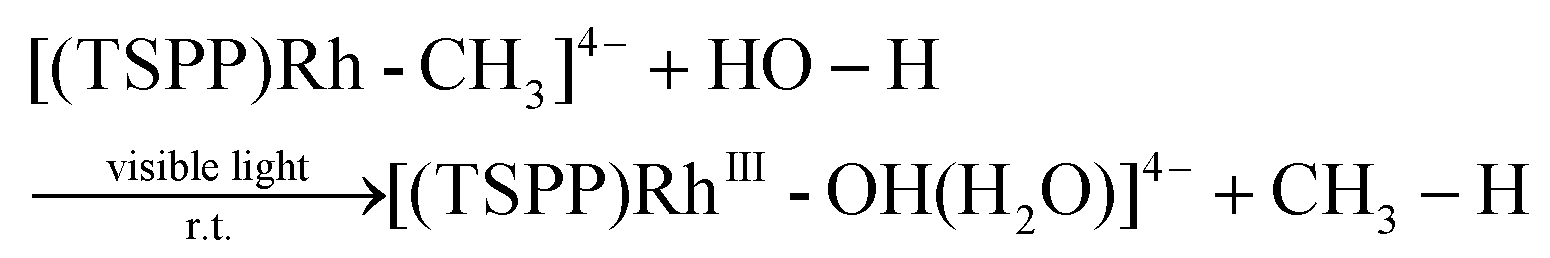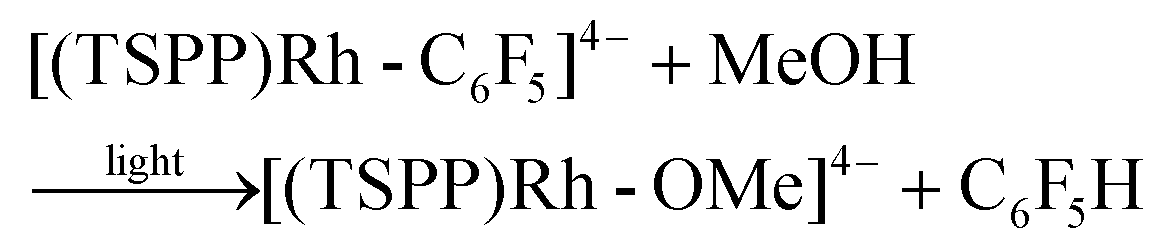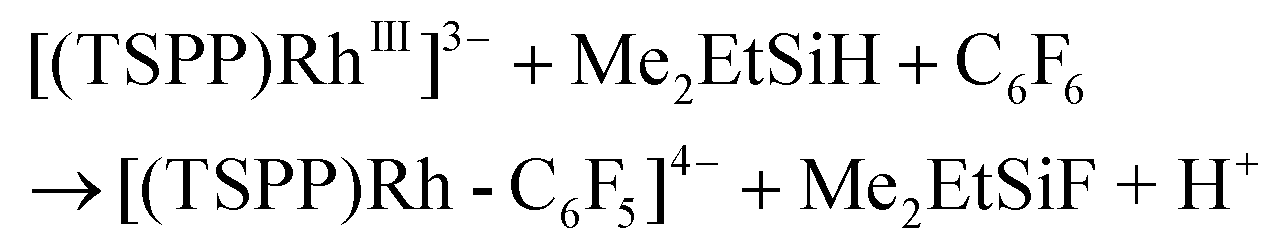DOI:
10.1039/C5QI00269A
(Research Article)
Inorg. Chem. Front., 2016,
3, 861-865
Light induced catalytic hydrodefluorination of perfluoroarenes by porphyrin rhodium†
Received
1st December 2015
, Accepted 31st March 2016
First published on 5th April 2016
Abstract
Photocatalytic hydrodefluorination of perfluoroarenes by rhodium porphyrin complexes with high tolerance to various functional groups has been developed. Mechanistic studies reveal that the rhodium aryl complex, (por)Rh-C6F4R, is the key intermediate.
 Xuefeng Fu | Xuefeng Fu received a Ph.D. in inorganic chemistry in 2006 from the University of Pennsylvania under the supervision of Prof. Bradford B. Wayland. After around 2 years post-doctoral research with Prof. Bradford B. Wayland, she joined the faculty of Peking University in Nov. 2007. Currently, she is an Associate Professor in the program titled “Hundred Talents Program of PKU” in the College of Chemistry and Molecular Engineering at Peking University in China. Her research interests can be broadly defined as being in the area of inorganic/organometallic chemistry with focus on fundamental inorganic and organometallic transformations and mechanistic studies. |
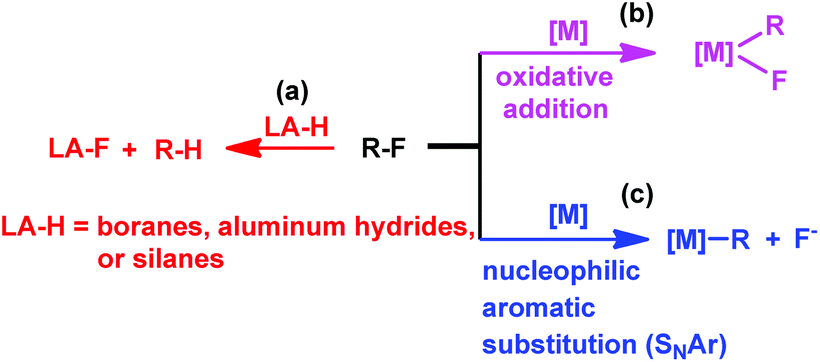
Fluorine-containing organic compounds are essential building blocks that have been extensively employed in materials chemistry, medicinal chemistry as well as agrochemistry.1 Simple perfluorinated bulk chemicals are easily accessible on an industrial scale, whereas the selective cleavage of C–F bonds of perfluorinated compounds is regarded as a promising approach to access partially fluorinated compounds.1d–h However, this defluorination process has been considered as one of the greatest challenges for synthetic chemistry due to the relative inertness of the C–F bond.2
Transition metal complexes have been primarily used in catalyzing C–F bond activation,3 typical metals include Ti,4 Zr,5 Fe,6 Ru,7 Rh,8 Ir,9 Ni,10 Pd,11 Pt,12 Cu,13 Au,14 and Zn.15 In addition, fluorophilic reagents, such as boranes,16 aluminum hydrides17 and silanes,18 also gave hydrodefluorination products where the fluorine atom is replaced by hydrogen (Scheme 1(a)).
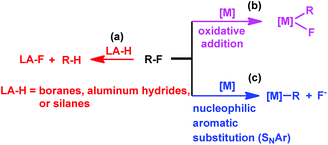 |
| | Scheme 1 Approaches to C–F bond activation. | |
The transition metal catalyzed C–F bond activation through an oxidative addition reaction often led to the formation of strong metal–fluorine bonds which impeded catalyst regeneration (Scheme 1(b)). However, rational design of alternate routes to avoid the formation of the catalyst–fluorine bond would give catalytic C–F bond activation with improved efficiency. Photocatalysis has recently been attracting increasing attention as a unique pathway to overcome high thermal barriers at relatively low energy and environmental costs.19 Weaver's group previously reported photocatalytic hydrodefluorination of perfluoroarenes mediated by the Ir(ppy)3 complex (tris[2-phenypyridinato-C2,N]iridium(III)).9c The 18 e−, coordination saturated Ir(ppy)3 complex would circumvent the formation of the catalyst–fluorine bond for catalytic turnover.
We recently reported the photocatalytic hydration of Si–C bonds20 and alkynes21 using the tetra(p-sulfonatophenyl)porphyrin rhodium ((TSPP)Rh) complex. Mechanistic studies indicated that one of the key steps was the visible light triggered hydration of Rh–C bonds to form the corresponding Rh–OH complex, which afforded a novel strategy to accomplish unusual photo-processes (eqn (1)).
| | 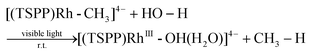 | (1) |
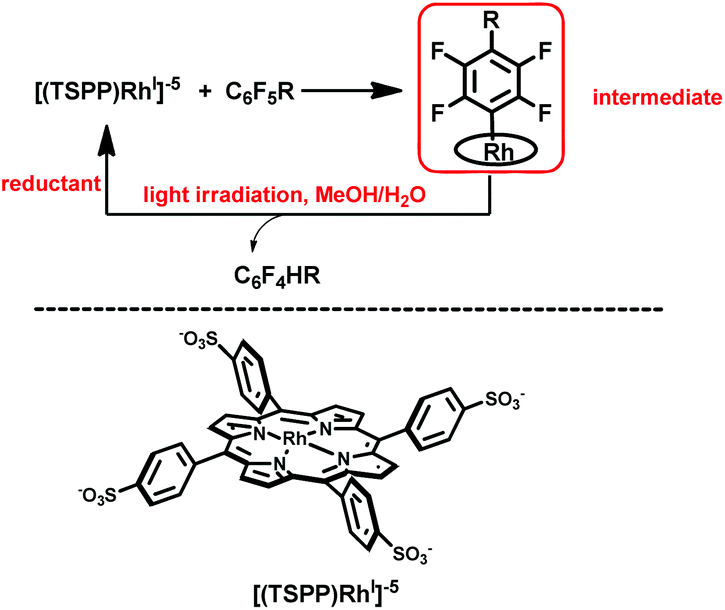
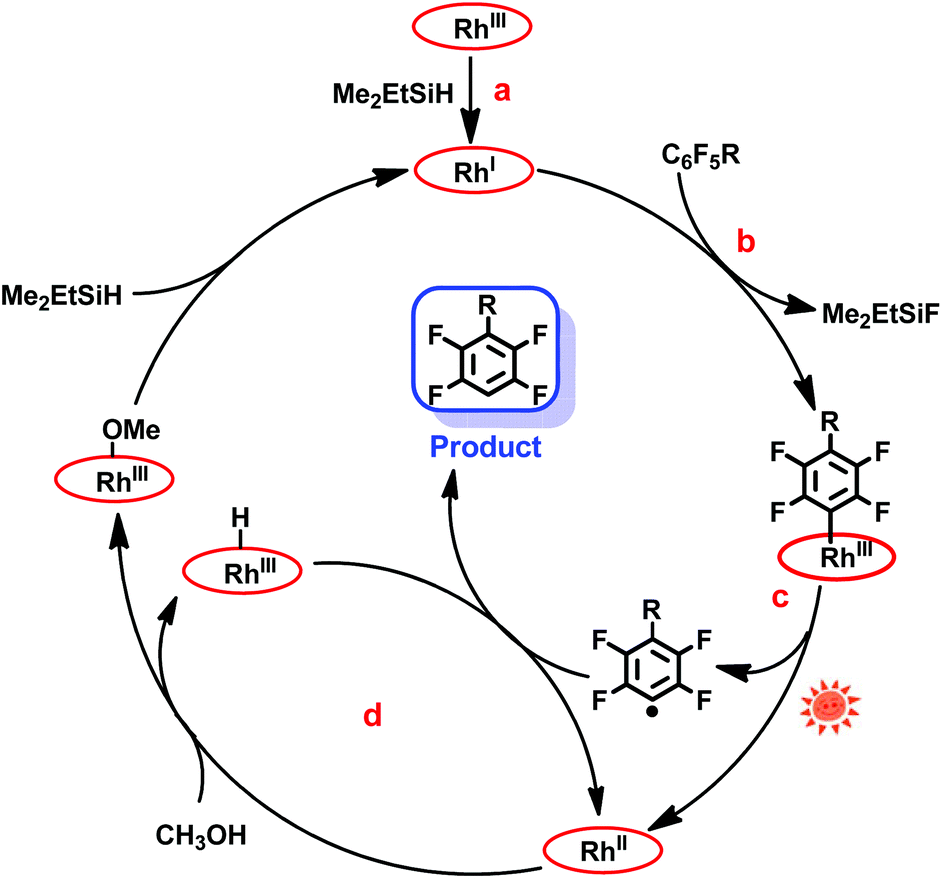
Herein, we report on light promoted catalytic hydrodefluorination of perfluoroarenes catalyzed by a highly reactive nucleophile, [(TSPP)RhI]5−, through a nucleophilic aromatic substitution pathway (SNAr) with a turnover number (TON) of 880 for octafluorotoluene. The [(TSPP)RhI]5− attacks the C–F bond in the perfluoroarenes to form the [(TSPP)Rh-C6F4R]4− complex (Scheme 1(c)). The polyfluorine-substituted organo–metal bond is quite inert so no facile thermal pathways are available for further transformation to complete the catalytic cycle.8h,22 However, in this study, a photo-hydration strategy is applied to [(TSPP)Rh-C6F4R]4− producing the hydrodefluorinated product and [(TSPP)RhIII-OH]4−. The facile reduction of the resulting RhIII-OH species regenerates [(TSPP)RhI]5− completing the catalytic cycle (Scheme 2).
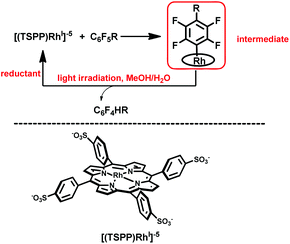 |
| | Scheme 2 Hydrodefluorination of perfluoroarenes catalyzed by [(TSPP)RhI]5−. | |
Stoichiometric hydrodefluorination reaction
Stirring the methanol solution of [(TSPP)RhI]5− (2.5 mM), sodium borate (0.012 M) and hexafluorobenzene (0.1 mmol) at 25 °C for 2 hours produced [(TSPP)Rh-C6F5]4− quantitatively. ESI-MS of [(TSPP)Rh-C6F5]4− gave a peak at m/z = 621.95265, corresponding to the anion [(TSPP)Rh-C6F5]Na22− (Fig. 1S†). In addition, the structure of [(TSPP)Rh-C6F5]4− was also verified by 1H NMR and 19F NMR (Fig. 2S and 3S†).
Heating the methanol-d4 solution of [(TSPP)Rh-C6F5]4− at 110 °C for 24 hours only produced a trace amount of pentafluorobenzene detected by GC-MS through the thermal dissociation of the Rh–C bond. However, with irradiation of the methanol solution of [(TSPP)Rh-C6F5]4− for 2 hours using a mercury lamp, over 95% conversion was achieved together with the formation of C6F5H detected by GC-MS, and [(TSPP)RhIII]3−, as evidenced by 1H NMR (eqn (2)–(5)). As expected, irradiation shunted the thermally disfavored reaction to a viable and efficient photo-process for the cleavage of Rh–C bonds. Furthermore, the carbon centered radicals resulting from the photo-cleavage of Rh–C bonds were trapped by TEMPO (TEMPO = 2,2,6,6-tetramethylpiperidine-1-oxyl) indicating that the reaction occurred through a radical pathway (Fig. 4S†).
| |  | (2) |
| | | 2[(TSPP)Rh˙II]4− + MeOH ⇌ [(TSPP)Rh-OMe]4− + [(TSPP)Rh-H]4− | (3) |
| | | [(TSPP)Rh-H]4− + ˙C6F5 → [(TSPP)Rh˙II]4− + C6F5H | (4) |
| | 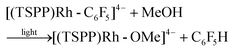 | (5) |
Catalytic hydrodefluorination reaction
The [(TSPP)Rh-OMe]4− generated from the photolysis of [(TSPP)Rh-C6F5]4− in methanol can be readily reduced to [(TSPP)RhI]5− by various reductants.23 Dihydrogen, commonly recognized as one of the cleanest and most environmentally friendly reductants, reduced [(TSPP)RhIII]3− at 25 °C (PH2 ≈ 0.5–0.8 atm).23b To our delight, 10 TONs were obtained and detected by GC when irradiating the 0.4 mL methanol-d4 solution containing hexafluorobenzene (0.1 mmol) and [(TSPP)RhIII]3− (2.5 mM) at 60 °C for 24 hours under one atmosphere of dihydrogen.| | | [(TSPP)RhIII]3− + H2 ⇌ [(TSPP)Rh-H]4− + H+ | (6) |
| | | [(TSPP)Rh-H]4− ⇌ [(TSPP)RhI]5− + H+ | (7) |
| | | [(TSPP)RhIII]3− + H2 + C6F6 → [(TSPP)Rh-C6F5]4− + HF + H+ | (8) |
The relatively low TON was ascribed to the shift of the equilibrium to the reactants in the reducing step (eqn (6)) with accumulation of HF acid. Considering both the reducing ability of the reductant and the affinity to F−, silane would be a good candidate for driving the catalytic hydrodefluorination reaction. By a careful screening of different silanes (Table 1S†), Me2EtSiH was found to show the best performance with 69 TONs under the same conditions (eqn (9)–(11)). The intermediate [(TSPP)Rh-C6F5]4− (eqn (11)) was characterized by ESI-MS, and 1H NMR in situ. The regeneration of [(TSPP)RhI]5− (eqn (10)) was confirmed by the formation of [(TSPP)Rh-CH3]4− upon trapping with CH3I (eqn (12)), which is the signature reaction of nucleophilic [(TSPP)RhI]5−, with characteristic 1H NMR methyl resonance (δ(Rh–CH3) = −6.59 ppm, 2J(Rh–CH) = 2.4 Hz in CD3OD)23b as well as ESI-MS results (m/z = 545.96830 corresponding to [(TSPP)Rh-CH3] Na22−) (Fig. 5S†). Furthermore, the resulting Me2EtSiF was detected by GC-MS.
| | | [(TSPP)RhIII]3− + Me2EtSiH → [(TSPP)Rh-H]4− + Me2EtSi+ | (9) |
| | | [(TSPP)Rh-H]4− ⇌ [(TSPP)RhI]5− + H+ | (10) |
| | 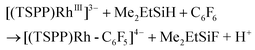 | (11) |
| | | [(TSPP)RhI]5− + CH3I → [(TSPP)Rh-CH3]4− + I− | (12) |
In order to increase the solubility of Me2EtSiH in methanol solution to further improve the catalytic efficiency, mixed solvents were examined, and the results of the experiments for solvent screening are illustrated in Table 1. Using a volume ratio of methanol/THF 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 30 TONs were obtained, and for methanol/1,4-dioxane 1:1, the TON was 45 (Table 1, entries 1 and 2). Generally, dimethylsulfoxide (DMSO) and N,N-dimethylformamide (DMF) are considered as two commonly used aprotic polar solvents for SN2 reactions. However, the TONs were not improved significantly for methanol/DMSO (1:1) as the solvent (Table 1, entry 3) while methanol/DMF (1:1) gave TONs of 350, and methanol/ethylene glycol (1:1) gave the highest TON (Table 1, entry 5). Apparently, the protic solvents facilitated nucleophilic C–F bond activation (Table 1, entry 5 vs. entries 1–4) due to their ability to stabilize the forming fluoride anion by hydrogen bonding, which agrees with Paquin's observation.24 Employing pure glycol as the solvent, however, did not lead to further improvement (Table 1, entry 6).
:1, 30 TONs were obtained, and for methanol/1,4-dioxane 1:1, the TON was 45 (Table 1, entries 1 and 2). Generally, dimethylsulfoxide (DMSO) and N,N-dimethylformamide (DMF) are considered as two commonly used aprotic polar solvents for SN2 reactions. However, the TONs were not improved significantly for methanol/DMSO (1:1) as the solvent (Table 1, entry 3) while methanol/DMF (1:1) gave TONs of 350, and methanol/ethylene glycol (1:1) gave the highest TON (Table 1, entry 5). Apparently, the protic solvents facilitated nucleophilic C–F bond activation (Table 1, entry 5 vs. entries 1–4) due to their ability to stabilize the forming fluoride anion by hydrogen bonding, which agrees with Paquin's observation.24 Employing pure glycol as the solvent, however, did not lead to further improvement (Table 1, entry 6).
Table 1 Solventa screening for the light induced catalytic hydrodefluorination of C6F6 catalyzed by [(TSPP)RhI]5−
| Entry |
Solvent |
Selectivityb |
TON |
|
Reaction conditions: 60 °C under light irradiation (500 W Hg lamp, 15 cm distance) for 36 hours.
Determined by 19F NMR resonances, selectivity = yield/conversion.
|
| 1 |
MeOH/THF = 1:1 |
25% |
30 |
| 2 |
MeOH/1,4-dioxane = 1:1 |
45.9% |
45 |
| 3 |
MeOH/DMSO = 1:1 |
32.4% |
27.5 |
| 4 |
MeOH/DMF = 1:1 |
97.2% |
350 |
| 5 |
MeOH/glycol = 1:1 |
99.5% |
418 |
| 6 |
1 ml glycol |
87% |
252.5 |
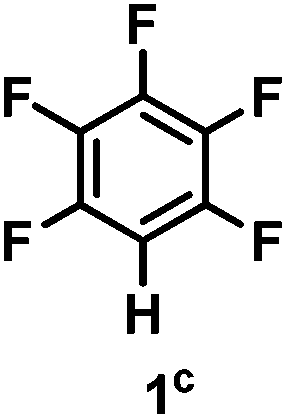
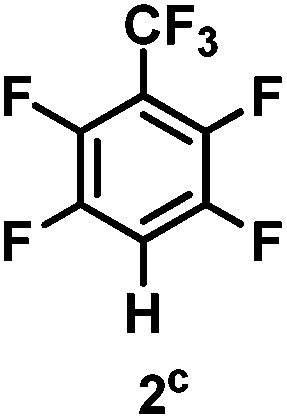
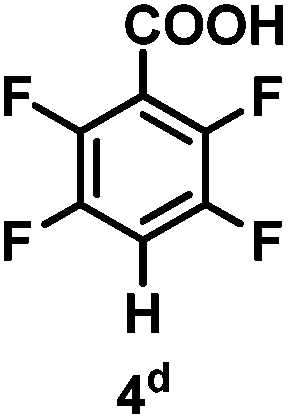
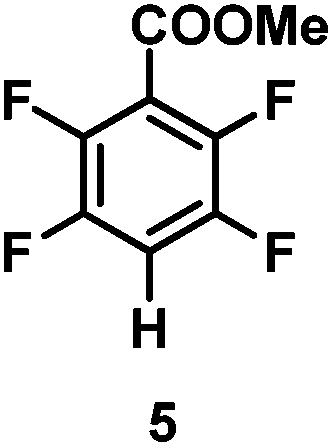
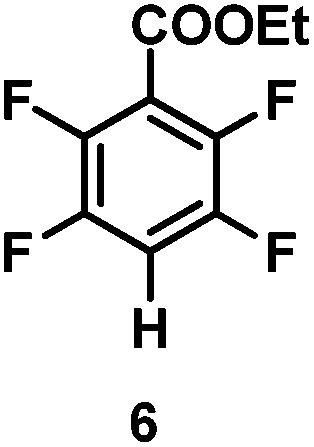
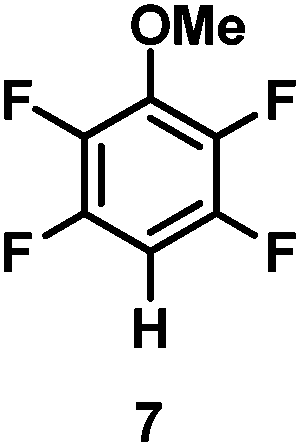
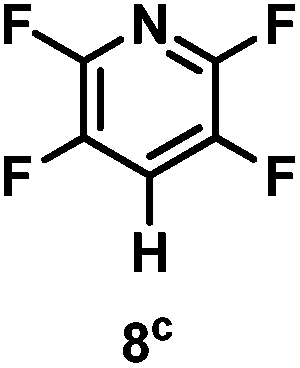
The catalytic hydrodefluorination of various perfluoroarenes under 1:1 of MeOH/ethylene glycol is listed in Table 2. Hexafluorobenzene underwent hydrodefluorination to give pentafluorobenzene (1) with a TON of 418, and the selectivity was 99.5%. Moreover, when one fluorine atom of the hexafluorobenzene was substituted by an electron withdrawing group such as trifluoromethyl (2), aldehyde (3), carboxyl (4) and esters (5 and 6), the hydrodefluorination products were obtained with good TON and high selectivity. However, when an electron-donating group (i.e. MeO, 7) was present, no C–F bond activation occurred. This method could also be applied to pentafluoropyridine, which was smoothly converted to the tetrafluoro-derivative (8) with a TON of 420. The remarkable selectivity of this approach (>90% in all cases) was manifested in the lack of over-reduction, suggesting the high tolerance of different functional groups. Additionally, control experiments under the standard conditions revealed that both the catalyst and light irradiation were necessary.
Table 2 Scope of the perfluoroarene substratesa
|
Reaction conditions: 60 °C under light irradiation (500 W Hg lamp, 15 cm distance) for 36 hours.
Determined by 19F NMR, selectivity = yield/conversion.
GC results.
1 mmol NaOH was added.
|
|
|
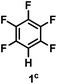
|
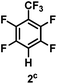
|
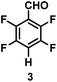
|
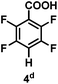
|
| Selectivityb: |
99.5% |
98% |
96% |
93% |
| TON: |
418 |
880 |
565 |
355 |
|
|
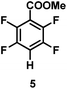
|
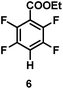
|
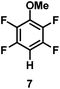
|
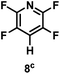
|
| Selectivityb: |
92% |
91% |
— |
95% |
| TON: |
838 |
757 |
0 |
420 |
Proposed mechanism
The mechanism of the hydrodefluorination of perfluoroarenes catalyzed by [(TSPP)RhI]5− was envisioned to proceed through a four-step cycle depicted in Scheme 3: (a) the reaction of [(TSPP)RhIII]3− with Me2EtSiH to produce [(TSPP)RhI]5− under basic conditions; (b) the nucleophilic aromatic substitution of [(TSPP)RhI]5− at the para-carbon of the perfluoroarene giving the key intermediate; (c) photolysis of the [(TSPP)Rh-C6F4R]4− complex yielding the C6F4R radical and [(TSPP)RhII]4−; (d) subsequently, [(TSPP)RhII]4− reacted with methanol rapidly producing [(TSPP)Rh-H]4− and [(TSPP)Rh-OMe]4−,21,25 followed by the hydrogen atom abstraction from [(TSPP)Rh-H]4− by the C6F4R radical yielding the final product HC6F4R. The high efficiency of C–F bond activation was attributed to the formation of Si–F and Rh–C bonds which provided sufficient thermodynamic driving force for the reaction.
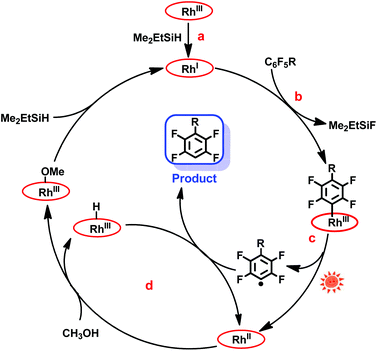 |
| | Scheme 3 Proposed mechanism of photo-induced hydrodefluorination of perfluoroarenes catalyzed by rhodium porphyrins. | |
Considering that the reduction potential of C6F6 was as negative as −2.22 eV in 75% dioxane/water,26 and the oxidation potential of (TPP)RhI to (TPP)RhII in DMSO was −1.1691 eV (TPP = tetraphenylporphyrin),27 a single electron transfer mechanism is probably too endothermic to be operative, although the redox potentials cannot be compared quantitatively due to the differences in ligands (TSPP vs. TPP) and solvents (dioxane/water vs. DMSO) used. The nucleophilic aromatic substitution pathway for the C–F bond activation was preferred in this system, however, the single electron transfer pathway could not be firmly ruled out.
In addition, the mechanism is quite different from Weaver's work.9c In our system, the Rh–C bond cleavage of the intermediate ([(TSPP)Rh-C6F4R]4−) only occurs with the assistance of light irradiation to complete the catalytic cycle, while in Weaver's system, light was used to promote the catalyst (Ir(ppy)3) to the excited state, releasing an electron to the LUMO of perfluoroarenes to trigger the photocatalytic hydrodefluorination.
Conclusions
In summary, we have developed a new strategy for catalytic hydrodefluorination of perfluoroarenes utilizing [(TSPP)RhI]5− through a nucleophilic aromatic substitution pathway. The key intermediate [(TSPP)Rh-C6F4R]4− was observed, which underwent light-induced Rh–C bond cleavage to produce hydrodefluorination products. By integrating the light harvesting ability of the porphyrin ligand with unique organometallic reactions of the rhodium metal center, we illustrated a novel photocatalytic approach which provides an alternate route to the design of efficient photocatalysts.
Acknowledgements
This work was supported by the National Science Foundation of China (21322108 and 21321001).
Notes and references
-
(a)
L. A. Paquette, Handbook of Reagents for Organic Synthesis: Fluorine-Containing Reagents, Wiley, New York, 2007 Search PubMed;
(b) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC;
(c) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed;
(d)
L. A. Paquette, Handbook of Reagents for Organic Synthesis: Fluorine-Containing Reagents, Wiley, New York, 2007 Search PubMed;
(e)
K. Uneyama, Organofluorine Chemistry, Blackwell, Oxford, 2006 Search PubMed;
(f)
J. P. Bégué and D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, Wiley, Hoboken, 2007 Search PubMed;
(g) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC;
(h) P. W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef PubMed.
-
B. E. Smart, in Chemistry of Functional Groups, Supplement D: Fluorocarbons, ed. S. Patai and Z. Rappoport, John Wiley and Sons, New York, 1983 Search PubMed.
-
(a) J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373 CrossRef CAS;
(b) J. Hu and J. L. Zhang, Top. Organomet. Chem., 2015, 52, 143 CrossRef;
(c) M. K. Whittlesey and E. Peris, ACS Catal., 2014, 4, 3152 CrossRef CAS;
(d) E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. Mcgrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333 CrossRef CAS PubMed;
(e) M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328 CrossRef PubMed;
(f) H. Dang, A. M. Whittaker and G. Lalic, Chem. Sci., 2016, 7, 505 RSC.
-
(a) M. F. Kühnel and D. Lentz, Angew. Chem., Int. Ed., 2010, 49, 2933 CrossRef PubMed;
(b) M. Klahn and U. Rosenthal, Organometallics, 2012, 31, 1235 CrossRef CAS.
-
(a) J. L. Kiplinger and T. G. Richmond, J. Am. Chem. Soc., 1996, 118, 1805 CrossRef CAS;
(b) B. L. Edelbach, B. M. Kraft and W. D. Jones, J. Am. Chem. Soc., 1999, 121, 10327 CrossRef CAS;
(c) B. M. Kraft, R. J. Lachicotte and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 10973 CrossRef CAS PubMed.
-
(a) J. Vela, J. M. Smith, Y. Yu, N. A. Ketterer, C. J. Flaschenriem, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2005, 127, 7857 CrossRef CAS PubMed;
(b) L. Wang, H. Sun and X. Li, Eur. J. Inorg. Chem., 2015, 2732 CrossRef CAS.
-
(a) M. K. Whittlesey, R. N. Perutz, B. Greener and M. H. Moore, Chem. Commun., 1997, 187 RSC;
(b) S. P. Reade, M. F. Mahon and M. K. Whittlesey, J. Am. Chem. Soc., 2009, 131, 1847 CrossRef CAS PubMed;
(c) J. A. Panetier, S. A. Macgregor and M. K. Whittlesey, Angew. Chem., Int. Ed., 2011, 123, 2835 CrossRef;
(d) T. Stahl, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 1248 CrossRef CAS PubMed;
(e) D. McKay, I. M. Riddlestone, S. A. Macgregor, M. F. Mahon and M. K. Whittlesey, ACS Catal., 2015, 5, 776 CrossRef CAS;
(f) L. M. Milner, L. M. Hall, N. E. Pridmore, M. K. Skeats, A. C. Whitwood, J. M. Lynam and J. M. Slattery, Dalton Trans., 2016, 45, 1717 RSC.
-
(a) S. T. Belt, M. Helliwell, W. D. Jones, M. G. Partridge and R. N. Perutz, J. Am. Chem. Soc., 1993, 115, 1429 CrossRef CAS;
(b) M. Aizenberg and D. Milstein, Science, 1994, 265, 359 CAS;
(c) M. Aizenberg and D. Milstein, J. Am. Chem. Soc., 1995, 117, 8674 CrossRef CAS;
(d) M. Ballhorn, M. G. Partridge, R. N. Perutz and M. K. Whittlesey, Chem. Commun., 1996, 961 RSC;
(e) M. D. Su and S. Y. Chu, J. Am. Chem. Soc., 1997, 119, 10178 CrossRef CAS;
(f) B. L. Edelbach and W. D. Jones, J. Am. Chem. Soc., 1997, 119, 7734 CrossRef CAS;
(g) M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947 CrossRef CAS PubMed;
(h) A. L. Raza, J. A. Panetier, M. Teltewskoi and S. A. Macgregor, Organometallics, 2013, 32, 3795 CrossRef CAS;
(i) S. D. Timpa, J. Zhou, N. Bhuvanesh and O. V. Ozerov, Organometallics, 2014, 33, 6210 CrossRef CAS;
(j) P. Tian, C. Feng and T. P. Loh, Nat. Commun., 2015, 6, 7472 CrossRef PubMed.
-
(a) S. A. Garratt, R. P. Hughes, I. Kovacik, A. J. Ward, S. Willemsen and D. Zhang, J. Am. Chem. Soc., 2005, 127, 15585 CrossRef CAS PubMed;
(b) R. P. Hughes, R. B. Laritchev, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2005, 127, 6325 CrossRef CAS PubMed;
(c) S. M. Senaweera, A. Singh and J. D. Weaver, J. Am. Chem. Soc., 2014, 136, 3002 CrossRef CAS PubMed;
(d) J. Choi, D. Y. Wang, S. Kundu, Y. Choliy, T. J. Emge, K. K. Jespersen and A. S. Goldman, Science, 2011, 332, 1545 CrossRef CAS PubMed;
(e) Y. Y. Qian, M. H. Lee, W. Yang and K. S. Chan, J. Organomet. Chem., 2015, 791, 82 CrossRef CAS.
-
(a) T. Braun and R. N. Perutz, Chem. Commun., 2002, 2749 RSC;
(b) T. Schaub, M. Backes and U. Radius, J. Am. Chem. Soc., 2006, 128, 15964 CrossRef CAS PubMed;
(c) T. Schaub, P. Fischer, A. Steffen, T. Braun, U. Radius and A. Mix, J. Am. Chem. Soc., 2008, 130, 9304 CrossRef CAS PubMed;
(d) S. A. Johnson, C. W. Huff, F. Mustafa and M. Saliba, J. Am. Chem. Soc., 2008, 130, 17278 CrossRef CAS PubMed;
(e) A. Arévalo, A. T. Aca, M. F. Alamo and J. J. García, J. Am. Chem. Soc., 2014, 136, 4634 CrossRef PubMed.
-
(a) J. Guihaumé, E. Clot, O. Eisenstein and R. N. Perutz, Dalton Trans., 2010, 39, 10510 RSC;
(b) D. Breyer, T. Braun and P. Kláring, Organometallics, 2012, 31, 1417 CrossRef CAS;
(c) S. Sabater, J. A. Mata and E. Peris, Nat. Commun., 2013, 4, 2553 Search PubMed;
(d) M. Ohashi, M. Shibata, H. Saijo, T. Kambara and S. Ogoshi, Organometallics, 2013, 32, 3631 CrossRef CAS.
-
(a) A. Nova, S. Erhardt, N. A. Jasim, R. N. Perutz, S. A. Macgregor, J. E. McGrady and A. C. Whitwood, J. Am. Chem. Soc., 2008, 130, 15499 CrossRef CAS PubMed;
(b) H. L. Buckley, T. Wang, O. Tran and J. A. Love, Organometallics, 2009, 28, 2356 CrossRef CAS.
-
(a) Y. C. Hou and C. M. Chiang, J. Am. Chem. Soc., 1999, 121, 8116 CrossRef CAS;
(b) H. Lv, Y. B. Cai and J. L. Zhang, Angew. Chem., Int. Ed., 2013, 125, 3285 CrossRef;
(c) R. Doi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2015, 54, 1 CrossRef.
-
(a) H. Lv, J. H. Zhan, Y. B. Cai, Y. Yu, B. Wang and J. L. Zhang, J. Am. Chem. Soc., 2012, 134, 16216 CrossRef CAS PubMed;
(b) J. H. Zhan, H. Lv, Y. Yu and J. L. Zhang, Adv. Synth. Catal., 2012, 354, 1529 CrossRef CAS.
-
(a) V. I. Krasnov, V. E. Platonov, I. V. Beregovaya and L. N. Shchegoleva, Tetrahedron, 1997, 53, 1797 CrossRef CAS;
(b) S. S. Laev and V. D. Shteingarts, J. Fluorine Chem., 1998, 91, 21 CrossRef CAS;
(c) G. A. Selivanova, A. V. Reshetov, I. V. Beregovaya and N. V. Vasil'eva, J. Fluorine Chem., 2012, 137, 64 CrossRef CAS.
- M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 122, 4039 CrossRef.
-
(a) U. J. Fiedler, M. Klahn, P. Arndt, W. Baumann, A. Spannenberg, V. V. Burlakov and U. Rosenthal, J. Mol. Catal. A: Chem., 2007, 261, 184 CrossRef;
(b) M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994 RSC.
-
(a) K. Uneyama and H. Amii, J. Fluorine Chem., 2002, 114, 127 CrossRef CAS;
(b) V. J. Scott, R. Ç. Çetin and O. V. Ozerov, J. Am. Chem. Soc., 2005, 127, 2852 CrossRef CAS PubMed;
(c) R. Panisch, M. Bolte and T. Müller, J. Am. Chem. Soc., 2006, 128, 9676 CrossRef CAS PubMed;
(d) C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188 CrossRef CAS PubMed;
(e) R. N. Perutz, Science, 2008, 321, 1168 CrossRef CAS PubMed;
(f) G. Meier and T. Braun, Angew. Chem., Int. Ed., 2009, 121, 1575 CrossRef;
(g) O. Allemann, S. Duttwyler, P. Romanato, K. K. Baldridge and J. S. Siegel, Science, 2011, 331, 574 CrossRef PubMed.
-
(a) Q. Liu, Y. Li, H. Zhang, B. Chen, C. Tung and L. Wu, Chem. – Eur. J., 2012, 18, 620 CrossRef CAS PubMed;
(b) F. Wang, W. Wang, X. Wang, H. Wang, C. Tung and L. Wu, Angew. Chem., Int. Ed., 2011, 50, 3193 CrossRef CAS PubMed;
(c) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- M. Yu and X. Fu, J. Am. Chem. Soc., 2011, 133, 15926 CrossRef CAS PubMed.
- X. Liu, L. Liu, Z. Wang and X. Fu, Chem. Commun., 2015, 51, 11896 RSC.
-
(a) P. K. Chan and W. K. Leong, Organometallics, 2008, 27, 1247 CrossRef CAS;
(b) M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947 CrossRef CAS PubMed;
(c) C. Segarra, E. M. Marzá, J. P. Lowe, M. F. Mahon, R. C. Poulten and M. K. Whittlesey, Organometallics, 2012, 31, 8584 CrossRef CAS.
-
(a) Z. Ling, L. Yun, L. Liu, B. Wu and X. Fu, Chem. Commun., 2013, 49, 4214 RSC;
(b) X. Fu and B. B. Wayland, J. Am. Chem. Soc., 2004, 126, 2623 CrossRef CAS PubMed.
- P. A. Champagne, J. Pomarole, M. Thérien, Y. Benhassine, S. Beaulieu, C. Y. Legault and J. F. Paquin, Org. Lett., 2013, 15, 2210 CrossRef CAS PubMed.
- S. Li, S. Sarkar and B. B. Wayland, Inorg. Chem., 2009, 48, 8550 CrossRef CAS PubMed.
- W. W. Bartle and B. R. Eggins, J. Polarogr. Soc., 1966, 12, 89 CAS.
- V. Grass, D. Lexa, M. Momenteau and J. M. Savéant, J. Am. Chem. Soc., 1997, 119, 3536 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedure for stoichiometric and catalytic reactions, and NMR data of typical organometallic compounds and organic products. See DOI: 10.1039/c5qi00269a |
|
| This journal is © the Partner Organisations 2016 |