
An iron porphyrin-based conjugated network wrapped around carbon nanotubes as a noble-metal-free electrocatalyst for efficient oxygen reduction reaction†
Hongxing
Jia
,
Zijun
Sun
,
Daochuan
Jiang
,
Shangfeng
Yang
and
Pingwu
Du
*
Key Laboratory of Materials for Energy Conversion, Chinese Academy of Sciences, Department of Materials Science and Engineering, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), University of Science and Technology of China, 96 Jinzhai Road, Hefei, Anhui Province 230026, P. R. China. E-mail: dupingwu@ustc.edu.cn; Fax: +86-551-63606207; Tel: +86-551-63606207
First published on 24th February 2016
Abstract
Developing an efficient, robust, and noble-metal-free catalyst for the oxygen reduction reaction (ORR) is crucial for the large-scale commercialization of fuel cells and metal–air batteries. Herein, we report a structurally well-defined iron porphyrin-based conjugated network on carbon nanotubes ((FeP)n-CNTs) as a novel electrocatalyst for the ORR. Its superior electrocatalytic activity toward the ORR is demonstrated by the high-performance catalytic activity of (FeP)n-CNTs with a positive ORR onset potential and half-wave potential (E1/2 ∼ 0.76 V vs. RHE) values as well as outstanding durability and methanol tolerance in alkaline media. In addition, the low H2O2 yield illustrates that the ORR occurs mainly via the direct four-electron (4e−) pathway, and testing shows that the small amount of produced H2O2 can be rapidly consumed through both electrochemical reduction and oxidation. Our results demonstrate that the as-prepared (FeP)n-CNT catalyst is a promising noble-metal-free catalyst for potential applications in fuel cells and metal–air batteries.
 Pingwu Du | Professor Pingwu Du received a B.S. degree from Wuhan University (2001) and a Ph.D. degree from the University of Rochester (2009) under the direction of Professor Richard Eisenberg. After postdoc training at MIT and Argonne National Laboratory (Director's Fellow), he joined the University of Science and Technology of China (USTC) as a faculty member in 2012. His current research focuses on photocatalysis and inorganic chemistry. He is a recipient of the Young Investigator Award (Inorganic Division, 2008) and the Nobel Laureate Signature Award (2011) from the American Chemical Society. |
Introduction
The oxygen reduction reaction (ORR) plays a crucial role in various life processes and in electrochemical energy technologies such as fuel cells and metal–air batteries.1–3 Given the sluggish kinetics of the ORR,4,5 tremendous efforts have been devoted to search for and design highly efficient electrocatalysts for it. To date, the state-of-the-art ORR catalysts have been predominantly Pt-based materials,6–9 but several drawbacks have seriously hindered their large-scale commercialization, including high-cost, scarce reserves, weak durability, and low tolerance to fuel crossover. Thus, developing a noble-metal-free catalyst with excellent performance as an alternative has attracted much attention.3,10–20In the literature, various types of electrocatalysts made of earth-abundant elements have been synthesized and investigated for the ORR, including metal oxides,21–23 metal chalcogenides,24 metal alloys,5,25,26 carbon materials functionalized by polyelectrolytes,11,27 and heteroatom doped carbon materials.10,12,28–30 In particular, transition metal nitrogen-coordinated macrocycles supported on carbon materials (M–N/C, M = Fe, Co) are considered among the most promising catalysts due to their low cost, facile preparation, and excellent performance toward the ORR.3,20 Notably, porphyrins and phthalocyanines with a metal (Fe, Co) are widely used as precursors to prepare ORR catalysts.31–33 Despite the significant progress made in the past few decades, it still remains a great challenge for researchers to develop an efficient, robust, and noble-metal-free electrocatalyst for the ORR.
Conjugated mesoporous polymers (CMPs) and covalent organic frameworks (COFs) are a novel class of polymeric materials with a micromesoporous structure and extended network.34,35 The advantages of CMPs and COFs, including tunable porous structures, their large surface areas, facile functionality, promising capacity for catalysis, and recyclability as a heterogeneous catalyst, have drawn much attention in recent years.36–38 Moreover, the noncovalent grafting approach has showed an enhanced catalytic performance for the ORR in a cobalt-porphyrin-based network, which can preserve the excellent electronic properties of CNTs without the loss of its stability.13 Inspired by the studies of CMPs and COFs, herein we report the use of an iron porphyrin-based organic framework on carbon nanotubes ((FeP)n-CNTs) as an excellent electrocatalyst for the ORR. This iron-incorporated porphyrin framework was synthesized via a facile template-polymerization reaction. The resulting (FeP)n-CNT material displays an outstanding catalytic activity toward the ORR in alkaline media, such as a positive half-wave potential (0.76 V vs. RHE), a nearly 4e− transfer pathway, and excellent stability (∼97% retention after 1000 cycles). All of these results suggest that the (FeP)n-CNT material is among the best Fe-based electrocatalysts for the ORR.
Results and discussion
Fig. 1 shows the synthesis procedure for the (FeP)n-CNT material. The 5,10,15,20-tetrakis-(triisopropylsilylethynyl)porphyrin (1) monomer was obtained following a reported procedure in the literature with a yield of ∼34% (see details in the ESI†).39,40 Then, iron metal ions were inserted into porphyrin monomers by a typical metal-porphyrin complexation reaction to produce iron 5,10,15,20-tetrakis-(triisopropylsilylethynyl)porphyrin (2, FeP-TIPS).41 The cleavage of TIPS protecting groups was performed in the presence of tetrabutylammonium fluoride (TBAF) to yield 5,10,15,20-tetrakisethynylporphyrin (3, FeP) as a green solid, which was confirmed by mass spectrometry (Fig. S1 and S2†), elemental analysis, and UV-vis absorption spectroscopy. Thereafter, the as-prepared FeP was used to grow a covalent porphyrin framework wrapped around carbon nanotubes (CNTs) via Hay-coupling to produce (FeP)n-CNTs.18,42 The formation of iron porphyrin frameworks wrapped around CNTs can be ascribed to π–π stacking interactions and van der Waals forces between FeP monomers and CNTs.43,44 Then, Hay-coupling between these adsorbed porphyrins took place in situ to produce butadiyne linkers between FeP monomers. During polymerization, the CNTs served as a template. As a consequence, a stable and large porphyrin framework shell was formed around the CNTs. The successful synthesis of the (FeP)n-CNT hybrid material was further characterized by scanning electron microscopy (SEM), high-resolution transmission electron microscopy (HRTEM), Raman spectroscopy, and X-ray photoelectron spectroscopy (XPS). The material's catalytic properties and reaction kinetics toward the ORR were investigated by a series of electrochemical measurements in alkaline media at room temperature.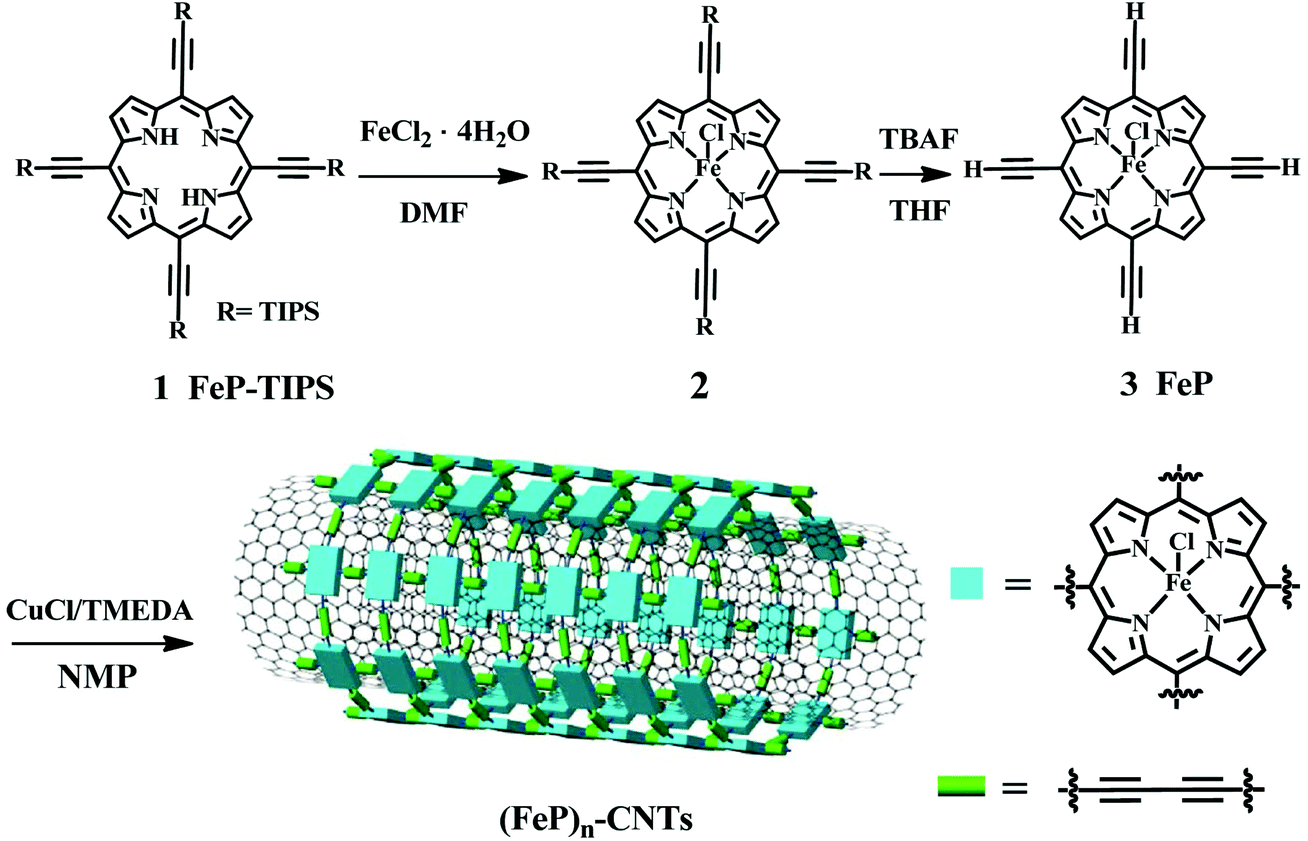 | ||
| Fig. 1 Schematic representation of the chemical synthesis of an iron porphyrin-based conjugated network, forming (FeP)n-CNTs. TIPS = Triisopropylsilyl. | ||
The morphologies of CNTs, FeP-TIPS/CNTs (FeP-TIPS mixed with CNTs), and (FeP)n-CNTs were investigated by SEM (Fig. 2a–c) and HRTEM (Fig. 2d–f). Fig. 2a shows an image of pristine CNTs. The tubular structure of CNTs appears as bundles with an average diameter of 1–3 nm. The tube-like feature remains in the SEM image of (FeP)n-CNTs but its diameter is increased (Fig. 2b). The FeP-TIPS/CNT image in Fig. 2c shows many nanoparticles on CNTs, indicating the aggregation of porphyrin monomers on the substrate upon drying.45,46 Compared to the HRTEM image of pristine CNTs (Fig. 2d), a thin layer with a thickness of 0.5–1.5 nm is clearly observed in (FeP)n-CNTs, wrapped around CNTs (Fig. 2e). No appreciable lattice fringes appeared, indicating that the coating layer around CNTs is probably an amorphous iron porphyrin-based conjugated network.
 | ||
| Fig. 2 SEM images of (a) CNTs, (b) (FeP)n-CNTs, and (c) FeP-TIPS/CNTs; HRTEM images of (d) CNTs, (e) (FeP)n-CNTs, and (f) FeP-TIPS/CNTs. | ||
Fig. 3a shows the Raman spectra of CNTs, FeP-TIPS, FeP-TIPS/CNTs, and (FeP)n-CNTs to further confirm the formation of an iron porphyrin conjugated network on CNTs. Except for FeP-TIPS, all other materials have typical D and G bands of carbon nanotubes at ∼1350 and ∼1590 cm−1, respectively. The spectra of FeP-TIPS and FeP-TIPS/CNTs show triple bond stretching modes of the protected acetylene moieties in FeP-TIPS at ∼2143 cm−1.18 The same signal is not observed in (FeP)n-CNTs, suggesting complete deprotection of the TIPS group. For (FeP)n-CNTs, the presence of the weak radial breathing mode (RBM) observed in the 200–300 cm−1 region indicated that the CNTs used here are no more than triple-walled,13 while the presence of a porphyrin scaffold is evidenced by the peak at ∼1230 cm−1. Notably, compared with CNTs and FeP-TIPS/CNTs, (FeP)n-CNTs exhibit a higher intensity of the D band and a lower intensity of RBM, the G band, and the G′ band (∼2683 cm−1), indicating their enhanced degree of disorder. This effect can be explained by the formation of the hybrid structure of the porphyrin covalent framework and CNTs, which is in good agreement with the SEM and HRTEM observations.
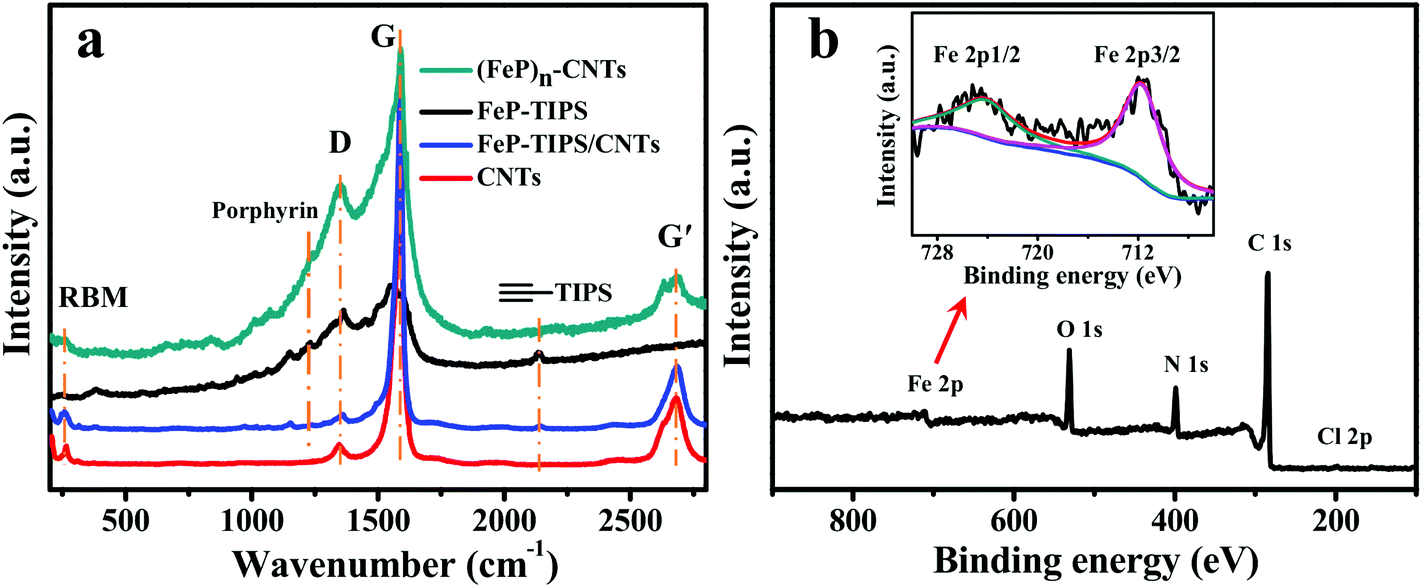 | ||
| Fig. 3 (a) Raman spectra of CNTs (red), FeP-TIPS/CNTs (blue), FeP-TIPS (black), and (FeP)n-CNTs (dark green). (b) XPS survey spectrum of (FeP)n-CNTs. The inset image is the high resolution XPS spectrum of Fe 2p. | ||
To identify the surface chemical composition and valence states of (FeP)n-CNTs, XPS spectra were further recorded (Fig. 3b). The survey spectrum reveals that the (FeP)n-CNT material is mainly composed of C, N, O, Fe, and Cl elements and the presence of Cl can be further confirmed by a high resolution XPS spectrum (Fig. S3†), indicative of the successful loading of iron porphyrin frameworks on CNTs. The high resolution XPS spectrum of Fe 2p shows two peaks at 711.7 eV and 724.0 eV, which are assigned to Fe 2p3/2 and Fe 2p1/2 in iron porphyrin, respectively (Fig. 3b, inset).47–50 In addition, the ratio of Fe and C was estimated to be approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :45, indicating that there is an average of 17 carbon atoms from CNTs and 28 carbon atoms from FeP for an area of each FeP. According to a reported calculation method,18 the CNT surface with an area of one FeP molecule contains about 40 carbon atoms. The above analysis indicates that a large number of FeP was stacked on CNTs, resulting in a multilayer organic framework species. For comparison, XPS data of FeP-TIPS/CNTs were also collected (Fig. S4†). The survey XPS spectrum displays silicon peaks (from TIPS in FeP-TIPS) at 102.0 eV and 153.0 eV. In addition, the estimated ratio of Fe and C was approximately 1:91, corresponding to an average of 27 carbon atoms on CNTs for each FeP-TIPS molecule. This result shows that the number of iron centers is not equivalent in (FeP)n-CNTs and FeP-TIPS/CNT hybrids. Therefore, different amounts of the catalyst material should be drop-cast onto the working electrode when we compare the catalytic properties of FeP-TIPS/CNTs with (FeP)n-CNTs.
:45, indicating that there is an average of 17 carbon atoms from CNTs and 28 carbon atoms from FeP for an area of each FeP. According to a reported calculation method,18 the CNT surface with an area of one FeP molecule contains about 40 carbon atoms. The above analysis indicates that a large number of FeP was stacked on CNTs, resulting in a multilayer organic framework species. For comparison, XPS data of FeP-TIPS/CNTs were also collected (Fig. S4†). The survey XPS spectrum displays silicon peaks (from TIPS in FeP-TIPS) at 102.0 eV and 153.0 eV. In addition, the estimated ratio of Fe and C was approximately 1:91, corresponding to an average of 27 carbon atoms on CNTs for each FeP-TIPS molecule. This result shows that the number of iron centers is not equivalent in (FeP)n-CNTs and FeP-TIPS/CNT hybrids. Therefore, different amounts of the catalyst material should be drop-cast onto the working electrode when we compare the catalytic properties of FeP-TIPS/CNTs with (FeP)n-CNTs.
Cyclic voltammetry (CV) in O2-saturated and Ar-saturated 0.1 M KOH solutions was first employed to examine the (FeP)n-CNT hybrid material for ORR catalytic activity at a scan rate of 10 mV s−1 (Fig. S5†). In an Ar-saturated solution, a featureless slope for the cathodic current was observed within the whole potential range of the CV curve (0.38 V to 1.18 V vs. RHE). In sharp contrast, a prominent cathodic peak emerging at 0.88 V (vs. RHE) in the CV curve was observed in an O2-saturated solution, suggesting a pronounced ORR activity for (FeP)n-CNTs.
Moreover, the ORR peak potential (ECORR) and the corresponding current density of (FeP)n-CNTs are superior to FeP-TIPS, CNTs, and FeP-TIPS/CNTs, as shown in Fig. 4a. An ECORR of 0.73 V is achieved for (FeP)n-CNTs, which is higher than that of FeP-TIPS (0.49 V), CNTs (0.72 V), and FeP-TIPS/CNTs (0.69 V). In addition, (FeP)n-CNTs also display a much higher ORR current density than the other three materials. All of these results suggest that the (FeP)n-CNT material has a highly enhanced electrocatalytic activity toward the ORR in alkaline media. The polarization curves for (FeP)n-CNTs and FeP-TIPS/CNTs were recorded with rotating disk electrode (RDE) measurements in 0.1 M KOH solution (Fig. 4b). The onset ORR potential was almost constant at varying rotating speeds for both materials, while the limiting current densities increased with the rotation rate. In contrast to the physisorbed blend of FeP-TIPS/CNTs, (FeP)n-CNTs displayed a positively shifted onset potential (0.88 V vs. RHE), half-wave potential (E1/2, 0.76 V vs. RHE based on the curve at a rotating speed of 1600 rpm) and a much higher limiting current density at low potential, confirming their superior catalytic performance toward the ORR. The E1/2 value for the ORR on the (FeP)n-CNT electrode reported in this work is much more positive than those obtained over many other Fe-based catalysts reported in the literature, such as the Fe3O4/N-graphene catalyst (0.59 V vs. RHE, 0.1 M KOH),51 Fe-phthalocyanine catalyst (0.61 V vs. RHE, 0.1 M KOH),19 Fe-porphyrin-like carbon nanotube catalyst (0.67 V vs. RHE, 0.1 M KOH),52 and FePc-MWCNT catalyst (0.75 V vs. RHE, 0.1 M KOH).53 Therefore, the as-prepared (FeP)n-CNT hybrid is among the best Fe-based electrocatalysts for ORR catalysis in alkaline media.
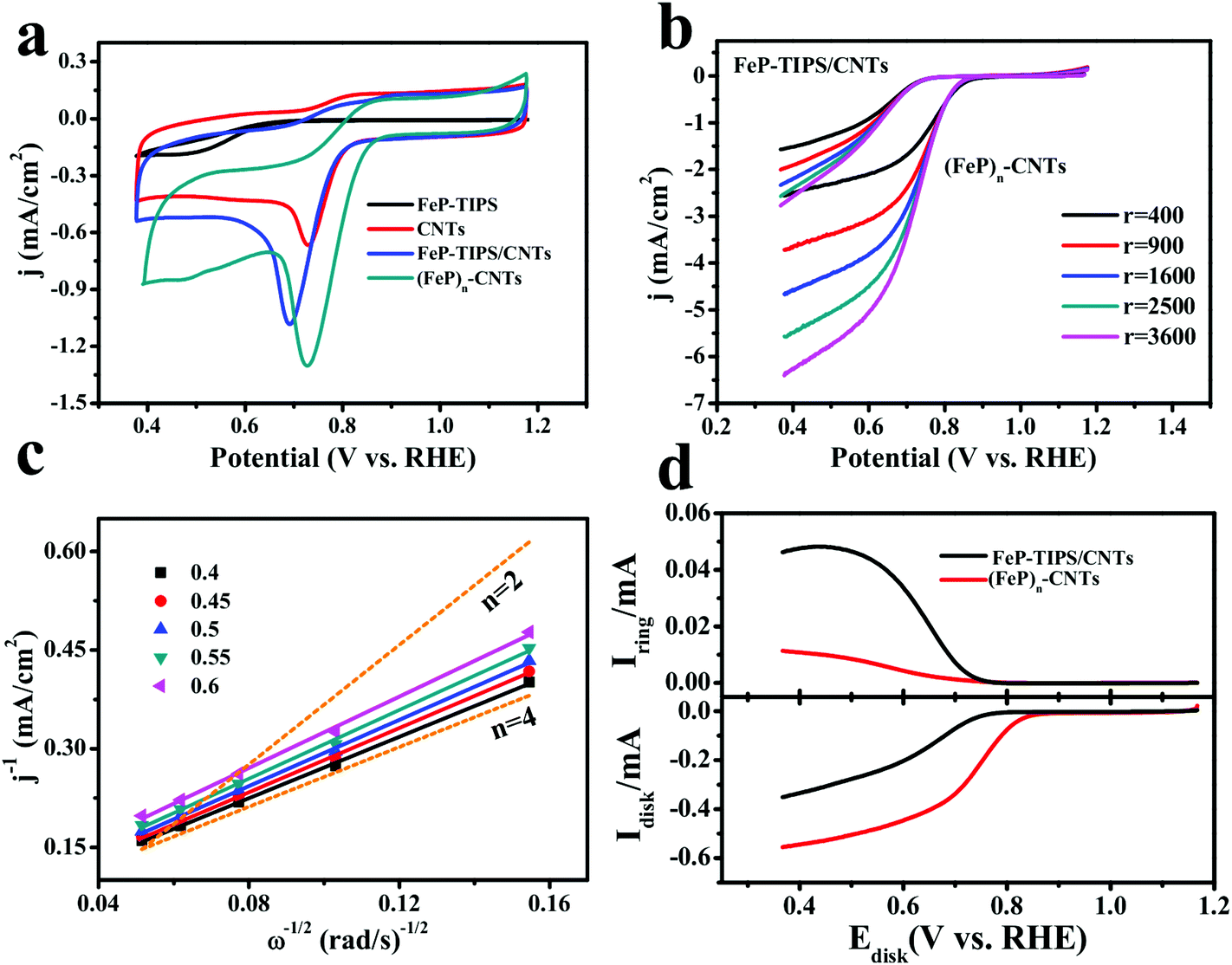 | ||
| Fig. 4 (a) CVs of CNTs (red), FeP-TIPS/CNTs (blue), FeP-TIPS (black), and (FeP)n-CNTs (dark green) in O2-saturated 0.1 M KOH. Scan rate: 10 mV s−1. (b) RDE polarization curves of (FeP)n-CNTs and FeP-TIPS/CNTs in O2-saturated 0.1 M KOH at different rotation rates. Scan rate: 10 mV s−1. (c) K–L plots of (FeP)n-CNTs, derived from data in Fig. 4b under different potentials. (d) RRDE measurements for (FeP)n-CNTs (red) and FeP-TIPS/CNTs (black) in O2-saturated 0.1 M KOH solution with the background capacitive current corrected. In the RRDE experiment, the ring electrode was polarized at 1.57 V vs. RHE to completely oxidize H2O2 to O2. Rotation rate: 400 rpm. Scan rate: 10 mV s−1. Catalyst loading: 0.5 mg cm−2 for (FeP)n-CNTs, CNTs, FeP-TIPS and 0.75 mg cm−2 for FeP-TIPS/CNTs. | ||
The Koutecky–Levich (K–L) plots for (FeP)n-CNTs (Fig. 4c) were then calculated in the mass transport limiting ORR region (0.40–0.60 V vs. RHE) on the basis of the Koutecky–Levich equation (see the ESI†). The well-fitted linearity of the plots indicates that the reaction is controlled by the mass transport of oxygen and follows first-order kinetics. In addition, the electron transfer number (n) involved in the ORR was estimated to be about 4 from the slopes of the K–L plots, which was further confirmed by RRDE measurements (Fig. 4d). The ring current for FeP-TIPS/CNTs was detected to be much higher than that for (FeP)n-CNTs, suggesting that more H2O2 was produced on the electrode modified with FeP-TIPS/CNTs. The n values calculated from the RRDE measurements are 2.95 for FeP-TIPS/CNTs and 3.79 for (FeP)n-CNTs, corresponding to yields of 52.5% and 10.5% for H2O2, respectively (see the ESI†). The electron transfer number based on the K–L plots is slightly larger than the value calculated from the RRDE measurements, which is probably due to the presence of the carbon material that increases the surface roughness and subsequently changes the local concentration of O2.54,55 All these results indicate that the ORR on the (FeP)n-CNT hybrid is dominated by the four-electron (4e−) direct pathway, although an indirect two-electron (2e−) pathway leading to the production of a H2O2 intermediate could occur.
The stability of the (FeP)n-CNT catalyst during the ORR was assessed in an O2-saturated 0.1 M KOH solution through continuous potential cycling from 0.27 V to 1.17 V vs. RHE at a scan rate of 10 mV s−1 for more than 5000 cycles. As shown in Fig. 5a, the deterioration of (FeP)n-CNTs resulted in a very slight current decrease with a retention of ∼97% (after 1000 cycles) and ∼92% (after 5000 cycles) (at 0.50 V vs. RHE) in alkaline solution. In contrast, the attenuation of current density for FeP-TIPS/CNTs after 5000 cycles is much more pronounced (Fig. 5b). These results demonstrate that the (FeP)n-CNT material possesses great durability for ORR catalysis in alkaline solution, which could be explained by its more positive onset potential and higher electron transfer number.
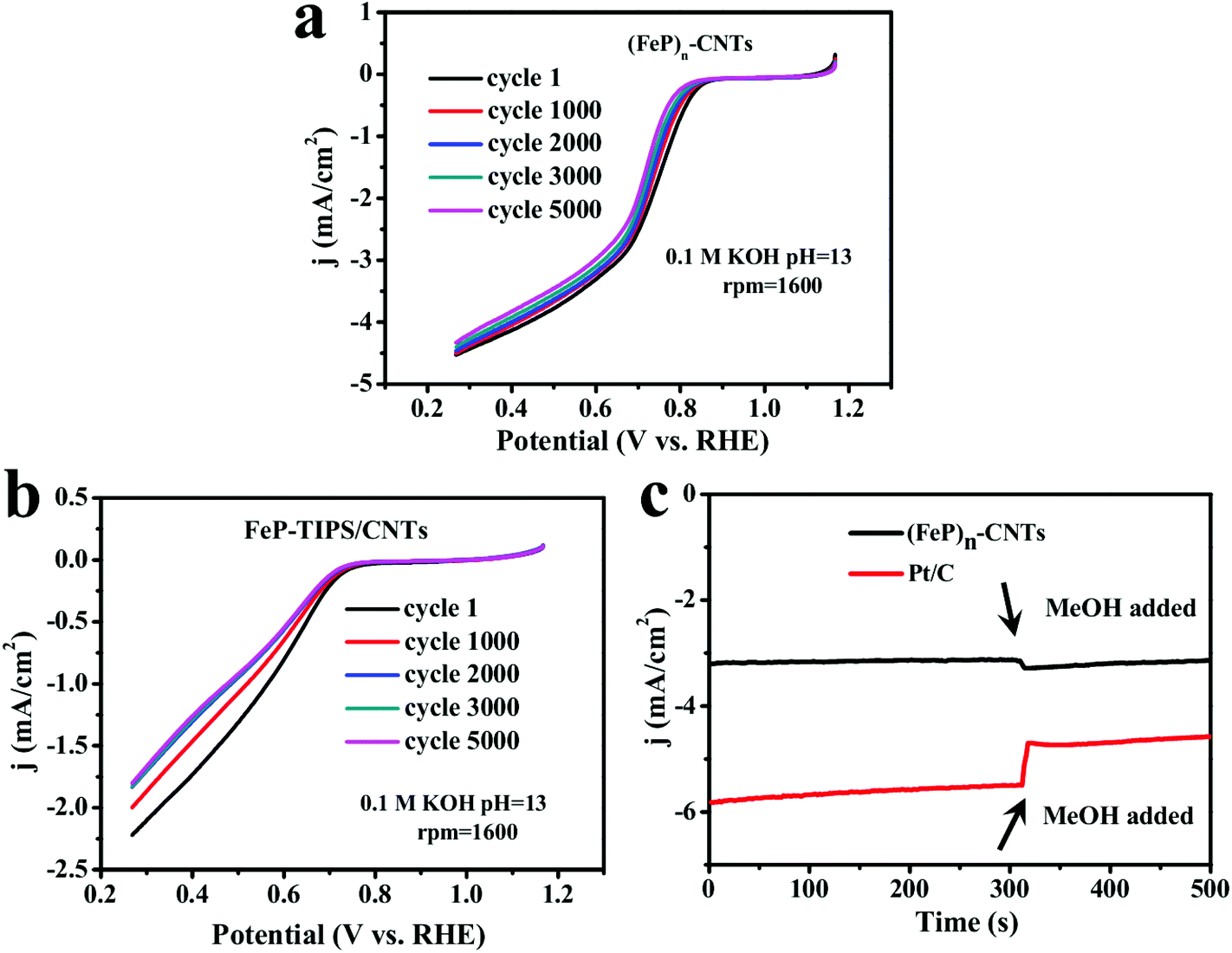 | ||
| Fig. 5 Long-term cycling CV test of (a) (FeP)n-CNTs and (b) FeP-TIPS/CNTs for the ORR in O2-saturated 0.1 M KOH. Rotation rate: 1600 rpm. Scan rate: 10 mV s−1. (c) Methanol crossover test on (FeP)n-CNTs (black) and Pt/C (red) in an O2-saturated 0.1 M KOH. Rotation rate: 900 rpm. | ||
To check the methanol-tolerant properties of the (FeP)n-CNT catalyst, chronoamperometric measurements were performed (Fig. 5c). No appreciable loss in the current density after the introduction of methanol was recorded. For comparison, there was a significant current decrease on the addition of methanol when using a Pt/C catalyst, as also seen in Fig. 5c. The results indicate that (FeP)n-CNTs may be used as catalysts in direct-methanol fuel cells, owing to their excellent capability to resist methanol poisoning.
Since the activity and durability of ORR catalysts may suffer from the produced H2O2 intermediate, more CV measurements in O2-saturated and Ar-saturated 0.1 M KOH solutions were performed to investigate the performance of (FeP)n-CNTs in the presence of 10 mM H2O2 (Fig. 6a). Remarkably, the current density recorded in both O2-saturated and Ar-saturated solutions was greatly increased after the addition of H2O2, demonstrating their great ability to reduce H2O2. To gain further insight into the catalytic activity of (FeP)n-CNTs towards H2O2, 5 mM H2O2 was added to an Ar-saturated 0.1 M KOH solution and the CV curves were immediately recorded, as shown in Fig. 6b. There was an evident H2O2 reduction current and an oxidation peak of H2O2 to O2 in the first CV curve. As for the second cycle, an enhanced reduction current appeared, which is ascribed to the reduction of the remaining H2O2 in solution, as well as the in situ produced O2. These results clearly demonstrate that the (FeP)n-CNT material efficiently reduces H2O2 in alkaline media via both electrochemical reduction and oxidation.
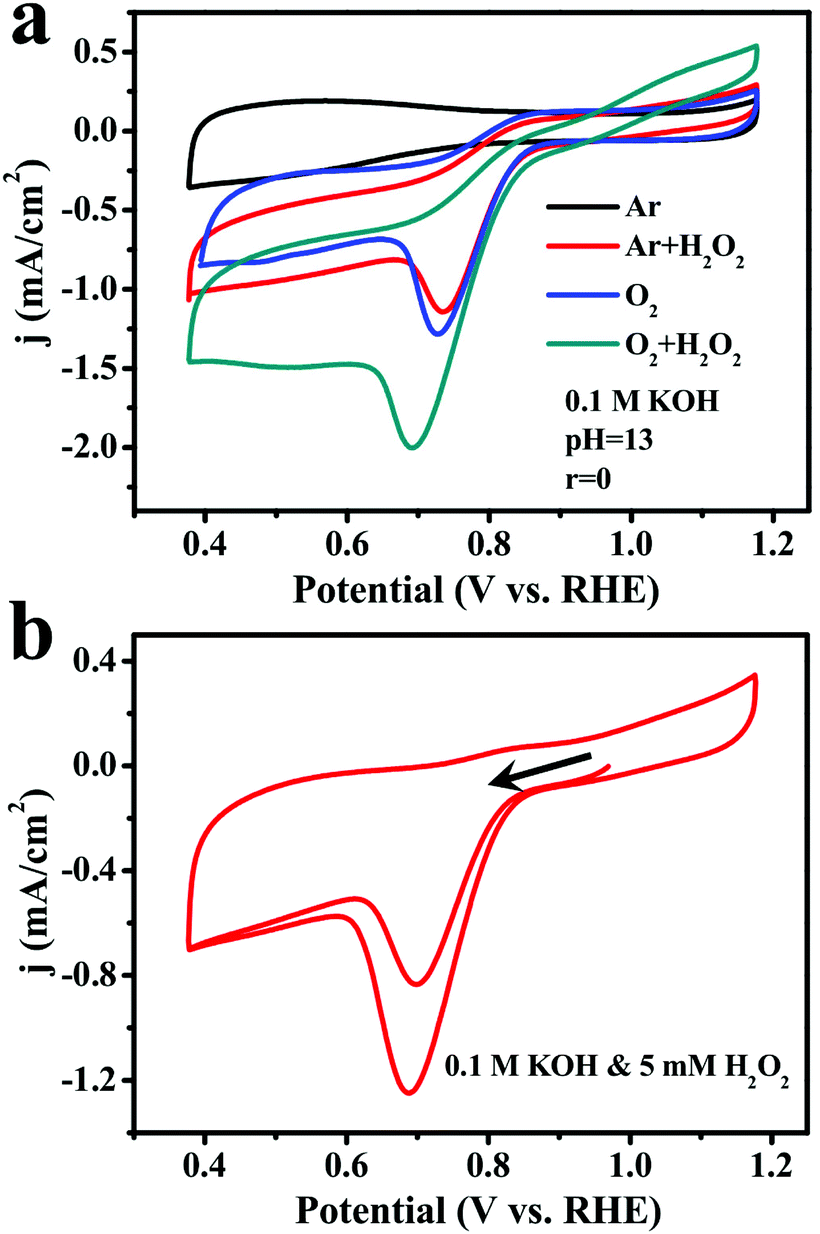 | ||
| Fig. 6 (a) CVs of (FeP)n-CNTs in Ar-saturated 0.1 M KOH (black), and upon addition of 10 mM H2O2 (red); in O2-saturated 0.1 M KOH (blue), and upon addition of 10 mM H2O2 (dark green). Scan rate: 10 mV s−1. (b) CVs of (FeP)n-CNTs in Ar-saturated 0.1 M KOH upon addition of 5 mM H2O2. Scan rate: 10 mV s−1. Catalyst loading: 0.5 mg cm−2. | ||
Conclusions
In conclusion, a novel noble-metal-free electrocatalyst has been successfully synthesized by wrapping iron porphyrin frameworks around a CNT template. The resulting hybrid material demonstrates high-performance ORR activity with great stability. The results show that: (i) (FeP)n-CNTs exhibited a positive E1/2 value (0.76 V vs. RHE), outstanding long-term durability, and high tolerance towards methanol, suggesting their excellent and robust activity for the ORR. (ii) The electron transfer number (n) involved in the ORR was tested to be 3.79, illustrating a high selectivity in the direct 4e− pathway. (iii) H2O2 was decomposed through both electrochemical reduction and oxidation. All these results indicate that (FeP)n-CNTs are a promising substitute for Pt-based catalysts for applications in fuel cells and metal–air batteries.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21271166, 21473170), the Fundamental Research Funds for Central Universities, the Program for New Century Excellent Talents in University (NCET), and the Thousand Young Talents Program.References
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- E. G. Kovaleva and J. D. Lipscomb, Nat. Chem. Biol., 2008, 4, 186–193 CrossRef CAS PubMed.
- P. J. Wei, G. Q. Yu, Y. Naruta and J. G. Liu, Angew. Chem., Int. Ed., 2014, 53, 6659–6663 CrossRef CAS PubMed.
- Z. M. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542–7543 CrossRef CAS PubMed.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. F. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- Y. H. Bing, H. S. Liu, L. Zhang, D. Ghosh and J. J. Zhang, Chem. Soc. Rev., 2010, 39, 2184–2202 RSC.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- D. L. Wang, S. F. Lu and S. P. Jiang, Chem. Commun., 2010, 46, 2058–2060 RSC.
- D. L. Wang, H. L. L. Xin, R. Hovden, H. S. Wang, Y. C. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- S. Y. Wang, D. S. Yu, L. M. Dai, D. W. Chang and J. B. Baek, ACS Nano, 2011, 5, 6202–6209 CrossRef CAS PubMed.
- S. Y. Wang, E. Iyyamperumal, A. Roy, Y. H. Xue, D. S. Yu and L. M. Dai, Angew. Chem., Int. Ed., 2011, 50, 11756–11760 CrossRef CAS PubMed.
- Y. Cheng, C. W. Xu, L. C. Jia, J. D. Gale, L. L. Zhang, C. Liu, P. K. Shen and S. P. Jiang, Appl. Catal., B, 2015, 163, 96–104 CrossRef CAS.
- M. Bourourou, K. Elouarzaki, M. Holzinger, C. Agnès, A. L. Goff, N. Reverdy-Bruas, D. Chaussy, M. Party, A. Maaref and S. Cosnier, Chem. Sci., 2014, 5, 2885–2888 RSC.
- X. P. Han, T. R. Zhang, J. Du, F. Y. Cheng and J. Chen, Chem. Sci., 2013, 4, 368–376 RSC.
- R. McGuire Jr., D. K. Dogutan, T. S. Teets, S. Jin, S.-H. Yang and D. G. Nocera, Chem. Sci., 2010, 1, 411–414 RSC.
- P. Su, H. Xiao, J. Zhao, Y. Yao, Z. Shao, C. Li and Q. Yang, Chem. Sci., 2013, 4, 2941–2946 RSC.
- I. Hijazi, T. Bourgeteau, R. Cornut, A. Morozan, A. Filoramo, J. Leroy, V. Derycke, B. Jousselme and S. Campidelli, J. Am. Chem. Soc., 2014, 136, 6348–6354 CrossRef CAS PubMed.
- W. M. Li, A. P. Yu, D. C. Higgins, B. G. Llanos and Z. W. Chen, J. Am. Chem. Soc., 2010, 132, 17056–17058 CrossRef CAS PubMed.
- A. J. Olaya, D. Schaming, P.-F. Brevet, H. Nagatani, T. Zimmermann, J. Vanicek, H.-J. Xu, C. P. Gros, J.-M. Barbe and H. H. Girault, J. Am. Chem. Soc., 2012, 134, 498–506 CrossRef CAS PubMed.
- S. J. Guo, S. Zhang, L. H. Wu and S. H. Sun, Angew. Chem., Int. Ed., 2012, 51, 11770–11773 CrossRef CAS PubMed.
- Y. Y. Liang, Y. G. Li, H. L. Wang, J. G. Zhou, J. Wang, T. Regier and H. J. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- Y. Y. Liang, H. L. Wang, J. G. Zhou, Y. G. Li, J. Wang, T. Regier and H. J. Dai, J. Am. Chem. Soc., 2012, 134, 3517–3523 CrossRef CAS PubMed.
- M. R. Gao, J. Jiang and S. H. Yu, Small, 2012, 8, 13–27 CrossRef CAS PubMed.
- C. Chen, Y. J. Kang, Z. Y. Huo, Z. W. Zhu, W. Y. Huang, H. L. L. Xin, J. D. Snyder, D. G. Li, J. A. Herron, M. Mavrikakis, M. F. Chi, K. L. More, Y. D. Li, N. M. Markovic, G. A. Somorjai, P. D. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- C. Wang, M. F. Chi, D. G. Li, D. Strmcnik, D. van der Vliett, G. F. Wang, V. Komanicky, K. C. Chang, A. P. Paulikas, D. Tripkovic, J. Pearson, K. L. More, N. M. Markovic and V. R. Stamenkovic, J. Am. Chem. Soc., 2011, 133, 14396–14403 CrossRef CAS PubMed.
- S. Y. Wang, D. S. Yu and L. M. Dai, J. Am. Chem. Soc., 2011, 133, 5182–5185 CrossRef CAS PubMed.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- Y. J. Sa, C. Park, H. Y. Jeong, S. H. Park, Z. Lee, K. T. Kim, G. G. Park and S. H. Joo, Angew. Chem., Int. Ed., 2014, 53, 4102–4106 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. G. Jin and S. Z. Qiao, Small, 2012, 8, 3550–3566 CrossRef CAS PubMed.
- C. T. Carver, B. D. Matson and J. M. Mayer, J. Am. Chem. Soc., 2012, 134, 5444–5447 CrossRef CAS PubMed.
- A. Vanderputten, A. Elzing, W. Visscher and E. Barendrecht, J. Electroanal. Chem., 1986, 214, 523–533 CrossRef CAS.
- Z. S. Wu, L. Chen, J. Z. Liu, K. Parvez, H. W. Liang, J. Shu, H. Sachdev, R. Graf, X. L. Feng and K. Müllen, Adv. Mater., 2014, 26, 1450–1455 CrossRef CAS PubMed.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef CAS PubMed.
- X. M. Liu, Y. H. Xu, Z. Q. Guo, A. Nagai and D. L. Jiang, Chem. Commun., 2013, 49, 3233–3235 RSC.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Cote, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS PubMed.
- H. Xu and D. L. Jiang, Nat. Chem., 2014, 6, 564–566 CrossRef CAS PubMed.
- T. Lange, J. D. vanLoon, R. R. Tykwinski, M. Schreiber and F. Diederich, Synthesis, 1996, 537–550 CrossRef CAS.
- M. J. Plater, S. Aiken, T. Gelbrich, M. B. Hursthouse and G. Bourhill, Polyhedron, 2001, 20, 3219–3224 CrossRef CAS.
- S. Nakagaki, F. L. Benedito and F. Wypych, J. Mol. Catal. A: Chem., 2004, 217, 121–131 CrossRef CAS.
- A. S. Hay, J. Org. Chem., 1962, 27, 3320–3321 CrossRef CAS.
- D. Q. Yang, J. F. Rochette and E. Sacher, J. Phys. Chem. B, 2005, 109, 4481–4484 CrossRef CAS PubMed.
- P. Yu, J. Yan, H. Zhao, L. Su, J. Zhang and L. Q. Mao, J. Phys. Chem. C, 2008, 112, 2177–2182 CAS.
- C. S. Huang, L. P. Wen, H. B. Liu, Y. L. Li, X. F. Liu, M. J. Yuan, J. Zhai, L. Jiang and D. B. Zhu, Adv. Mater., 2009, 21, 1721–1725 CrossRef CAS.
- Y. J. Li, X. F. Li, Y. L. Li, H. B. Liu, S. Wang, H. Y. Gan, J. B. Li, N. Wang, X. R. He and D. B. Zhu, Angew. Chem., Int. Ed., 2006, 45, 3639–3643 CrossRef CAS PubMed.
- R. Cao, R. Thapa, H. Kim, X. Xu, M. G. Kim, Q. Li, N. Park, M. L. Liu and J. Cho, Nat. Commun., 2013, 4 Search PubMed.
- M. E. Lipinska, J. P. Novais, S. L. H. Rebelo, B. Bachiller-Baeza, I. Rodriguez-Ramos, A. Guerrero-Ruiz and C. Freire, Polyhedron, 2014, 81, 475–484 CrossRef CAS.
- M. E. Lipinska, S. L. H. Rebelo and C. Freire, J. Mater. Sci., 2014, 49, 1494–1505 CrossRef CAS.
- Z. D. Liu, H. X. Zhao and C. Z. Huang, PLoS One, 2012, 7, e50367, DOI:10.1371/journal.pone.0050367.
- Z. S. Wu, S. B. Yang, Y. Sun, K. Parvez, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 9082–9085 CrossRef CAS PubMed.
- D. H. Lee, W. J. Lee, W. J. Lee, S. O. Kim and Y. H. Kim, Phys. Rev. Lett., 2011, 106, 175502, DOI:10.1103/PhysRevLett.106.175502.
- S. A. Mamuru and K. I. Ozoemena, Electrochem. Commun., 2010, 12, 1539–1542 CrossRef CAS.
- P.-C. Li, C.-C. Hu, H. Noda and H. Habazaki, J. Power Sources, 2015, 298, 102–113 CrossRef CAS.
- D. Eisenberg, W. Stroek, I. N. J. Geels, C. S. Sandu, A. Heller, N. Yan and G. Rothenberg, Chem. – Eur. J., 2016, 22, 501–505 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details are available including synthetic details, characterization data, XPS data, cyclic voltammetry data of (FeP)n-CNTs. See DOI: 10.1039/c5qi00198f |
| This journal is © the Partner Organisations 2016 |