–N4 2.112(14), Fe1–N5 2.103(13), Fe1–O1 2.212(10); 6: Zn1–N1 2.136(2), Zn1–N2 2.216(2), Zn1–N3 2.267(2), Zn1–N4 2.096(2), Zn1–O2 2.087(2), Zn1–O7 2.578(2), Zn1–O1 2.554(4); 7: Zn1–N1 2.169(2), Zn1–N2 2.133(2), Zn1–N3 2.379(2), Zn1–N4 2.045(2), Zn1–O2 2.057(2), Zn1–O1 2.273(4). | ||
 | ||
| Scheme 4 Examples of chelating ligands previously reported for the study of seven-coordinate pentagonal bipyramidal iron and zinc complexes (A10; B21; C22; D23; E24). | ||
| 4·2CH3CN | 5·CH3CN | 6·CH2Cl2 | 7 | |
|---|---|---|---|---|
| Empirical formula | C43H52F12FeN8OP2 | C43H48F12FeN6OP2 | C38H42Cl2F6N4O7S2Zn | C43H45F6N5O7S2Zn |
| fw | 1042.72 | 1010.66 | 981.14 | 987.33 |
| Cryst. syst. | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | P21/c | P21/c |
| a (Å) | 11.8732(14) | 17.6546(17) | 10.361(3) | 18.0907(15) |
| b (Å) | 12.7552(15) | 14.2997(14) | 15.945(4) | 14.3660(12) |
| c (Å) | 16.3941(18) | 18.8804(19) | 25.761(6) | 18.9329(15) |
| α (°) | 76.579(5) | 90 | 90 | 90 |
| β (°) | 83.331(5) | 92.641(5) | 93.772(14) | 91.824(4) |
| γ (°) | 82.122(5) | 90 | 90 | 90 |
| Volume (Å3) | 2383.0(5) | 4761.4(8) | 4246.7(17) | 4918.0(7) |
| Z | 2 | 4 | 4 | 4 |
| ρ calc (mg mm−3) | 1.453 | 1.410 | 1.535 | 1.333 |
| μ (mm−1) | 0.474 | 0.471 | 0.881 | 0.657 |
| F(000) | 1076 | 2080 | 2016 | 2040 |
| Reflns collected | 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 160 160 |
203262 |
88748 |
138041 |
| Indep. reflns/Rint | 8733/0.0443 | 13923/0.0410 |
8356/0.0568 | 9693/0.0357 |
| Data/restraints/param. | 8733/85/684 | 13924/21/659 |
8356/84/549 | 9693/248/805 |
| GOF on F2 | 1.042 | 1.024 | 1.019 | 1.056 |
| Final R1 indexes [I ≥ 2σ(I)] | 0.0340 | 0.0387 | 0.0339 | 0.0409 |
| Final wR2 indexes (all data) | 0.0900 | 0.1105 | 0.0444 | 0.0508 |
| Δρmin/max (e Å−3) | 0.736/−0.302 | 0.807/−0.569 | 0.637/−0.729 | 0.622/−0.423 |
External ligands in these systems reside in the axial positions, in a trans-arrangement. iXa provides a similar framework to the aforementioned pentadentate molecules. However, one of the pyridine groups (N4) occupies an apical position with respect to the N1–N2–O1–N3 plane; the acetonitrile molecules thus adopt an unusual cis-configuration. The two planes comprised of the α-iminopyridine group and of the adjoining xanthene aromatic ring diverge by ∼17°. Conjugation of the two groups, combined with the rigidity of the xanthene scaffold, likely prevent a close approach of the two opposing pyridine ligands, forcing the N4-donor atom into the axial position.
The extra pyridine group in iXa-2 leads to the formation of the six-coordinate 5. However, the equatorial ligands, likewise comprised of the analogous N1,N2,O1,N3 atoms, are far from square planar, with a 139.37° N1–Fe–N3 angle. The geometry of 5 is thus also best described by a distorted pentagonal bipyramid, absent one vertex of the equatorial pentagon (the relevant N–Fe–N and N–Fe–O angles are 71°–76°, approximating the 72° expected for an ideal pentagon; Table S1†). The α-iminopyridine moiety is nearly co-planar with the neighbouring aromatic ring of the xanthene backbone (6.5° angle between the two planes), and the N4 and N5 atoms consequently take up the axial positions. Steric encumbrance presumably precludes further coordination of a solvent molecule to the ‘seventh’ position.
Surprisingly, a single Zn ion is likewise encapsulated by the iXa and iXa-2 scaffolds, despite the smaller ionic radius and general tendency of this d10 metal to adopt a tetrahedral geometry. The geometries of 6 and 7, including M–N bond distances and first coordination sphere bond angles (Fig. 1 and Table S1†), are remarkably similar to those of their sister Fe-(iXa) and Fe-(iXa-2) complexes. The triflate ligands of 6 occupy equivalent positions to the acetonitrile ligands in 4. The most noticeable difference among the structures is a substantial buckling of the xanthene backbone in 6 to accommodate the smaller zinc ion.
DFT calculations
DFT calculations (B3LYP) were carried out on 4 and 5, providing a more complete electronic structure description of the ferrous compounds. As expected for the S = 2, high-spin configuration, the spin density is primarily localized on the iron atoms, with small contributions from the O- and N-atoms in the first coordination sphere (Fig. 2). The approximate ordering of the orbitals (Fig. 3) is also as anticipated for the pentagonal bipyramidal complexes, with loss of degeneracy of the e′′ (xz, yz) and e′ (xy, x2 − y2) set, given the unsymmetric ligand environment (with respect to the orbital description and notation for a fully symmetric complex with D5h symmetry).25 The lowest energy d-orbitals of 4, the doubly occupied yz and singly occupied xz, can be considered essentially ‘non-bonding’ (88 and 96.8% d-orbital character based on analysis of the quasi-restricted orbital descriptions). The yz orbital of 5, however, overlaps with π* orbitals of the xanthene moiety, which may account for the shorter Fe–O bond distance observed in the solid state structure of this complex. The diimine π* character of the LUMOs of 4 and 5 are also noteworthy. One-electron reduction of the ferrous compound would place an electron in the metal-based SOMOs, or alternatively, in the ligand-based LUMO.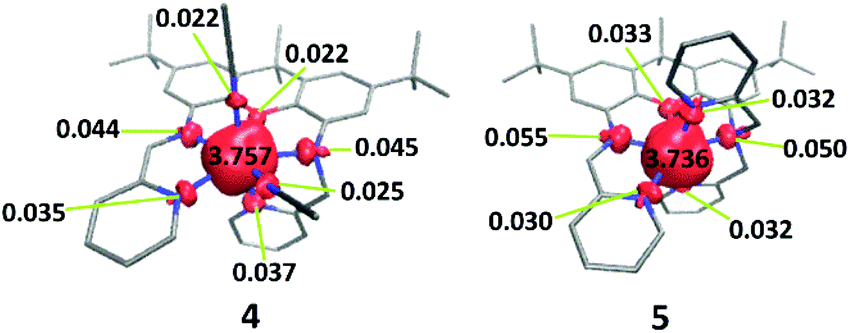 | ||
| Fig. 2 DFT-derived (B3LYP) spin density plots for 4 and 5 based on Löwdin population analysis. | ||
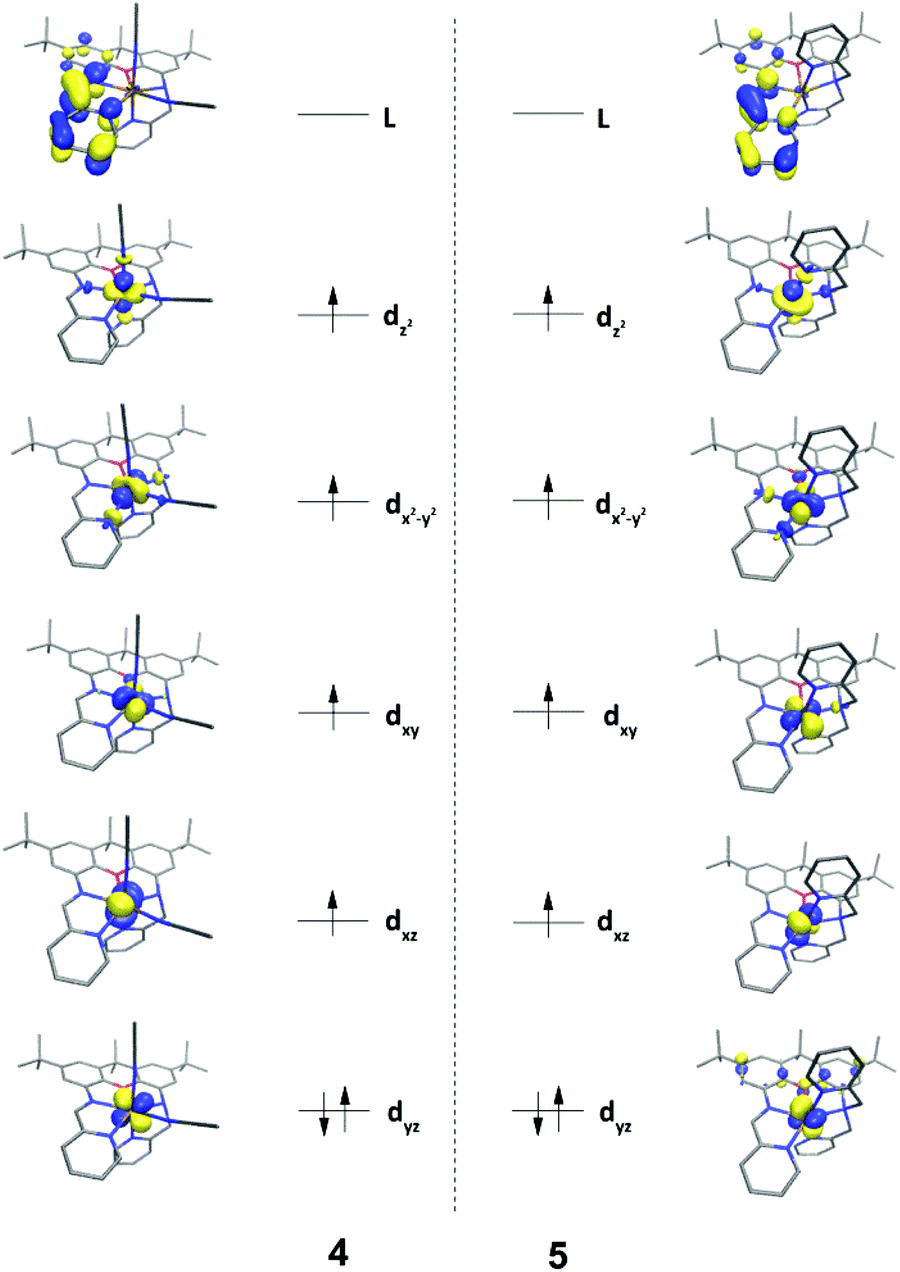 | ||
| Fig. 3 DFT-derived (B3LYP) qualitative MO diagrams for 4 and 5. | ||
Solution state characterization
The electronic spectra of 4 and 5 each feature several intense transitions in the visible region (4: 328 and 381 nm; 5: 329 and 377 nm, Fig. 4). The absorption spectra of the uncomplexed ligands, as well as of 6 and 7, also exhibit bands in this region (Fig. S31†), such that the transitions in the spectra of 4 and 5 likely encompass π → π* components. The spectra of both ferrous compounds additionally display weaker absorption bands at lower energy, which account for the red-brown (4, λmax = 493 nm) and green (5, λmax = 548 nm) colours of the complexes in solution.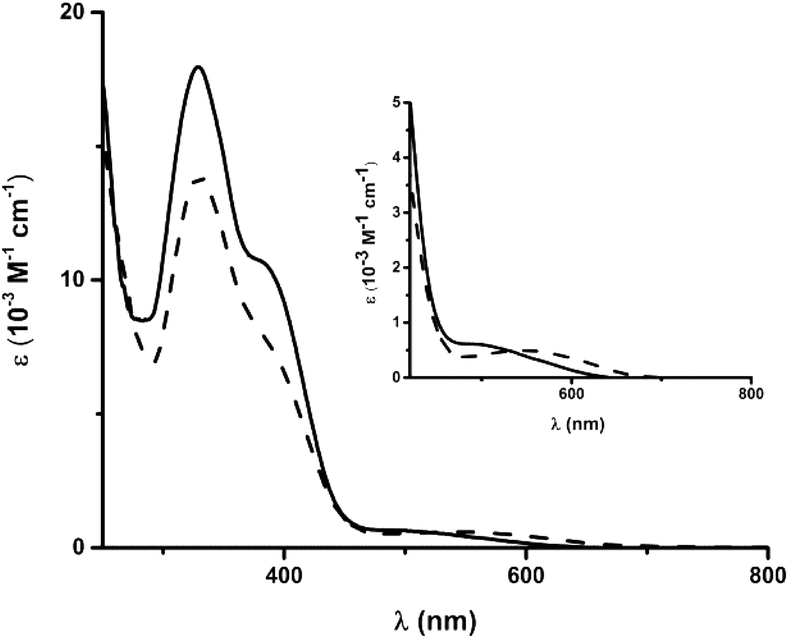 | ||
| Fig. 4 Electronic spectra of 4 (solid line) and 5 (dashed line) in CH3CN. Inset: magnified region from 400 to 800 nm. | ||
The redox-activity of the α-iminopyridine functionality in the iXa and iXa-2 ligands is well-recognized; the molecule is able to accept at least one-electron into its diimine π* system (as illustrated by the LUMOs in Fig. 3), and offers a competing reduction site in metal-complexes thereof. Electrochemical studies were therefore carried out to examine the available redox processes. An irreversible reduction is observed in the CVs of both ferrous compounds at −1.0 to −1.2 V vs. Fc+/0 (Fig. 5), which could be attributed to either a FeII/I couple or to a ligand-centred reduction corresponding to [iXa]/[iXa˙]− and [iXa-2]/[iXa-2˙]−.
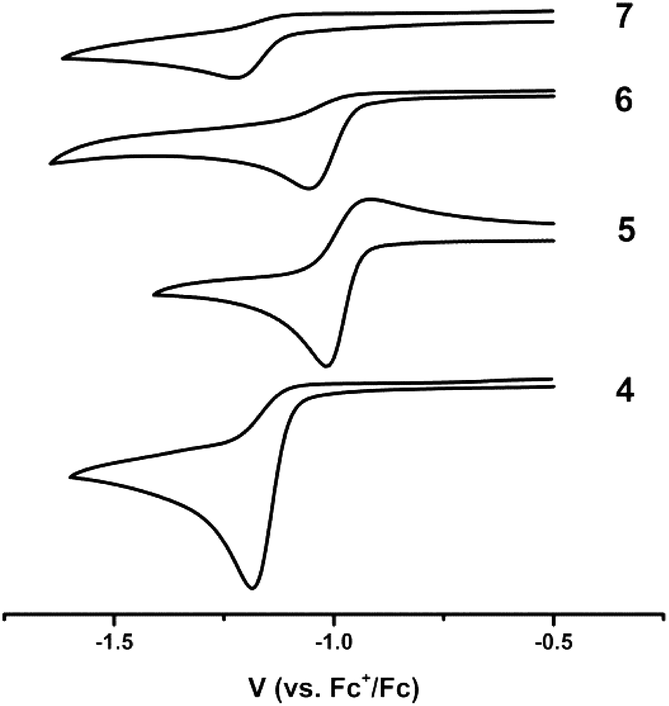 | ||
| Fig. 5 Cyclic voltammograms of 4–7 depicting the first reductive event; CH3CN, 0.1 V s−1, 0.1 M [N(n-Bu)4]PF6. | ||
The CVs of the uncomplexed iXa and iXa-2 do not exhibit reductive events at potentials more positive than −2.0 V (Fig. S32†). However, reduction of 6 and 7 also occurs between −1.0 and −1.2 V, suggesting that the redox couple seen in the CV of the iron complexes is in fact ligand-centred. Attempts to chemically generate and isolate reduced forms of the complexes have thus far been unsuccessful. Irreversible oxidations also are apparent in the CVs of iXa, iXa-2 and 5, in the region of 0.2–1.0 V, but are absent in the CV of 4 even up to a potential of +1.5 V (Fig. S32†). The coordination environment of the seven-coordinate 4 appears to strongly disfavour the oxidized, ferric form of this compound.
NMR studies
The resonances in the 1H NMR spectra of the ligands iXa and iXa-2, as well as of the zinc-containing 6 and 7, could be fully assigned from the complement of 1H, 13C, COSY, HSQC and HMBC NMR spectra. Assignment of the individual protons is depicted in Fig. 6 and 7 (see ESI† for additional NMR spectra). The singlets at 8.75 (iXa) and 8.79 ppm (iXa-2) correspond to the imine proton (He) of each ligand. The methylene protons (Hn) are located at 4.54 (iXa) and 4.65 (iXa-2) ppm. The amine proton of iXa (Hm) is observed as a broad signal at 5.27 ppm. The signals of the pyridine ring that forms part of the redox-active diimine moiety (pyr′) are generally shifted to low frequency with respect to the pyridine protons belonging to the innocent (2-pyridylmethyl)amino groups (pyr′′).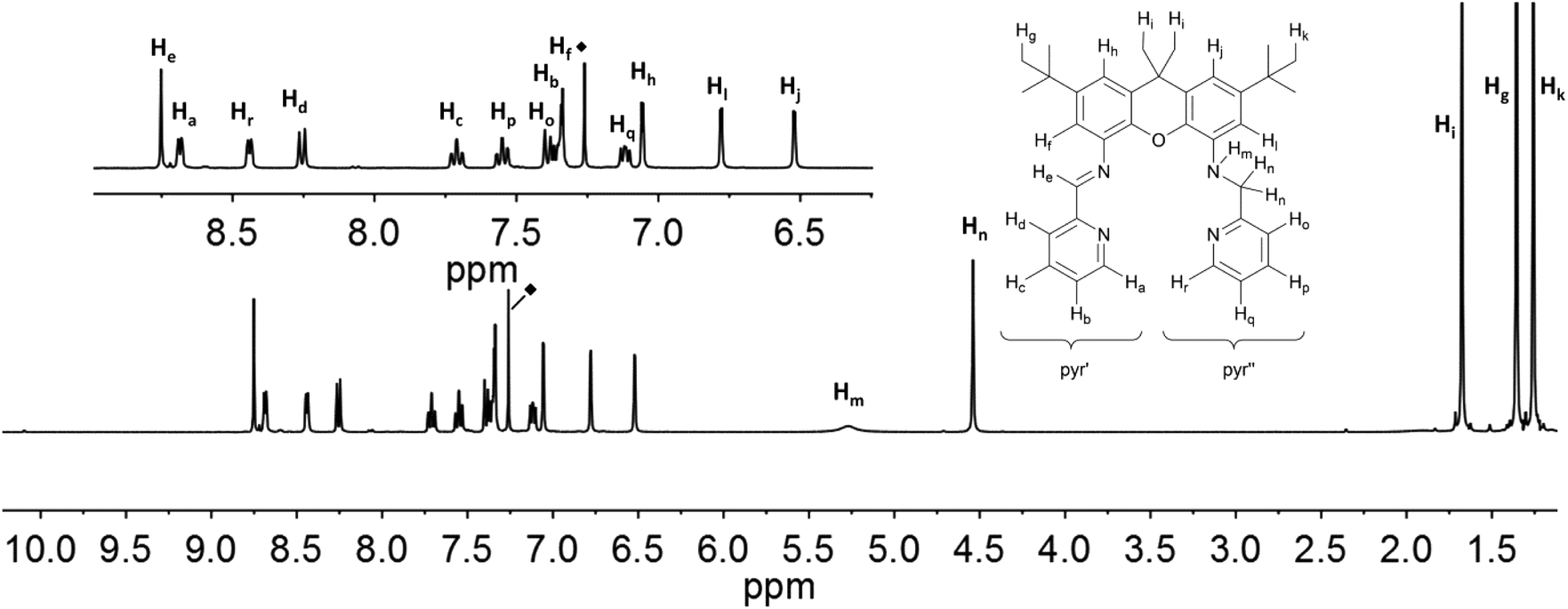 | ||
| Fig. 6 1H NMR-spectra of iXa (600 MHz, CDCl3) with labelling scheme and magnified aromatic region (inset); ◆ = residual solvent signal. | ||
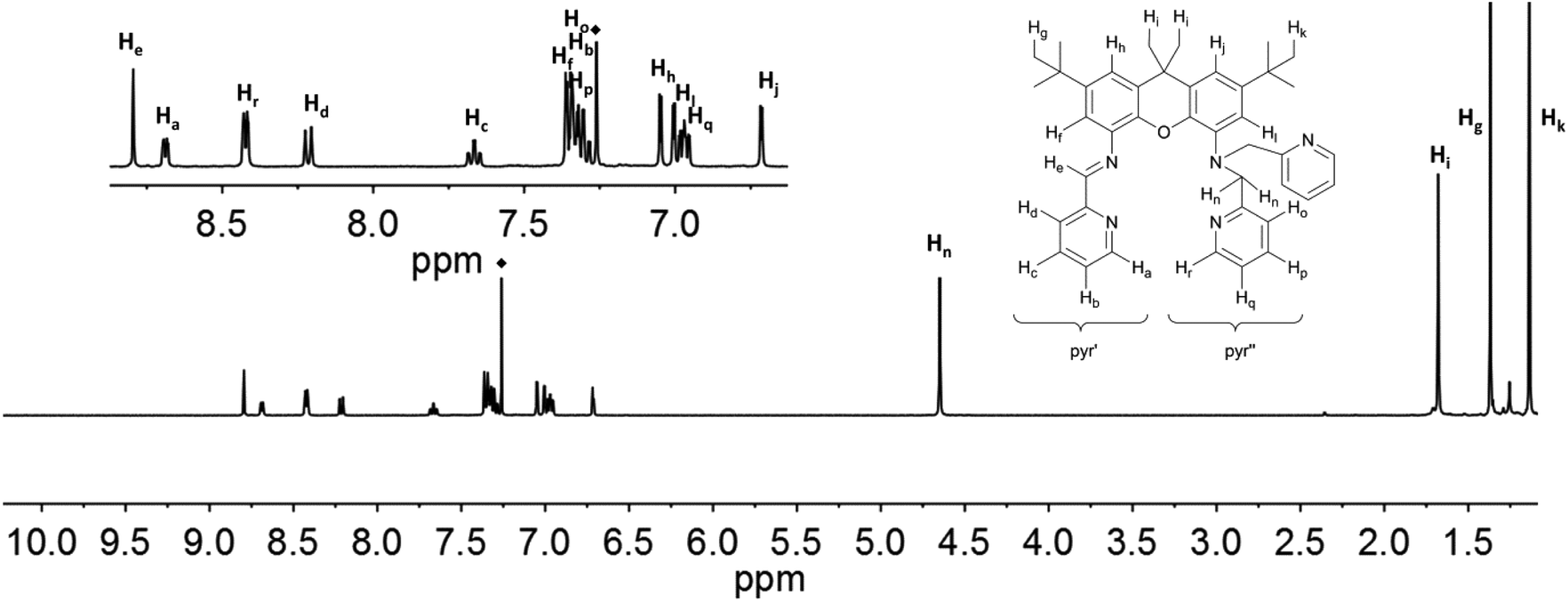 | ||
| Fig. 7 1H NMR-spectra of iXa-2 (600 MHz, CDCl3) with labelling scheme and magnified aromatic region (inset); ◆ = residual solvent signal. | ||
The proton signals in the spectra of 6 and 7 (Fig. 8 and 9) are all shifted to higher frequency in comparison to the analogous resonances of the uncomplexed ligand. This is expected given the deshielding effect of the zinc ions. The imine protons, He, are distinguished by a substantial shift to 9.39 (6) and 9.83 (7) ppm. The presence of the zinc ion in 6 leads to the splitting of the signals corresponding to the two methylene protons (Hn: 5.08 ppm, J = 7.6 Hz and 17.0 Hz; Hn′: 4.38 ppm, J = 17.0 Hz), as well as those of the methyl protons on the xanthene backbone (Hi: 1.68 ppm; Hi′: 1.39 ppm). The signal of the proton closest to the metal ion is assigned to the resonance with the largest shift (Zn⋯Hn = 3.336; Zn⋯Hn′ = 3.903; Zn⋯Hi(avg) = 5.930 Å; Zn⋯Hi′(avg) = 7.105), in fact by analogy to the shift changes seen on passing from the free ligands to the Zn derivatives. The additional pyridine moiety of 7 restores the equivalence of the xanthene methyl protons, which are observed as a single resonance at 1.53 ppm. Distinct signals are still observed for Hn (5.11 ppm) and Hn′ (5.06 ppm), though neither the relative shifts, nor their relative proximity to the Zn ion, differ as markedly as in 6. The protons of the xanthene backbone (Hj, Hl, Hf and Hh) and of the individual pyr′ and pyr′′ protons also could be distinguished in the spectrum of each Zn-containing compound.
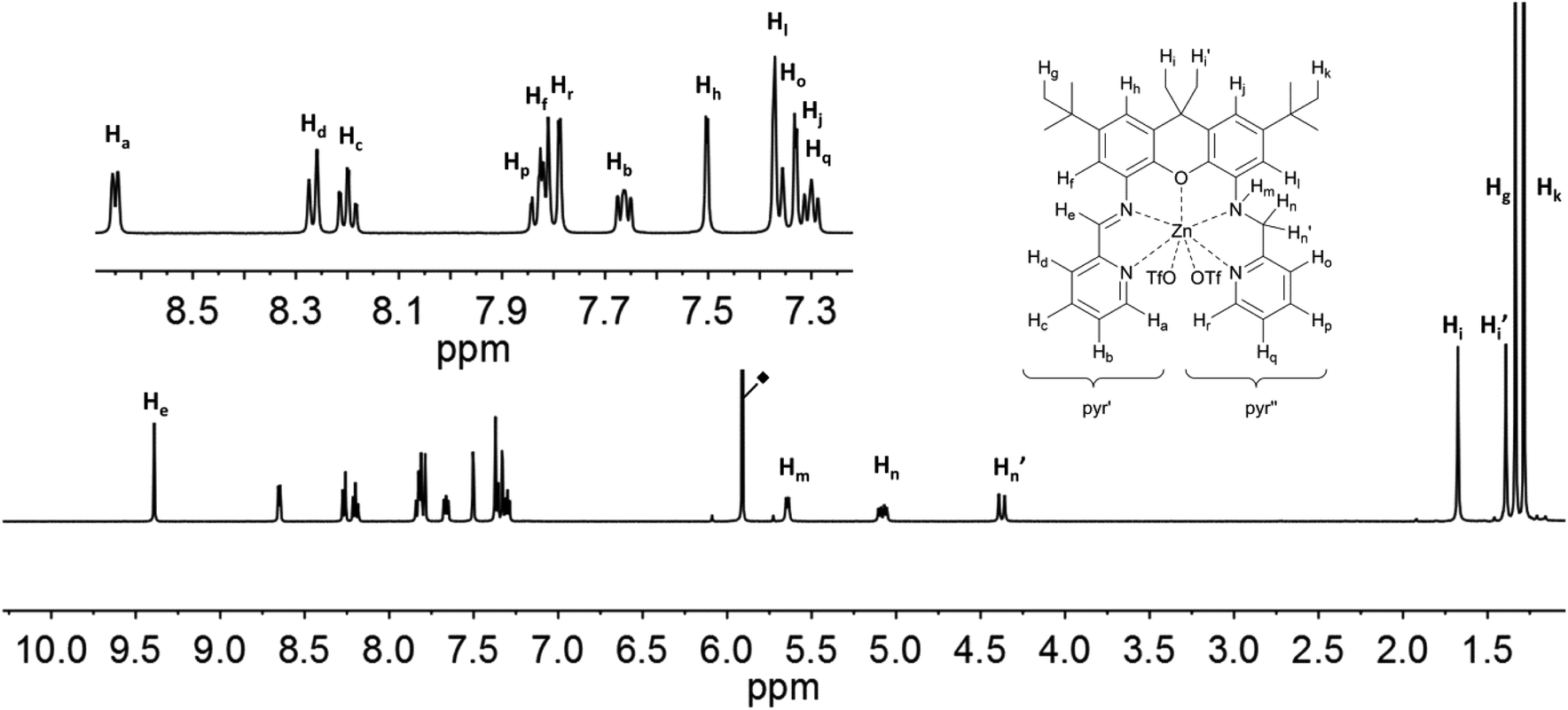 | ||
| Fig. 8 1H NMR-spectra of 6 (500 MHz, C2D2Cl4) with labelling scheme and magnified aromatic region shown in inset; ◆ = residual solvent signal. | ||
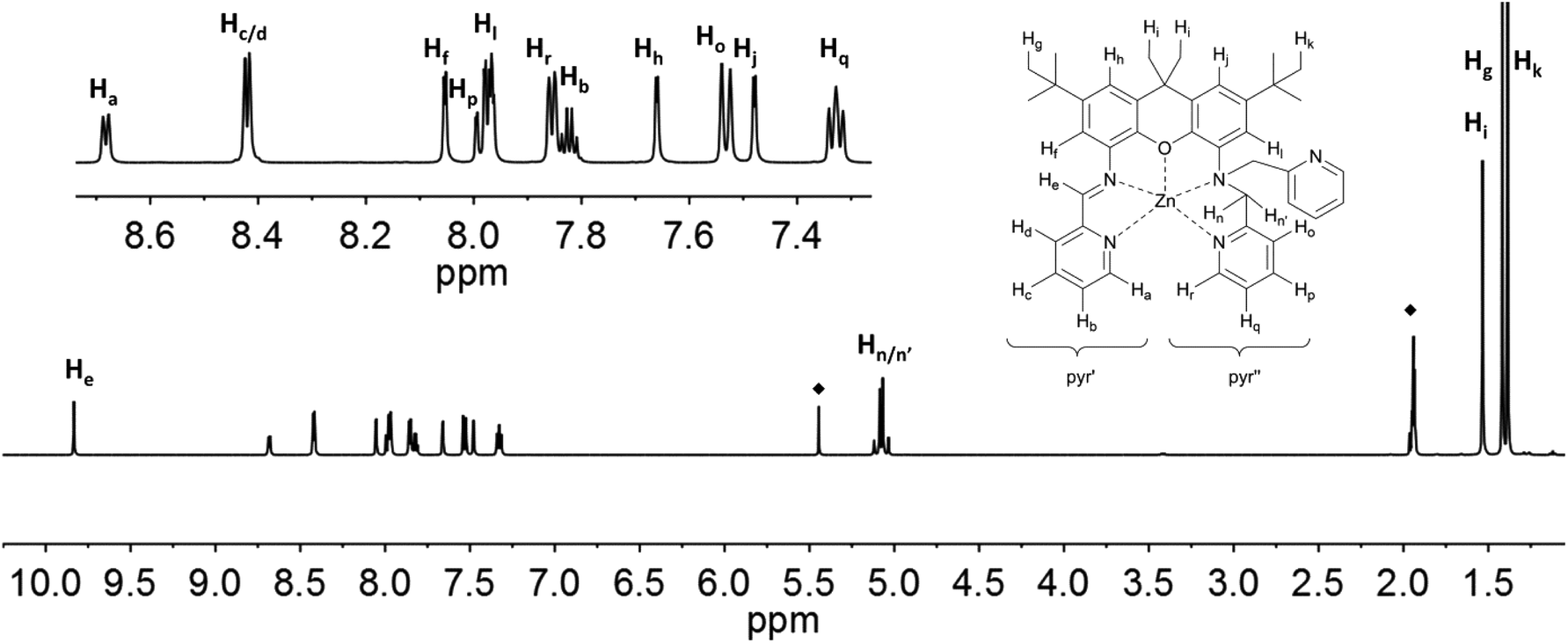 | ||
| Fig. 9 1H NMR-spectra of 7 (500 MHz, CD3CN) with labelling scheme and magnified aromatic region shown in inset; ◆ = solvent signals: CH3CN and CH2Cl2. | ||
The solution 1H NMR spectra of 4 and 5 clearly show that both compounds are paramagnetic. Near room temperature the signal shifts of 4 span a range of approximately −60 to 300 ppm (Fig. 10); the signals are broadened up to a half width, Δ, of about 15 Hz to 2.9 kHz so that no proton couplings are observed. The corresponding data for 5 are −3 to 226 ppm and up to Δ = 1 kHz (Fig. 11). Since the expected symmetry of the cation of 4 in solution (C1) is lower than that of 5 (Cs), the spectrum of the latter is simpler.
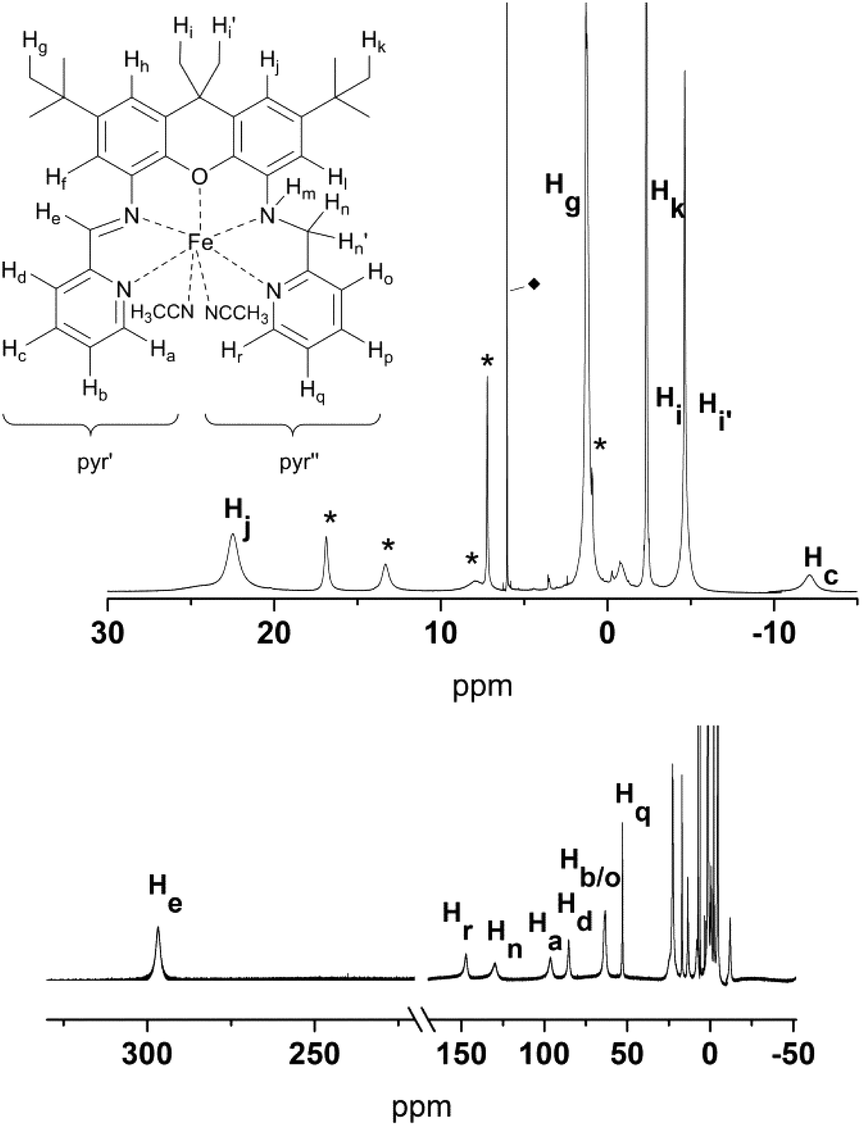 | ||
| Fig. 10 1H NMR-spectra of 4 (400 MHz, C2D2Cl4), with labelling scheme and magnified regions between 30 and −15 ppm depicting the individual proton assignments; * denotes signals that correspond to 4 but that could not be individually assigned. ◆ = residual solvent signal. | ||
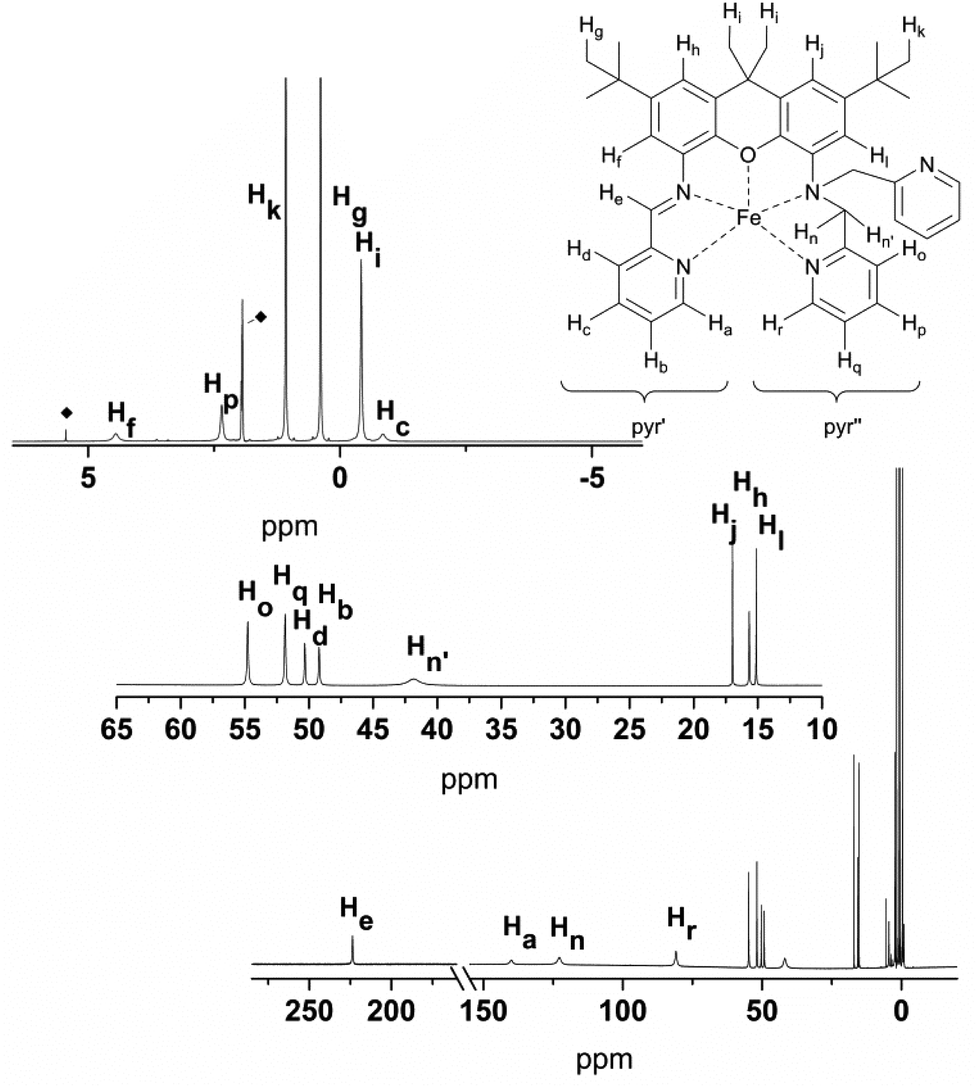 | ||
| Fig. 11 1H NMR-spectra of 5 (400 MHz, CD3CN), with labelling scheme, and magnified regions between 60 and 10 ppm, and 8 and −4 ppm, depicting the individual proton assignments. ◆ = solvent signals: CH3CN and CH2Cl2. | ||
The signal assignment typically is based on the mechanism that transfers spin density from the metal d orbitals to the ligand nuclei.26 The xanthene and pyridine moieties of the iXa and iXa-2 ligands are expected to receive spin in their π systems, such that typical patterns of negatively and positively shifted signals should be seen. However, the four SOMOs (Fig. 3) with dominant d-orbital character are not only π-faced but also σ-faced, such that the signal patterns are unreliable. Consequently, and due to the substitution pattern of the xanthene moiety, only three signals with a negative shift are observed in the spectrum of 5.
The signal assignment may also be derived from shift calculations.27–30 Here we approximate the theoretical shifts by converting the calculated spin densities into contact shifts, δconT, at the measuring temperature T (cf. ESI† for further details). The contact shift is a component of the experimental shift, δexpT:
| δconT = δexpT − δdipT− δdia, |
The NMR spectra complement the visualization of the DFT-derived spin data in Fig. 2, as they reflect data that correlates with spin delocalization onto the ligand protons. For example, the pyridine moieties of 5 accommodate much more spin (strongly shifted signals of Ha–d and Ho–r) than does the xanthene moiety (weakly shifted signals of Hf, Hh, Hj, and Hl). The signals of the diastereotopic protons Hn and Hn′ are separated by 81 ppm, which is three orders of magnitude larger than for the zinc analogue 7 (0.05 ppm). The striking difference is due to the fact that nuclear shielding is in operation in diamagnetic compounds, while in paramagnetic compounds, the electron-nuclear coupling is in effect (after correcting for the shielding via δdia). The coupling across the path Fe–N–C–Hn/n′ is determined by the spin transfer from the metal to protons in the γ-position26 (second-order Fe–Hn/n′ hyperconjugation). Hence the signal shift of Hn/n′ depends on the dihedral angle between the Fe–N and the N–C–Hn/n′ bonds as well as on the angle between the C–Hn/n′ bonds and some π-faced metal-centred SOMO component.
The partial signal assignment of compound 4 is given in Fig. 10 and Table S3.† In the shift range between 23 and −2 ppm one of the tert-butyl groups is clearly visible, but the assignment of the other signals is too speculative. Unlike 5, compound 4 shows a very broad signal at −58 ppm which obviously belongs to the NH. The signal separation of the diastereotopic protons Hn/n′ is similar to that of 5 (Hn at 131 ppm, Hn′ between 23 and −2 ppm). Another pair of diastereotopic protons is Hi/i′; however at 400 MHz and ambient temperature the signals are not resolved. Further detailed NMR studies to assign the resonances of 4, and to analyze the temperature dependence of the resonances of both complexes, are currently underway.
Magnetic susceptibility measurements
Magnetic susceptibility data were collected on both ferrous complexes (Fig. 12). Compounds 4 and 5 exhibit room temperature magnetic moments of 6.1μB and 5.6μB (0.1 T), respectively, verifying the high spin, S = 2 configuration at each iron centre. However, the magnetic moment of 4 increases sharply below 100 K to a maximum value of 7μB at 12 K. An increase in the magnetic moment of 5 also is observed upon cooling, albeit more gradual, to a value of 6.1μB at 25 K. The data suggest long-range ordering of the molecules and a ferromagnetic transition, upon cooling. The best fit of the Curie–Weiss equation to the 1/χM data confirms the deviation from Curie-behaviour of both compounds at low temperature, and yields values of C = 4.52 cm3 mol−1, θ = 0.57 K for 4, and C = 3.80 cm3 mol−1, θ = 3.33 K for 5 (Fig. S33 and S34†). To assess whether the magnetic behaviour could be attributed to inter- or intra-molecular interactions, we additionally carried out magnetic susceptibility measurements on solutions of 4 and 5 (∼6 mM, CH3CN).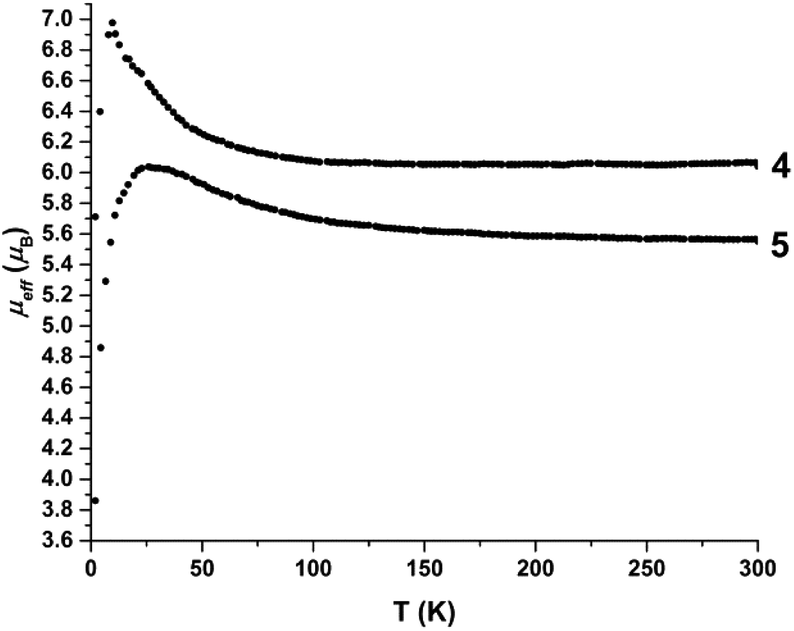 | ||
| Fig. 12 Temperature dependence of the magnetic moment μeff (μB), of solid samples of 4 and 5. | ||
Any long range, ferromagnetic interactions would not persist in dilute solutions of the complexes. The solution state magnetic susceptibility data (Fig. S35 and S36†) show temperature independent magnetic moments between ∼20–175 K, yielding room temperature magnetic moments of 4.1 and 3.6μB for 4 and 5, respectively. In contrast to the solid state behaviour, the magnetic moment decreases below 25 K.
The best fit of the data provided an estimate of the zero field splitting values, |D| ≈ 5 cm−1, for both complexes. The room temperature magnetic moments and g-values (4: g = 1.7; 5: g = 1.5) are lower than expected for a high spin ferrous ion, but are a consequence of the solvent correction applied for these measurements. We note that the paramagnetic NMR shifts, which depend on g2, were reproduced reasonably well with g = 2.7 (4) and 2.1 (5), providing a better estimate for these values.
The solution state data indicate that the solid state magnetic properties of the ferrous compounds arise from intermolecular interactions. However, the nearest Fe⋯Fe distances in the crystal structures of 4 and 5 are 7.52 and 8.37 Å. π-stacking interactions involving the xanthene backbone or pyridine rings also are not apparent in the structures and cannot account for the intermolecular exchange interactions. A Hirshfeld surface analysis reveals that in both iron complexes, the closest contacts between two molecules are mediated by the PF6− ions (Fig. 13). Spin delocalisation mediated by PF6− ions has been reported in the literature32 and offer a plausible explanation for the magnetic behaviour of our systems. We therefore carried out 31P NMR spectroscopy studies to assess interactions with the counterions.
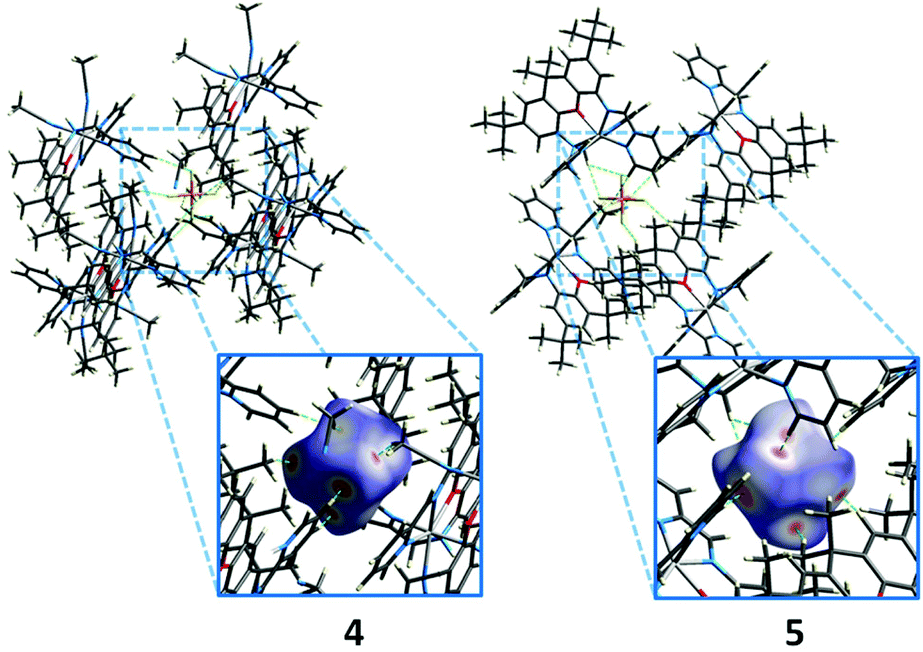 | ||
| Fig. 13 Packing of the PF6− ions in the crystal structure (extract) of 4 (left) and 5, and Hirshfeld surface analysis of the closest contacts (H⋯F, green dashes). Red areas on the Hirshfeld surface indicate close contacts. Only the interactions of one of the two PF6− ions of each structure are shown. Non-interacting molecules are omitted for clarity. | ||
The solution 31P NMR spectra of 4 and 5 each exhibit a septet at −144.6 and −144.3 ppm (Fig. 14), whereas the corresponding signal in the solid state spectrum of each compound is substantially broadened and shifted to low frequency due to the presence of the paramagnetic iron centre (−140.8 and −129.5 ppm, Fig. 14). These features are consistent with intermolecular spin density transfer from the cation to the anion,32,33 and describes the pathway for intermolecular interactions between the ferrous centres.
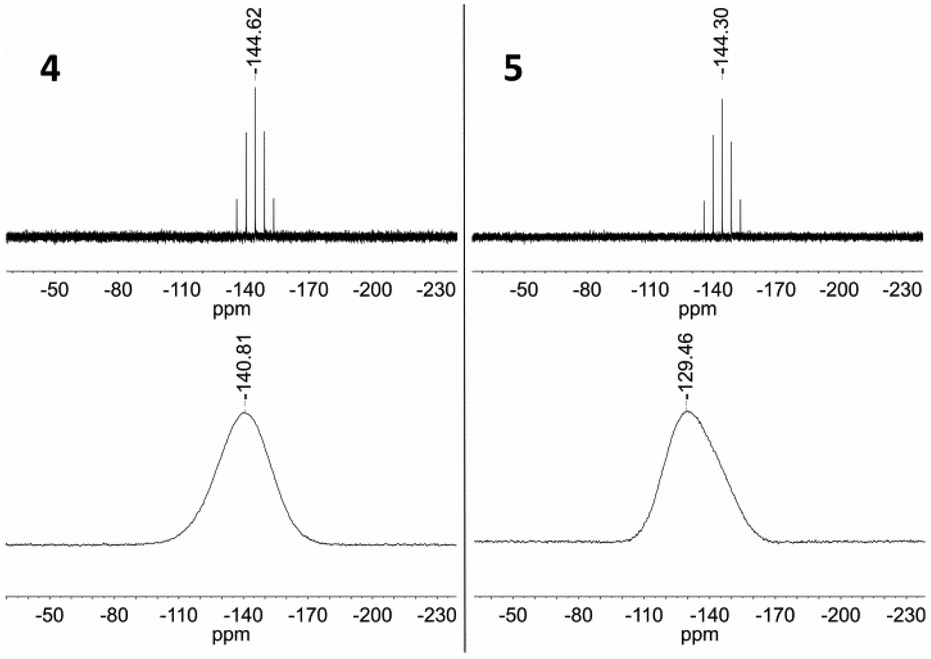 | ||
| Fig. 14 Top: 31P NMR (162 MHz, CD3CN) spectra of 4 and 5. Bottom: 31P MAS NMR (121 MHz) spectra of 4 and 5. | ||
Conclusions
The unsymmetric functionalization of the xanthene molecule affords strong chelating ligands, iXa and iXa-2, that favour mononuclear complex formation. This feature contrasts with related symmetric analogues of iXa, which support bimetallic complexes. However, the flexible methylpyridine groups included in our systems readily assume positions perpendicular to the diimine group upon metal binding. The resultant metal centres of 4–7 coordinate either six or seven donor atoms, but in all four complexes, the structure resembles a pentagonal bipyramid.As noted previously, few reports discuss the reactivity of seven-coordinate iron compounds. The atom transfer chemistry displayed by the iron complexes reported by Chang and Che stand out among the examples.7,8 The unique reactivity of these systems, leading to the formation of oxido and amido products, occurs via a labile axial ligand in the starting Fe(II) complexes. The sterically encumbered 5 may not be suited for reactivity: the iron centre is ligated by the N- and O-atoms supplied by iXa-2, and a further solvent molecule is not accommodated. The Fe ion in 4, however, coordinates two acetonitrile molecules in addition to the iXa scaffold, offering two potential reaction sites. The cis-arrangement of the acetonitrile groups could also be advantageous in this regard. Furthermore, the electrochemistry data points to the ability to generate formally low-valent forms of 4–7; in fact, the one-electron reduction observed in the CV of 5 is fully reversible. Ligand-centred reduction would place an electron in the π* orbitals of the redox active diimine group, coincident with formation of an FeII(L˙) species, whereas four SOMOs are available for metal-centred reduction to generate Fe(I) forms of 4 and 5. The former description is implied by the electrochemical data. We are unaware of other studies pertaining to seven coordinate complexes coordinated by redox-active ligands, or of the existence of mono-valent seven coordinate iron complexes with Werner-type ligands. The reactivity of the metal-iXa and -iXa-2 complexes, including the formation of reduced forms, will be a focus of further studies in our group. Information regarding the solution structural dynamics of these compounds will be provided by ensuing detailed NMR studies.
The low-temperature ferromagnetism of 4 and 5 is a unique attribute of these complexes, given the absence of any covalent interactions between the high spin ferrous molecules. Magnetic exchange coupling in other seven-coordinate metal complexes is fostered by covalently linked bridging molecules or hydrogen bonds.3,14,34–36 Our data, instead, implicate the PF6− counterions as intermediaries for the intermolecular interactions between the ferrous ions in 4 and 5. The iXa and iXa-2 scaffolds may thus be well-suited for magnetic materials. The results warrant further investigation of such aspects in these and other metal-containing complexes supported by our new ligands.
Experimental
Materials and methods
Chemicals were purchased from Sigma Aldrich and used as received unless otherwise noted. Metal compounds were synthesized in an inert atmosphere glove box (N2), using anhydrous solvents. Solvents were dried by passage over activated alumina columns from MBraun and stored over 3 Å or 4 Å molecular sieves. [Fe(CH3CN)6](PF6)2,37 4,5-dibromo-2,7-di-tert-butyl-9,9-dimethylxanthene,38 2,7-di-tert-butyl-9,9-dimethyl-4,5-xanthene dicarboxylic acid38 and 2,7-di-tert-butyl-9,9-dimethyl-4,5-xanthene diamine39 were prepared as described in the literature.Solution state NMR spectra were recorded on Bruker Avance-400 or Bruker Avance Ultrashield (400 MHz 1H, 100 MHz 13C, 162 MHz 31P), Bruker Avance Ultrashield (500 MHz 1H, 126 MHz 13C), Varian VNMRS-600 (600 MHz 1H, 151 MHz 13C) or Varian VNMRS-500 (500 MHz 1H, 126 MHz 13C) spectrometers. Solid state NMR spectra for 31P were recorded using a Bruker Avance 300 (121 MHz 31P) spectrometer equipped with a 2.5 mm BBMAS probe head at a rotation frequency of 20 kHz and referenced to (NH4)H2PO4 (δ (31P) = 1.1 ppm) as an external standard at 298 K. ZrO2 rotors of 2.5 mm were packed under argon and sealed with Kel-F caps. The FIDs were sampled after applying single pulses. Electronic spectra were recorded on an Agilent Cary 60 UV-visible spectrophotometer. ESI mass spectra were measured using a Waters TQD instrument or on a Xevo QToF for high-resolution spectra (atmospheric pressure solids analysis probe ionization experiments (ASAP)). Microanalyses were carried out at the London Metropolitan University or at the Technische Universität München. Electrochemical measurements were carried out using an EmStat3+ potentiostat using a three-electrode cell equipped with a glassy carbon working electrode and Pt wire as counter and reference electrodes. Potentials are reported with reference to an internal standard of ferrocenium/ferrocene (Fc+/0).
Magnetic susceptibility data collected on solid samples were recorded using a MPMS XL 5 (Quantum Design) superconducting quantum interference device (SQUID) magnetometer with liquid Helium cooling in a temperature range of 1.8–300 K and magnetic field of 0.1 T. The samples were placed in a calibrated gelatine capsule, fixed in the centre of a plastic straw.
Magnetic susceptibility data (2–290 K) for solution samples were recorded using a SQUID magnetometer (MPMS7, Quantum Design) in a 1 T external field. Data were corrected for underlying diamagnetism using tabulated Pascal's constants, a solvent correction was applied, and the data fit using julX (Dr E. Bill, MPI for Energy Conversion). The samples were dissolved in acetonitrile (∼0.5 mM) and sealed in glass tubes under Argon.
Crystallographic data were collected on an X-ray single crystal diffractometer equipped with a CCD detector (Bruker APEX II, κ-CCD), a rotating anode (Bruker AXS, FR591) with Mo Kα radiation (λ = 0.71073 Å) and a MONTEL mirror optic by using the APEX software package.40 The measurements were performed on a single crystal coated with perfluorinated ether. The crystal was fixed on top of a glass fiber and transferred to the diffractometer. The crystal was frozen under a stream of cold nitrogen. A matrix scan was used to determine the initial lattice parameters. Reflections were merged and corrected for Lorenz and polarisation effects, scan speed, and background using SAINT.41 Absorption corrections, including odd and even ordered spherical harmonics were performed using SADABS.41 Space group assignments were based upon systematic absences, E statistics, and successful refinement of the structures. Structures were solved by direct methods with the aid of successive difference Fourier maps, and were refined against all data using SHELXLE42 in conjunction with SHELXL-2014.43 Hydrogen atoms were assigned to ideal positions and refined using a riding model with an isotropic thermal parameter 1.2 times that of the attached carbon atom (1.5 times for methyl hydrogen atoms). If not mentioned otherwise, non-hydrogen atoms were refined with anisotropic displacement parameters. Full-matrix least-squares refinements were carried out by minimising ∑w(Fo2 − Fc2)2 with SHELXL-9744 weighting scheme. Neutral atom scattering factors for all atoms and anomalous dispersion corrections for the non-hydrogen atoms were taken from International Tables for Crystallography.45 Images of the crystal structures were generated by PLATON.46,47
Density Functional Theory (DFT) calculations were performed with the ORCA program package.48 Geometry optimizations of the complexes were performed at the B3LYP49–51 level of DFT. The all-electron Gaussian basis sets were those developed by Ahlrich's group.52,53 Triple-ζ quality basis sets (TZV(P)) were used, with one set of polarization functions on the metals and on the atoms directly coordinated to the metal centre.53 For the carbon and hydrogen atoms, slightly smaller polarized split-valence SV(P) basis sets were used that were of double-ζ quality in the valence region and contained a polarizing set of d functions on the non-hydrogen atoms. Auxiliary basis sets used to expand the electron density in the resolution-of-the-identity (RI) approach were chosen,54,55 where applicable, to match the orbital basis. SCF calculations were tightly converged (1 × 10−8Eh in energy, 1 × 10−7Eh in the density change, and 1 × 10−7Eh in maximum element of the DIIS error vector). Geometry optimizations for all complexes were carried out in redundant internal coordinates without imposing symmetry constraints. In all cases the geometries were considered converged after the energy change was less than 5 × 10−6Eh, the gradient norm and maximum gradient element were smaller than 1 × 10−4 and 3 × 10−4Eh Bohr−1, respectively, and the root-mean square and maximum displacements of all atoms were smaller than 2 × 10−3 and 4 × 10−3 Bohr, respectively. For 4, the acetonitrile ligands were constrained in the geometry optimizations. Single-point calculations on the non-optimised structures, using the coordinates obtained crystallographically, also were carried out using the B3LYP functional, for comparison. Orbital and spin density plots were created using VMD.56
Synthetic procedures
:hexane = 1:4, Rf = 0.45) to give a colourless solid (1.2 g, 63% yield). 1H NMR (400 MHz, CDCl3): δ 7.95 (1 H, s), 7.10 (1 H, d, J = 2.2), 6.91 (1 H, s), 6.86 (1 H, d, J 2.2), 6.72 (1 H, d, J 2.2), 3.87 (2 H, s), 1.62 (6 H, s), 1.56 (9 H, s), 1.35 (9 H, s), 1.31 (9 H, s); 13C NMR (100 MHz, CDCl3): δ 153.0, 146.2, 145.6, 137.5, 136.4, 133.6, 129.9, 129.4, 125.7, 116.5, 115.0, 112.7, 111.5, 80.7, 34.9, 34.8, 34.6, 32.1, 31.7, 31.6, 31.1, 28.6; IR (cm−1, neat) 3360, 2962, 1688, 1626, 1494, 1429, 1359, 1285, 1227, 1159, 1063, 856, 768; LRMS (ESI+) m/z: 453.24 [M + H]+, 475.14 [M + Na]+; HRMS (ESI+) m/z: 453.2410 [M + H]+.
:2 ethanol/water mixture (45 mL). The reaction mixture was heated to 100 °C for 4 h. The suspension was cooled to room temperature, filtered over celite and the residue washed with ethyl acetate. The solvent was removed in vacuo and the crude product purified by column chromatography (ethyl acetate:hexane = 1:2 → 1:1, 2bRf (ethyl acetate:hexane = 1:2) = 0.41, 2aRf (ethyl acetate:hexane = 1:1) = 0.39) to give the two products as light yellow solids (2a: 1.7 g, 86%; 2b: 0.4 g, 14%). 2a. 1H NMR (400 MHz, CDCl3): δ 8.69 (1 H, d, J 4.5), 7.96 (1 H, s), 7.67 (1 H, td, J 7.7, 1.8), 7.38 (1 H, d, J 7.8), 7.20 (2 H, t, J 5.9), 7.11 (1 H, d, J 2.2), 6.81 (1 H, d, J 2.1), 6.60 (1 H, d, J 2.1), 4.57 (2 H, s), 1.63 (6 H, s), 1.56 (9 H, s), 1.36 (9 H, s), 1.29 (9 H, s); 13C NMR (100 MHz, CDCl3): δ 158.3, 153.3, 149.3, 146.2, 145.5, 137.9, 136.9, 136.4, 135.9, 129.7, 129.0, 125.8, 122.2, 121.9, 116.5, 115.4, 110.9, 107.5, 80.5, 49.7, 34.90, 34.88, 34.80, 32.0, 31.72, 31.68, 28.6; IR (cm−1, neat) 3456, 2963, 1732, 1626, 1537, 1422, 1364, 1283, 1224, 1151, 1075, 999, 884, 770; LRMS (ESI+) m/z: 544.4 [M + H]+, 566.3 [M + Na]+; HRMS (ESI+) m/z: 544.3539 [M + H]+. 2b. 1H NMR (400 MHz, CDCl3): δ 8.56–8.58 (2 H, m), 8.34 (1 H, s), 7.99 (1 H, d, J 1.8), 7.52 (2 H, td, J 7.7, 1.8), 7.29 (2 H, d, J 7.8), 7.07–7.10 (3 H, m), 7.03 (1 H, d, J 2.2), 6.86 (1 H, d, J 2.2), 4.51 (4 H, s), 1.62 (6 H, s), 1.50 (9 H, s), 1.37 (9 H, s), 1.16 (9 H, s); 13C NMR (100 MHz, CDCl3): δ 158.8, 153.7, 149.3, 145.7, 145.3, 142.6, 138.4, 137.8, 136.4, 130.6, 130.0, 126.6, 123.1, 122.0, 118.4, 116.5, 115.7, 115.4, 80.2, 59.1, 35.2, 35.0, 34.7, 31.8, 31.49, 31.46, 28.6; IR (cm−1, neat) 3455, 2965, 1618, 1534, 1435, 1364, 1231, 1159, 1066, 981, 866, 732; LRMS (ESI+) m/z: 635.4 [M + H]+, 657.4 [M + Na]+; HRMS (ESI+) m/z: 635.3978 [M + H]+.
:hexane = 1:1) 0.32; IR (cm−1, neat) 3349, 2952, 1632, 1520, 1494, 1445, 1422, 1360, 1286, 1224, 1167, 1093, 1046, 858, 840, 738; LRMS (ESI+) m/z: 444.3 [M + H]+, 466.1 [M + Na]+; HRMS (ESI+) 444.3014 [M + H]+.
960, 10700 and 650).
880, 8250 and 600), LRMS (ESI+m/z) [M]2+ 33996.
960 and 8450). 1H NMR (500 MHz, C2D2Cl4): δ 9.39 (1 H, s, He), 8.66 (1 H, d, J = 4.9, Ha), 8.27 (1 H, d, J = 7.7, Hd), 8.20 (1 H, td, J = 7.7, 1.4, Hc), 7.83 (1 H, d, J = 7.8, 1.4, Hp), 7.81 (1 H, d, J = 1.8, Hf), 7.79 (1 H, d, J = 5.3, Hr), 7.66 (1 H, td, J = 6.0, 0.7, Hb), 7.50 (1 H, d, J = 1.7, Hh), 7.37–7.33 (3 H, m, Hl/o/j), 7.30 (1 H, t, J = 6.5, Hq), 5.65 (1 H, d, J = 7.6, Hm), 5.08 (1 H, dd, J = 7.6, J = 17.0, Hn), 4.38 (1 H, d, J = 17.0, Hn′), 1.68 (3 H, s, Hi), 1.39 (3 H, s, Hi′), 1.34 (9 H, s, Hg), 1.28 (9 H, s, Hk); 13C NMR (126 MHz, C2D2Cl4): δ 154.8, 154.5, 150.4, 149.7, 149.3, 147.3, 146.8, 142.7, 142.0, 141.7, 141.4, 132.9, 132.9, 131.7, 131.4, 130.2, 129.8, 128.7, 126.1, 126.0, 124.5, 122.3, 122.3, 122.0, 121.4, 118.9, 113.6, 55.2, 36.2, 35.5, 35.3, 32.3, 31.7, 31.6, 28.3.
860 and 8470). 1H NMR (500 MHz, CD3CN): δ 9.83 (1 H, s, He), 8.69 (1 H, d, J = 4.9, Ha), 8.42 (2 H, m, Hc/d), 8.06 (1 H, d, J = 2.0, Hf), 7.96–8.00 (3 H, m, Hp/l), 7.86 (2 H, d, J = 5.2, Hr), 7.83 (1 H, q, J = 4.7, Hb), 7.66 (1 H, d, J = 1.9, Hh), 7.53 (2 H, d, J = 7.9, Ho), 7.48 (1 H, d, J = 2.1, Hj), 7.33 (2 H, t, J = 6.5, Hq), 5.11 (2 H, d, J = 16.7, Hn), 5.06 (2 H, d, J = 16.7, Hn′), 1.53 (6 H, s, Hi), 1.42 (9 H, s, Hg), 1.39 (9 H, s, Hk); 13C NMR (126 MHz, CD3CN): δ 159.7, 156.2, 151.3, 150.6, 149.2, 148.5, 148.4, 143.2, 142.9, 141.4, 140.7, 137.0, 131.9, 131.3, 131.16, 131.13, 129.2, 127.8, 126.4, 125.5, 124.6, 123.3, 122.6, 120.8, 115.0, 63.9, 55.3, 35.96, 35.94, 35.90, 32.1, 31.40, 31.39.
Acknowledgements
We thank Prof. Thomas Fässler for the use of his instrument for solid state magnetic susceptibility measurements, and Dr Viktor Hlukhyy for technical assistance. We are grateful to Dr Eckhard Bill (Max Planck Institute for Energy Conversion) for the solution state magnetic susceptibility measurements and for helpful discussions. Our thanks go as well to Dr Gabriele Raudaschl-Sieber for assistance with solid state NMR measurements. We thank Dmitry Yufit (Durham University) for crystallography contributions. Funding by an EPSRC grant for MA is gratefully acknowledged.Notes and references
- Current status in CCDC on the Dec 2nd 2015.
- D. Casanova, P. Alemany, J. M. Bofill and S. Alvarez, Chem. – Eur. J., 2003, 9, 1281–1295 CrossRef CAS PubMed.
- B. Drahoš, R. Herchel and Z. Trávníček, Inorg. Chem., 2015, 54, 3352–3369 CrossRef PubMed.
- D. Zhang, D. H. Busch, P. L. Lennon, R. H. Weiss, W. L. Neumann and D. P. Riley, Inorg. Chem., 1998, 37, 956–963 CrossRef CAS.
- G.-F. Liu, M. Filipovic, F. W. Heinemann and I. Ivanovic-Burmazovic, Inorg. Chem., 2007, 46, 8825–8835 CrossRef CAS PubMed.
- C. T. Gutman and T. C. Brunhold, Inorg. Chem., 2012, 51, 12729–12737 CrossRef CAS PubMed.
- H. S. Soo, M. T. Sougrati, F. Grandjean, G. J. Long and C. J. Chang, Inorg. Chim. Acta, 2011, 369, 82–91 CrossRef CAS.
- G. S. M. Tong and C.-M. Che, Eur. J. Inorg. Chem., 2010, 2010, 5113–5123 CrossRef.
- I. Morgenstern-Badarau, F. Lambert, J. P. Renault, M. Cesario, J.-D. Maréchal and F. Maseras, Inorg. Chim. Acta, 2000, 297, 338–350 CrossRef CAS.
- M. S. Nelson, P. D. A. Mcllroy and C. S. Stevenson, J. Chem. Soc., Dalton Trans., 1986, 991–995 RSC.
- E. König, G. Ritter, J. Dengler and S. M. Nelson, Inorg. Chem., 1987, 26, 3582–3588 CrossRef.
- S. Bonhommeau, T. Gullion, L. M. L. Daku, P. Demont, J. S. Costa, J.-F. Létard, G. Molnàr and A. Bousseksou, Angew. Chem., Int. Ed., 2006, 45, 1625–1629 CrossRef CAS PubMed.
- G. A. Craig, L. A. Barrios, J. S. Costa, O. Roubeau, E. Ruiz, S. J. Teat, C. C. Wilson, L. Thomas and G. Aromí, Dalton Trans., 2010, 39, 4874–4881 RSC.
- A. K. Bar, C. Pichon, N. Gogoi, C. Duhayon, S. Ramasesha and J.-P. Sutter, Chem. Commun., 2015, 51, 3616–3619 RSC.
- S. Hayami, Z.-Z. Gu, Y. Einaga, Y. Kobayasi, Y. Ishikawa, Y. Yamada, A. Fujishima and O. Sato, Inorg. Chem., 2001, 40, 3240–3242 CrossRef CAS PubMed.
- J. S. Costa, C. Balde, C. Carbonera, D. Denux, A. Wattiaux, C. Desplanches, J.-P. Ader, P. Gütlich and J.-F. Létard, Inorg. Chem., 2007, 46, 4114–4119 CrossRef PubMed.
- S. Lindsay, S. K. Lo, O. R. Maguire, E. Bill, M. R. Probert, S. Sproules and C. R. Hess, Inorg. Chem., 2013, 52, 898–909 CrossRef CAS PubMed.
- A. Panunzi, F. Giordano, I. Orabona and F. Ruffo, Inorg. Chim. Acta, 2005, 358, 1217–1224 CrossRef CAS.
- A. Bheemaraju, J. W. Beattie, Y. Danylyuk, J. Rochford and S. Groysman, Eur. J. Inorg. Chem., 2014, 2014, 5865–5873 CrossRef CAS.
- S. Takano, D. Takeuchi, K. Osakada, N. Akamatsu and A. Shishido, Angew. Chem., Int. Ed., 2014, 53, 9246–9250 CrossRef CAS PubMed.
- M. Grau, J. England, R. T. Martin de Rosales, H. S. Rezepa, A. J. P. White and G. J. P. Britovsek, Inorg. Chem., 2013, 52, 11867–11874 CrossRef CAS PubMed.
- S. Gosiewska, J. J. L. M. Cornelissen, M. Lutz, A. L. Spek, G. van Koten and R. J. M. Klein Gebbink, Inorg. Chem., 2006, 45, 4214–4227 CrossRef CAS PubMed.
- Z. P. Haque, D. C. Liles, M. McPartlin and P. A. Tasker, Inorg. Chim. Acta, 1977, 23, L21–L22 CrossRef CAS.
- A. Bencini, E. Berni, A. Bianchi, P. Fornasari, C. Giorgi, J. C. Lima, C. Lodeiro, M. J. Melo, J. Seixas de Melo, A. J. Parola, F. Pina, J. Pina and B. Valtancoli, Dalton Trans., 2004, 2180–2187 RSC.
- R. Hoffmann, B. F. Beier, E. L. Muetterties and A. R. Rossi, Inorg. Chem., 1977, 16, 511–522 CrossRef CAS.
- F. H. Köhler, in Magnetism: Molecules to Materials, Wiley-VCH, Weinheim, Germany, 2001, vol. 1, ch. 12 Search PubMed.
- J. Vaara, Phys. Chem. Chem. Phys., 2007, 9, 5399–5418 RSC.
- F. Aquino, B. Pritchard and J. Autschbach, J. Chem. Theory Comput., 2012, 8, 598–609 CrossRef CAS PubMed.
- F. Rastrelli and A. Bagno, Chem. – Eur. J., 2009, 15, 7990–8004 CrossRef CAS PubMed.
- M. Kaupp and F. H. Köhler, Coord. Chem. Rev., 2009, 253, 2376–2386 CrossRef CAS.
- Referencing relative to the various proton signals of the zinc analogues would tremendously complicate Fig. 10 and 11, where the scales are therefore plotted relative to the solvent signal.
- H. Heise, F. H. Köhler, M. Herker and W. Hiller, J. Am. Chem. Soc., 2002, 124, 10823–10832 CrossRef CAS PubMed.
- J. Blümel, N. Hebendanz, P. Hudeczek, F. H. Köhler and W. Strauss, J. Am. Chem. Soc., 1992, 114, 4223–4230 CrossRef.
- T. S. Venkatahrishnan, S. Sahoo, N. Brefuel, C. Duhayon, C. Paulsen, A.-L. Barra, S. Ramasesha and J.-P. Sutter, J. Am. Chem. Soc., 2010, 132, 6047–6056 CrossRef PubMed.
- L. D. Slep, R. Calvo, O. R. Nascimento, R. Baggio, M. T. Garland, O. Peña and M. Perec, Inorg. Chim. Acta, 2007, 360, 2911–2916 CrossRef CAS.
- J. Larionova, O. Kahn, S. Golhen, L. Ouahab and R. Clerac, Inorg. Chem., 1999, 38, 3621–3627 CrossRef CAS PubMed.
- C. J. Barbour, J. H. Cameron and J. M. Winfield, J. Chem. Soc., Dalton Trans., 1980, 2001–2005 RSC.
- J. S. Nowick, P. Ballester, F. Ebmeyer and J. Rebek, J. Am. Chem. Soc., 1990, 112, 8902–8906 CrossRef CAS.
- P. Bühlmann, S. Nishizawa, K. P. Xiao and Y. Umezawa, Tetrahedron, 1997, 53, 1647–1654 CrossRef.
- APEX suite of crystallographic software, APEX 2, version 2008.4, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- SAINT, version 7.56a, SADABS, version 2008.1, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, SHELXLE, J. Appl. Crystallogr., 2011, 44, 1281–1284 CrossRef PubMed.
- G. M. Sheldrick, SHELXL-2014, University of Göttingen, Göttingen, Germany, 2014 Search PubMed.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Göttingen, Germany, 1998 Search PubMed.
- A. J. C. Wilson, International Tables for Crystallography, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1992 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1986, 84, 4524–4529 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- K. Eichkorn, O. Treutler, H. Ohm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS.
- K. Eichkorn, O. Treutler, H. Ohm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: CIF files, additional spectroscopic, magnetic susceptibility and DFT computational data, and further details concerning NMR spectra assignments. CCDC 1442708–1442711. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00298b |
| ‡ Current address: Institute of Chemistry, University of the Punjab, Quaid-i-Azam Campus, Lahore-54590, Pakistan. |
| This journal is © the Partner Organisations 2016 |