
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective enzymatic chemistry for polymer conjugation to protein lysine residues: PEGylation of G-CSF at lysine-41
A.
Mero
a,
A.
Grigoletto
a,
K.
Maso
 a,
H.
Yoshioka
b,
A.
Rosato
c and
G.
Pasut
*ac
a,
H.
Yoshioka
b,
A.
Rosato
c and
G.
Pasut
*ac
aDepartment of Pharmaceutical Sciences, University of Padua, via F. Marzolo 5, 35131 Padua, Italy
bNOF Corporation, 3-3 Chidori-Cho, Kawasaki-Ku, Kawasaki, Kanagawa 210-0865, Japan
cVeneto Institute of Oncology IOV – IRCCS, Padua, Italy. E-mail: gianfranco.pasut@unipd.it
First published on 4th October 2016
Abstract
Microbial transglutaminase (mTGase) is an enzyme that catalyzes site-specific protein derivatization at specific glutamines. mTGase-mediated conjugation with PEG-NH2 to granulocyte colony stimulating factor (G-CSF) yields a site selective mono-derivative conjugate involving Gln135. The same enzymatic reaction of mTGase, i.e. the transfer of the Gln acyl group to an amino donor, was investigated by reversing the substrates. A specific acyl donor PEG derivative was synthesized by coupling the Z-QG mTGase substrate to PEG. The mTGase-mediated conjugation of this PEG-ZQG in the presence of G-CSF generated a high-yield PEG-G-CSF conjugate in which the polymer was selectively coupled to Lys41 of the protein. The PEG-K41-G-CSF conjugate was compared with the PEG-Q135-G-CSF one obtained through mTGase conjugation of PEG-NH2 to Gln135. Biophysical characterization showed that the two positional isomers have similar behaviors, and pharmacokinetic studies in rats demonstrated that they have comparable half-life extensions. Overall, the study demonstrates that mTGase protein derivatization is linked to inherent advantages because it carries with it the possibility of targeting lysines or glutamines, in both cases with a high site-selective specificity.
Introduction
Methods of site-specific protein modification are important for the obtainment of homogeneous protein conjugates in several fields, such as polymer–protein and antibody–drug conjugates. In this direction, the approaches based on chemical methods, not involving genetic engineering, are mainly directed to an N-terminus, cysteine or disulphide bridge conjugation.1–4 Recently, new advances in polymer conjugation have been sought for the use of enzymes as a tool for site specific protein modification. Among the several proposed enzymes, we have focused our attention to microbial transglutaminase (mTGase) from Streptomyces mobaraensis. Just as other transglutaminases, mTGase catalyzes transamidation reactions of a protein substrate leading to a modification of the side-chains of Gln residues (R–CONH2, the acyl donor) and an amino group of an alkyl amine (R′–NH2, the amino donor) forming a new amide bond (R–CONH–R′).5 In nature, transglutaminases form intra- or inter-molecular protein cross-linkings involving the side chains of Gln and Lys residues. mTGase can, however, also catalyze the covalent attachment of the R′–NH2 amino-containing moiety in which R′ can be a fluorescent or a metal-chelating probe or even a polymer chain to specific amido groups (Gln residues) of the target proteins. Given this flexibility in the amino donor substrate and, at the same time, the strict site-specific requirements for the Gln substrate in a protein, mTGase has been used as a tool to generate well-defined protein–polymer conjugates. Several examples of mTGase-mediated polymer conjugation to the protein's Gln residues have been described.6–11 Interestingly, protein modification at the level of Lys residues by mTGase-mediated derivatization is also feasible, but it requires a ligand containing a Gln residue or a glutamine analogue. Being able to incorporate various chemical entities and/or functional groups at the level of protein-bound Gln or Lys residues seems to be an important feature of mTGase use that has enabled a variety of strategies and approaches for protein modification to be developed for basic research purposes and biotechnological applications.The suitability of mTGase for site selective mono-conjugates involving glutamines or lysines is investigated here using G-CSF, a protein, which regulates, in particular, neutrophil production, and is widely used to treat myelosuppressive chemo-therapy-induced neutropenia. As G-CSF is rapidly cleared from the body by both swift renal elimination and receptor mediated cell internalization, it requires repeated injections or continuous infusion to achieve sufficiently elevated neutrophil counts and stem cell mobilization in the peripheral blood.12 G-CSF is a widely studied protein for PEG conjugation, and site specific G-CSF PEGylation by N-terminal reductive amination,13 thiol PEGylation to Cys18,2 GlycoPEGylation14 and by exploiting a mTGase-mediated conjugation at Gln13515 has already been described. With regard to the last approach, a highly selective conjugation at Gln135 has been achieved despite the fact that the protein sequence contains a total of 17 Gln(s).16 The flexibility of the peptide chain embedding Gln135 protein permits recognition of the enzyme only at that site.17
The current study investigated the use of mTGase for site-specific derivatization of G-CSF at the level of Lys residues. G-CSF is a small monomeric protein of 175 amino acid residues that contains 4 Lys residues that can be potentially modified by mTGase. It is demonstrated here that it is possible to use mTGase to produce a mono-derivate isomer of G-CSF. The PEGylated G-CSF with a PEG20 kDa was characterized as far as its biophysical properties and pharmacokinetics were concerned and compared with a previously studied PEG20 kDa-G-CSF derivative in which the polymer was coupled to Gln135, PEG-Q135-G-CSF.
Results and discussion
In a precedential work we demonstrated that the mTGase-mediated selective PEGylation of several proteins is controlled by the high flexibility or local unfolding of the chain region that encompass the Gln substrate.17 Although some studies have demonstrated that the specificity for protein-bound Gln residues is fundamental, it is less important for protein-bound Lys residues.18 The current study focused on evaluating if the local environment of Lys residues in a protein can affect the selectivity of the mTGase for Lys residues.In the first attempt to generate a PEG substrate an as acyl donor, a simple PEG containing a γ-carboxyamide group was synthesized by reacting PEG-NH2 with glutaric anhydride, yielding a PEG-NHCO-(CH2)3-COOH derivative that in turn was treated with an ammonia solution in the presence of EDC/HOBt, forming PEG-NH-CO-(CH2)3-CO-NH2. Unfortunately, PEG-NH-CO-(CH2)3-CO-NH2 was not an acyl donor substrate for mTGase, probably because vicinal amino acids are necessary for a local rearrangement of the enzyme on the protein (data not shown). We thus chose to link PEG to the well-known ZQG enzyme substrate by activating the carboxylic group of glycine and in turn to couple it to PEG-NH2.
Synthesis of PEG-ZQG
The derivatization of PEG20 kDa-NH2 with the CBZ-QG substrate was carried out in the presence of organic solvents by activating the carboxylic group of the dipeptide with HOBT/EDC. Low molecular weight species were removed by dialysis. The absence of free PEG-NH2 in the PEG-ZQG was confirmed by the Snyder assay, which did not uncover any free amino groups. The identity of the PEG derivative was confirmed by 1H-NMR.mTGase-mediated PEGylation of G-CSF with PEG-ZQG
G-CSF was chosen as the model protein for two reasons: (i) the high selectivity of mTGase towards Q135 out of 17 Gln residues has already been reported by other authors as well as by us; (ii) it is an important pharmaceutical protein widely used in clinical practice and, in fact, a variety of effective G-CSF derivatives are presently available on the market.The conjugation reactions of G-CSF with PEG-ZQG were carried out by testing different G-CSF concentrations (1.0 and 2.0 mg ml−1), G-CSF/PEG molar ratios (1/5, 1/10 or 1/20) and pH values of reaction buffer (10 mM sodium phosphate buffer, pH 7.2 and 100 mM sodium borate buffer pH 9). mTGase/G-CSF ratios and incubation temperatures were unmodified in all cases (E/S 1/50 (w/w) and 25 °C). The reaction mixtures were analyzed by RP-HPLC after 0, 1, 2, 4, 24 and 48 hours of incubation and the peak areas of free and PEGylated G-CSF were compared in order to determine the highest yield. The best conditions were 2 mg ml−1 of G-CSF, 1/20 G-CSF/PEG molar ratio, 10 mM sodium phosphate buffer, pH 7.2 for 24 h of incubation, resulting in a conversion yield (based on the peak area of G-CSF and the conjugate in RP-HPLC) of 95%. The RP-HPLC analysis of the reaction performed under these optimized conditions is outlined in Fig. 1A. G-CSF eluting at 21.5 min decreased over time, while a new peak eluting at 19.4 min increased over time. The conjugate peak was characterized by MALDI-TOF mass spectrometry and SDS-PAGE detection was carried out with iodine and Coomassie Blue staining (Fig. 1B and C). The MALDI-TOF spectrum presents a product with a mass of 39![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 843.5 Da, which corresponds to the G-CSF conjugated to one chain of PEG20 kDa. SDS-PAGE showed a protein derivative at the apparent molecular weight of 60 kDa, while iodine staining confirmed the presence of PEG. The apparent MW in SDS-PAGE fits perfectly with that of monoPEGylated G-CSF; in fact, the PEG MW in SDS-PAGE is known to be overestimated.
843.5 Da, which corresponds to the G-CSF conjugated to one chain of PEG20 kDa. SDS-PAGE showed a protein derivative at the apparent molecular weight of 60 kDa, while iodine staining confirmed the presence of PEG. The apparent MW in SDS-PAGE fits perfectly with that of monoPEGylated G-CSF; in fact, the PEG MW in SDS-PAGE is known to be overestimated.
 | ||
| Fig. 1 Proof of conjugate formation. Panel (A) RP-HPLC profiles of PEG-ZQG and G-CSF reaction mixtures after 0, 2, 4 and 24 h. Panel (B) SDS-PAGE analysis, lane 1: markers, lane 2: G-CSF, lane 3: PEG-K41-G-CSF, (lanes 1, 2, 3 stained by using Coomassie Blue), lane 4: PEG-K41-G-CSF detected by iodine staining. Panel (C) MALDI-TOF mass spectrum of the peak at 19.5 min of the chromatogram reported in panel A. The peak at 39843 kDa is the PEGylated derivative and the peak at approximately 20 kDa is free PEG-ZQG. | ||
In order to preserve the native conformation of G-CSF, the PEGylated reaction mixtures were purified by cation exchange (CEX) chromatography (Fig. 2), dialyzed against 10 mM sodium acetate buffer at pH 4.7 containing 5% sorbitol and used for conformational and pharmacokinetic studies.
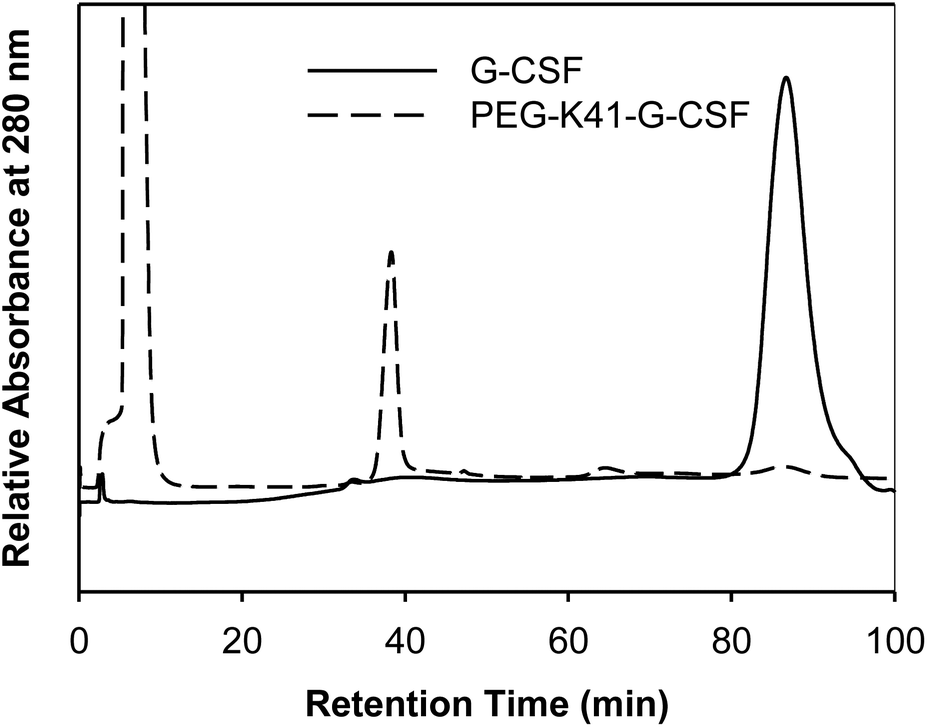 | ||
| Fig. 2 Purification of PEG-K41-G-CSF by CEX-HPLC. The peak eluting at 35 min corresponds to PEG-K41-G-CSF, the G-CSF eluted instead at 86 min. | ||
The purity of the two conjugates of G-CSF purified by CEX chromatography was verified by RP-HPLC (data not shown).
Identification of the sites of mTGase derivatization of G-CSF by mass spectrometry
In order to identify the Lys residues specifically modified by mTGase, the PEGylated G-CSF was purified by RP-HPLC; the peak corresponding to the conjugate was collected, lyophilized and then digested with trypsin. The native G-CSF also underwent the same procedure as a reference measure. The peptide mixtures of G-CSF and PEGylated G-CSF were analyzed by LC-MSE. The LC-MSE raw data were processed using BiopharmaLynx (Waters). ESI-LC-MSE peptide mapping of the digests made it possible to achieve a yield sequence coverage of 35.6% and 39.7% for PEGylated and free protein, respectively. This low sequence coverage can be attributed to the fact that trypsin digestion of G-CSF forms the 42–147 fragment (11.5 kDa) that was undetected by the ESI-LC-MSE method used. This fragment is nevertheless irrelevant because it does not contain free Lys residues. The fragments identified are outlined in Table 1. Three out of four fragments containing Lys residues (K17, K24, K35) were present in the digested mixture of the native and PEGylated proteins, while fragment 36–41 was not present in PEGylated G-CSF, thus confirming the selective modification of K41.| Tryptic peptide | Peptide sequence | Molecular mass (Da) | ||
|---|---|---|---|---|
| Found in G-CSFa | Found in PEG-G-CSFa | Calculatedb | ||
| a Experimental molecular mass determined by LC-MSE. b Molecular mass calculated by Expasy. c Fragment 36–41 was not detected in the mass spectrum of the tryptic digest of PEGylated G-CSF. | ||||
| 1–17 | MTPLGPASSLPQSFLLK | 1785.9698 | 1785.9728 | 1785.9698 |
| 18–23 | CLEQVR | 803.3960 | 803.3962 | 803.4032 |
| 24–35 | KIQGDGAALQEK | 1256.6725 | 1256.6776 | 1256.7633 |
| 36–41 | LCATYK | 745.3684 | —c | 754.3756 |
| 149–167 | AGGVLVASHLQSFLEVSYR | 2032.0741 | 2032.0825 | 2032.0776 |
| 149–170 | AGGVLVASHLQSFLEVSYRVLR | 2400.3276 | 2400.3342 | 2400.3345 |
| 171–175 | HLAQP | 546.3023 | 546.3020 | 564.3020 |
Providing further evidence of selective modification, MSE uncovered the presence of 1–17 and 24–35 fragments for both products with almost the complete b and y series, while the b/y fragment ions for the 36–41 fragments were present only in the G-CSF profile.
Although this protein presents four Lys residues (K17, K24, K35 and K41), we have demonstrated here that mTGase selectively recognizes only K41. This selectivity can be explained in connection with the structural features of the protein substrate. When the B-factor profile of G-CSF was being analysed, it became apparent that K41 is the most exposed Lys residue out of the four present in the protein (Fig. 3A and B). Indeed, K17 and K24 have low B-factor values. But although K35's B-factor value is comparable with that of K41's, it is preceded by E40 whose negative charge prevents the interaction with the mTGase catalytic site. Furthermore, the three-dimensional structure of G-CSF means that K17, K24, and K35 are embedded in an alpha-helix structure (Fig. 3C). Since the 39–45 fragment is a flexible region between the two alpha-helix bundles, it is more accessible to the catalytic site of mTGase.
 | ||
| Fig. 3 Sequence and three-dimensional structure of hG-CSF. (A) Amino acid sequence and secondary structure of hG-CSF (note that in this study rhG-CSF has been used, thus containing an additional methionine as the first amino acid). Alpha-helices are indicated by squiggled lines above the sequence. The B-turn is indicated in green segments. The dark lines indicate the disordered region; (B) profile of the B-factor of hG-CSF as determined from the X-ray structure of protein extracted by using PDB (code 1RHG). The regions 1–9, 61–71, 126–137, and 171–174 do not present B-factor values due to the fact that there is no electron density for them (very disordered regions); (C) 3D structure of hG-CSF derived from the X-ray structure of the protein (PDB code 1RHG). The approximate positions of lysines are labelled. | ||
Spectroscopy analyses
Far-UV CD of G-CSF shows the characteristic double minimum at 208 and 222 nm typical of an α-helical secondary structure. The protein's helical structure was not affected after modification with PEG20kDa according to the far-UV CD spectra of the G-CSF derivative, which was superimposable with that of the native protein. Melting studies demonstrated that protein stability was only slightly affected by PEGylation as the melting temperature decreased by 2 °C (Fig. 4A and B).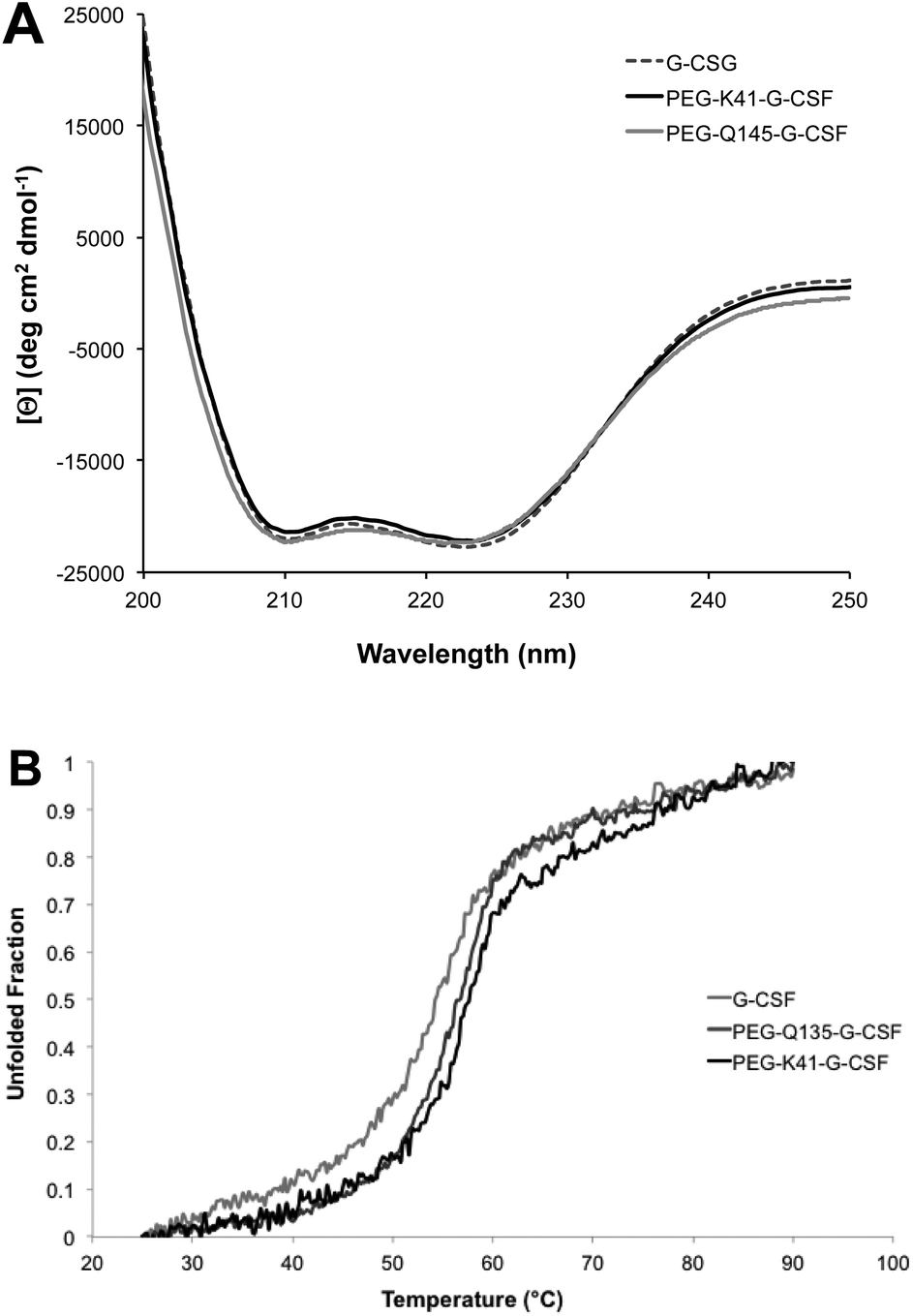 | ||
| Fig. 4 Characterization of the conjugate secondary structure. Far-UV CD (panel A) and melting CD (panel B) spectra of G-CSF, PEG-K41-G-CSF and PEG-Q135-G-CSF conjugates; the melting temperatures are shown in the insert of panel B. | ||
Intrinsic fluorescence due to the excitation of tryptophan showed superimposable spectra of free G-CSF and PEG-K41-G-CSF, with a slight, not significant, increase for PEG-Q135-G-CSF. Extrinsic fluorescence, instead, showed a significant decrease in the dye signal that can be attributed to a more compact structure of the conjugates with respect to free G-CSF. There was, however, no red or blue shift in the maximum extrinsic fluorescence peak, which suggests that protein hydrophobicity was not modified after the PEGylation process (Fig. 5A and B).
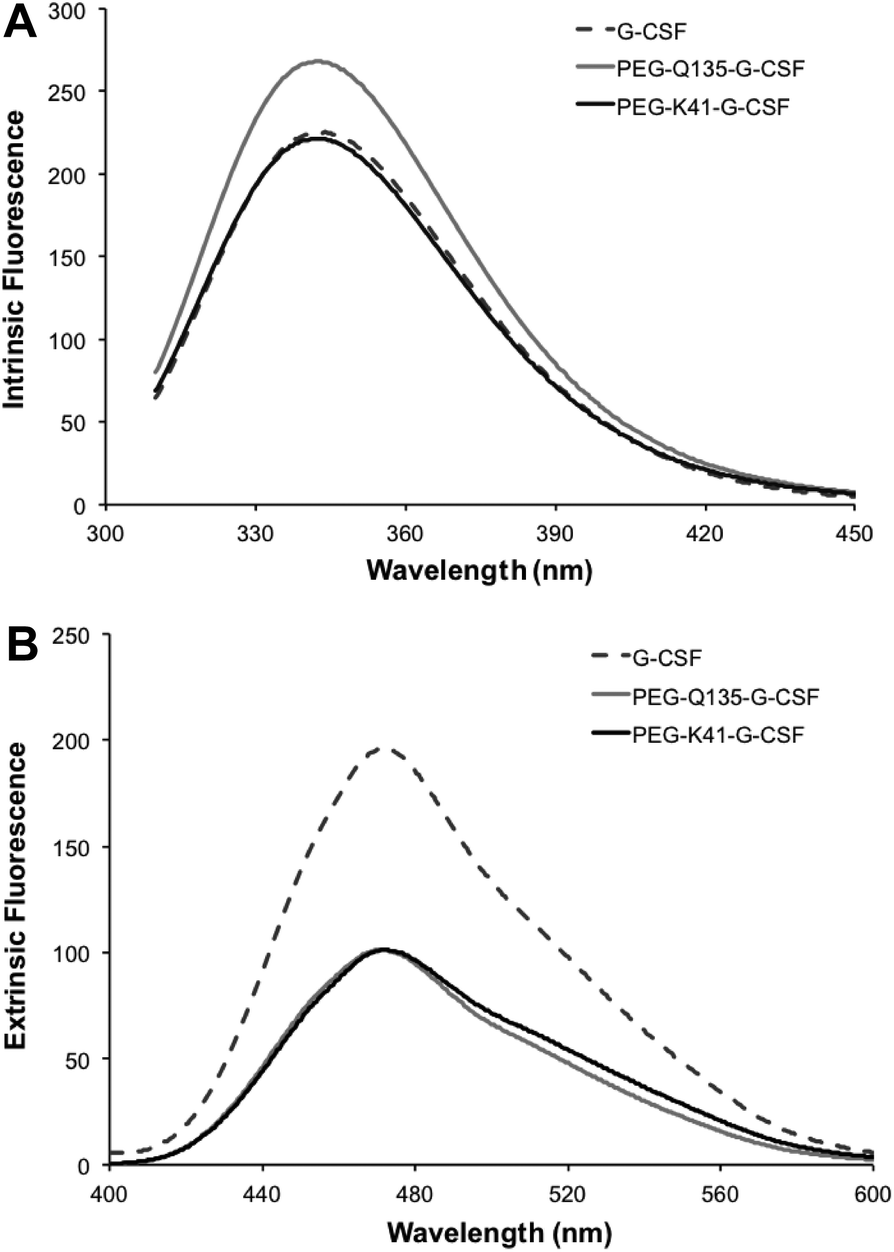 | ||
| Fig. 5 Investigation of the tertiary structure of the conjugate by fluorescence measurement. Intrinsic fluorescence (panel A) and extrinsic fluorescence (panel B) of G-CSF, PEG-Q135-G-CSF and PEG-K41-G-CSF. | ||
Stability studies
G-CSF stability after PEG conjugation was evaluated in 100 mM sodium phosphate, pH 7.4 at 37 °C. As shown in Fig. 6, PEG conjugates were more stable than the free protein, reducing G-CSF's aggregation propensity. Indeed after 48 h, 90% of the free protein was completely precipitated while more than 50% of both PEGylated G-CSF derivatives was still in solution. | ||
| Fig. 6 Aggregation tendency of free and conjugated G-CSF. The stability of G-CSF, PEG-K41-G-CSF and PEG-Q135-G-CSF incubated in 100 mM sodium phosphate, pH 7.4 after 0, 24 and 48 h at 37 °C. Data are presented as mean ± SD. *p < 0.05, conjugate vs. G-CSF; **p < 0.005, conjugate vs. G-CSF. | ||
Pharmacokinetic studies
Pharmacokinetic analysis of G-CSF, PEG-Q135-G-CSF and PEG-K41-G-CSF was carried out in rats after i.v. administration. The G-CSF content in serum samples was quantified by using a human G-CSF ELISA kit, and the pharmacokinetic data were analysed using PKSolver software after a bi-compartmental model was fitted.As shown in Fig. 7 and Table 2, PEGylation led to a prolonged residence time of modified G-CSF. The half-lives of PEG-K41-G-CSF and PEG-Q135-G-CSF were respectively 9.02 and 10.98 h, corresponding to about 3 and 3.7 fold with respect to the half-life of G-CSF (2.98 h). Bioavailability was likewise improved after PEGylation: PEG-K41-G-CSF and PEG-Q135-G-CSF showed an increase in the area under the curve (AUC) by approximately 2 to 2.4 fold respectively, compared to that of G-CSF. The clearance and the apparent volume of distribution of the PEGylated derivatives were reduced with respect to those of native G-CSF.
 | ||
| Fig. 7 Pharmacokinetic investigation in rats. Pharmacokinetic profiles of G-CSF, PEG-Q135-G-CSF and PEG-K41-G-CSF in Sprague Dawley rats (n = 3 per group) after the i.v. administration of 100 μg kg−1 G-CSF (protein equiv.). Data are presented as mean ± SD. The y-axis is presented as a logarithmic scale. | ||
| Product | t½ α (h) | t½ β (h) | AUC (0 → ∞) (μg min mL−1) | Cl (mL h−1) | VD (mL) |
|---|---|---|---|---|---|
| G-CSF | 2.98 | 2.98 | 7.04 | 2.83 | 10.28 |
| PEG-K41-G-CSF | 0.73 | 9.02 | 14.9 | 1.33 | 5.64 |
| PEG-Q135-G-CSF | 1.12 | 10.98 | 17.12 | 1.16 | 5.96 |
The study of the pharmacokinetics of PEG-K41-G-CSF and PEG-Gln135-G-CSF conjugates demonstrated that the site of PEG coupling did not affect the pharmacokinetic performance of the conjugates and prolonged the residence times in a similar manner with respect to G-CSF.
Experimental
Materials
G-CSF was a kind gift from Sandoz (Ljubljana, Slovenia). Trypsin of sequencing grade, carbobenzoxy-L-glutaminylglycine (ZQG), dansylcadaverine (DC), bis-ANS (4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid, dipotassium salt) and all other chemicals were purchased from Sigma-Aldrich (Milwaukee, WI, USA). PEG-NH2 was obtained from NOF Corporation (Tokyo, Japan). mTGase, of Streptomyces mobaraensis origin (ACTIVA M), was supplied by Ajinomoto Co. (Tokyo, Japan). Human G-CSF ELISA Kit was provided by Life Technologies (Waltham, MA, USA). Stock solutions of mTGase were prepared by dissolving the mTGase ACTIVA MP powder in 0.1 M sodium phosphate buffer pH 7.0 to obtain a protein concentration of approximately 1 mg ml−1, as determined by UV spectroscopy. Typically, 1 ml of phosphate buffer was added to 200 mg of mTGase powder and after vortexing for 5 min and centrifuging for 10 min at 13200 rpm, the protein concentration was measured using the supernatant. The solution was then divided into aliquots and stored at −80 °C until they could be used. The G-CSF concentration and the mTGase solutions were determined on the basis of the absorbance at 280 nm using the protein's extinction coefficient generated by ProtParam (http://www.expasy.org/tools/protparam.html; absorbance at 280 nm of a solution of 1 g l−1 is 0.78 for G-CSF and 1.89 for mTGase).
Synthesis of PEG-ZQG
PEG-ZQG was synthesized starting from PEG-NH2; 19.2 mg (0.1 mmol) of EDC (MW 191.7 Da) and 13.5 mg (0.1 mmol) of HOBT (MW 135.13 Da) were added to 13.5 mg (40 μmol) of ZQG (MW 337.33 Da) previously dissolved in 5 mL of a 0.1 M borate buffer/CH3CN (3:2) mixture pH 8.0. After 1 hour, 200 mg (10 μmol) of PEG-NH2 (MW 20 kDa) was added, and the pH of the solution was adjusted to 8. The reaction was allowed to stir at room temperature for 18 hours, and then 2.52 mg (25 μmol) of succinic anhydride (MW 100.08 Da) was added to the mixture. The solution was dialyzed for 24 hours against Milli-Q water and the product PEG20 kDa-Gln was then lyophilized (yield: 187 mg, 94%). The absence of free amino groups was verified using the TNBS-based test according to the Snyder and Sobocinski assays.19 The derivatization degree was evaluated by 1H-NMR: (CDCl3, δ ppm) PEG: 3.36 (s, 3H), 3.63 (s, 480H), ZQG: 7.5 (m, 5H), 5.20 (s, 2H), 2.06 (m, 4H).
mTGase-mediated conjugation of G-CSF with PEG-ZQG and PEG-NH2
PEG-Q135-G-CSF was prepared, as was reported previously,20,21 and then characterized by comparing it with the G-CSF conjugated PEG-ZQG.For PEG-ZQG conjugation to Lys residues of G-CSF: stock solution of G-CSF (∼4.6 mg ml−1) in 10 mM sodium acetate buffer pH 4.7 containing 5% sorbitol was diluted with 0.1 M phosphate buffer, pH 7.2 or 0.1 M borate buffer pH 9.3. A molar excess of PEG-ZQG of 1:5, 1:10 and 1:20 with respect to G-CSF was used. The reaction mixtures were prepared by dissolving the PEG polymer in the volume of buffer needed to dilute the G-CSF stock solution to 1 or 2 mg ml−1. The PEG solution was added under stirring to the G-CSF solution, and an aliquot of the reaction mixture was taken before mTGase was added at time 0 h. The stock solution of mTGase was added in order to obtain an E/S ratio of 1/50 by weight between mTGase/G-CSF. The reactions were incubated at 25 °C under magnetic stirring and were quenched after 1, 2, 4, 24 and 48 h of incubation upon addition of a stock solution of iodoacetamide at a molar ratio with mTGase of 30/1 (iodoacetamide/mTGase).
The aliquots of the reaction mixtures were analyzed by RP-HPLC on an Agilent series 1260 HPLC with online UV detection from Agilent Technologies. RP-HPLC analyses were performed using a C18 Jupiter column (300 Å, 5 μm, 250 × 4.60 mm) applying a gradient of acetonitrile (ACN), containing 0.1% trifluorocetic acid (TFA) and water, 0.1% TFA from 5 to 40% of ACN in 5 min and from 40 to 70% in 25 min. The column was eluted at a flow rate of 1 ml min−1 and the absorbance was read at 226 nm.
The reaction mixtures of G-CSF presenting the highest degree of protein PEGylation were loaded on a TSK-gel SP 5-PW column (300 Å, 7 μm, 7.5 × 0.75 cm) for purification. The chromatographic separation was performed using an Agilent series 1260 HPLC. The cation exchange column was eluted with a gradient of buffer A (10 mM sodium phosphate pH 4.7) and B (0.1 M sodium phosphate, 0.1 M NaCl pH 4.85). After the sample was injected, the column was eluted with 5% B for 10 min and then with a gradient of B from 5% to 65% in 50 min and from 65% to 100% in 20 min. The column was then washed for 10 min with 100% B and for 5 min with 5% B. The flow rate was 1 ml min−1 and the effluent absorbance was recorded at 226 nm.
The G-CSF conjugate peak was collected from the cation exchange column and dialyzed at 4 °C against 10 mM sodium acetate buffer, pH 4.7 containing 5% sorbitol using a dialysis membrane with a MWCO of 6–8000 Da (Spectra/Por Dialysis Membrane). Then the solution was concentrated using Amicon Ultra-4 10 K (Millipore). The purified PEGylated G-CSF was characterized as outlined below.
Using a native protein as reference, 2 mg of G-CSF was dialyzed at 4 °C against 10 mM sodium phosphate pH 4.7 and then subjected to cation exchange chromatography, dialysis, and ultrafiltration, as described above for PEGylated G-CSF.
Proteolytic digestion of PEG-G-CSF and LC-MSE analyses
Native and PEGylated proteins (200 μg) purified by RP-HPLC were lyophilized and dissolved in 6 M Gdn-HCl, 50 mM Tris-HCl, pH 9.0, to reach a protein concentration of approximately 1 mg mL−1. TCEP was added to the protein solution at a final concentration of 5 mM, and the reaction mixture was kept for 1 h at 37 °C. The reduced protein was then reacted with 25 mM iodoacetamide, and the S-alkylation was allowed to proceed for 30 min at 37 °C in the dark. Protein samples were purified by RP-HPLC on a Zorbax C18 column, as reported above. The reduced and S-carboxamidomethylated samples of G-CSF and PEG-G-CSF were dissolved in 8 M urea and diluted in 0.1 M ammonium carbonate buffer, pH 7.8, to reach a final protein concentration of 0.8 mg mL−1 and a urea concentration of 0.8 M. An aliquot from a stock solution of trypsin was then added at an E/S ratio of 1:50 (by weight) and the proteolysis was allowed to proceed at 37 °C overnight. The digestion mixtures, desalted by PepClean C-18 Spin columns (Pierce, Rockford, IL), were directly analyzed using an UPLC-Q-TOF mass spectrometry method with a Grace Vydac TP C18 column (150 × 1 mm; 5 μm), maintained at 32 °C, flow-rate 0.05 mL min−1, detection at 280 nm, eluted with a solvent gradient of water/ACN both containing 0.1% formic acid. Gradient 3′–3% ACN, 24′–80% ACN, 28′–80% ACN, 29′–3% ACN, 35′–3%. The Xevo G2-S Q-Tof was operated in the ESI positive ion, resolution mode and with a detection window between 50–2000 m/z. Source parameters were: capillary (kV) 1.5, sampling cone voltage 30.0 V and source offset of 80 V. MSE acquisition was performed by alternating two MS data functions: one for acquisition of the peptide mass spectra with the collision cell at low energy (6 V) and the other for the collection of the peptide fragmentation spectra with the collision cell at elevated energy (linear ramp 20 to 40 V). Analyses were performed with LockSpray™ using a solution of 1 ng μL−1 Leu-Enk in 50:50 acetonitrile/water containing 0.1% formic acid, sampled every 45 s. MSE data were processed with the BiopharmaLynx 1.3.4 Software (Waters) setting Glu-C as a digest reagent and 5 missed cleavages. The MS ion intensity threshold was set to 250 counts, and the MSE threshold was set to 100 counts. Both MS mass match tolerance and MSE mass match tolerance were set to 15 ppm. Conjugated peptides were confirmed by at least four MS/MS b/y fragment ions.
Mass spectrometry analysis
MALDI-MS analyses were performed using a REFLEX time-of-flight instrument (4800 Plus MALDI TOF/TOF, AB Sciex, Framingham, Massachusetts, USA) equipped with a SCOUT ion source operating in positive linear mode. The ions generated by a pulsed UV laser beam (nitrogen laser, λ 337 nm) were accelerated to 25 kV. A saturated solution of sinapinic acid in water/ACN (1:1, v/v) was used as a matrix and mixed with the samples dissolved in 0.1% TFA aqueous solution at a v/v ratio 1:1.
Spectroscopy analyses
| Θ = Θobs(MRW)/10L[C] |
Thermal denaturation of the free and conjugated G-CSF was monitored on the same samples used for CD analysis by recording the decrease in the ellipticity signal at 222 nm as a function of temperature. Denaturation experiments were carried out in a 0.1 cm cell path length heated between 25 and 90 °C at the protein concentration of 0.1 mg mL−1.
Fluorescence measurement
Intrinsic fluorescence emission spectra were measured with a Jasco FP-650 spectrofluorometer equipped with a thermostatted cell holder at 25 °C. G-CSF (final protein conc. 0.1 mg mL−1) tryptophan was excited at 290 nm and the emission was recorded in the 305–450 nm range. Data were collected at 1 nm increments with a slit width of 5 nm. Extrinsic fluorescence was measured using bis-ANS as a dye. Fifty mg mL−1 of bis-ANS was prepared in NaOH 0.1 M. One μL of the dye solution was added to 500 μL of protein solution (0.1 mg mL−1). The sample was excited at 380 nm, and the emission spectrum was in the 400–600 nm range.Stability study
G-CSF, PEG-K41-G-CSF and PEG-Q135-G-CSF were diluted from stock solutions to 100 mM sodium phosphate, pH 7.4 at a concentration of 1 mg mL−1. The protein concentrations were measured at 280 nm. Samples were incubated at 37 °C in a mixer rotating at 300 rpm, removed after 24 and 48 h of incubation, and centrifuged for 10 min (12000g) to remove insoluble aggregates. The samples were filtered through a 0.2 μm filter, and the soluble protein fractions were analyzed by size exclusion chromatography using an Agilent series 1260 HPLC. The GF-250 Zorbax column (300 A, 4.6 × 25 cm, 5 μm) was used for elution with 0.1 M sodium phosphate buffer, 0.1 M NaCl, pH 7.2. The flow rate was 0.3 ml min−1 and the absorbance was read at 226 nm. Peak areas in the chromatograms corresponding to the monomer or soluble aggregates were used to determine the type of aggregation.
Pharmacokinetic study
Pharmacokinetic profiles of PEG-K41-G-CSF, PEG-Q135-G-CSF and G-CSF were determined in Sprague-Dawley rats (250–350 g, three rats per group). A dose of 100 μg kg−1 G-CSF (equiv.) was administered via tail vein to the rats anesthetized with 5% isoflurane gas (mixed with O2 in enclosed cages). At predetermined times, blood samples were collected from the tail vein and centrifuged at 1500g for 15 min. The G-CSF content in the serum samples was quantified using a Human G-CSF ELISA Kit (Life Technologies). Pharmacokinetic data were analyzed using PkSolver software after a bi-compartimental model was fitted.22Ethics statement
The study protocol was approved by the Ethics Committee of the University of Padova and the Italian Ministry of Health (CEASA 24/2013), and the animals were handled in compliance with National (Italian) Legislative Decree 116/92 guidelines and the “Guide for the Care and Use of Laboratory Animals” by the National Research Council of the National Academies.Conclusions
mTGase has been demonstrated to be a valuable tool for double site-selective protein modification. In fact, depending on the substrate used in excess, i.e. PEG-NH2 as an amino donor or PEG-ZQG as an acyl donor, the selective derivatization of proteins can be directed to Gln or Lys residues. The presence of Gln and Lys residues in flexible protein loops is a specific requirement for being a mTGase substrate which allows the amino acids to interact with the catalytic site of the enzyme. mTGase can thus offer relevant opportunities in the field of protein derivatization and polymer conjugation offering at the same time flexibility to the potential site of conjugation (Lys or Gln residues) and high selectivity. We and other labs have already demonstrated that PEG is not the only polymer that can be used in mTGase mediated polymer conjugation. For example, poly(2-ethyl-2-oxazoline)21 and hydroxyl-ethyl-starch23 containing an amino group were also conjugated to glutamines of proteins. Therefore, it can be expected that these polymers and others, such as polysialic acid,24 can also be adapted to mTGase mediated conjugation to lysines, thus widening the possibilities of protein modifications.It will be relevant to investigate the feasibility of this new approach with other therapeutic proteins and especially monoclonal antibodies (MAbs) for the preparation of site specific conjugated ADC, although our preliminary results with MAbs seems to exclude the presence of lysines as a substrate of mTGase.
Acknowledgements
The research was supported in part by the Italian Ministry of Education, University and Research (MIUR; PRAT CPDA135898/13 and ex60% 60A04-9512), the Italian Ministry of Health (“Ricerca Finalizzata” GR2011 – 02351128) and AIRC (MFAG 15458).Notes and references
- O. B. Kinstler, D. N. Brems, S. L. Lauren, A. G. Paige, J. B. Hamburger and D. Treuheit, Pharm. Res., 1996, 13, 996 CrossRef CAS.
- F. M. Veronese, A. Mero, F. Caboi, M. Sergi, C. Marongiu and G. Pasut, Bioconjugate Chem., 2007, 18, 1824 CrossRef CAS PubMed.
- S. Balan, J. W. Choi, A. Godwin, I. Teo, C. M. Laborde, S. Heidelberger, M. Zloh, S. Shaunak and S. Brocchini, Bioconjugate Chem., 2007, 18, 61 CrossRef CAS PubMed.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, J. Am. Chem. Soc., 2012, 134, 1847 CrossRef CAS PubMed.
- J. Folk, Adv. Enzymol. Relat. Areas Mol. Biol., 1983, 54, 1 CAS.
- H. Sato, E. Hayashi, N. Yamada, M. Yatagai and Y. Takahara, Bioconjugate Chem., 2001, 12, 701 CrossRef CAS PubMed.
- J. Buchardt, H. Selvig, P. F. Nielsen and N. L. Johansen, Pept. Sci., 2010, 94, 229 CrossRef CAS PubMed.
- A. Mero, M. Schiavon, F. M. Veronese and G. Pasut, J. Controlled Release, 2011, 154, 27 CrossRef CAS PubMed.
- D. da Silva Freitas, A. Mero and G. Pasut, Bioconjugate Chem., 2013, 24, 456 CrossRef CAS PubMed.
- B. Spolaore, N. Damiano, S. Raboni and A. Fontana, Bioconjugate Chem., 2014, 25, 470 CrossRef CAS PubMed.
- A. Grigoletto, A. Mero, I. Zanusso, O. Schiavon and G. Pasut, Macromol. Biosci., 2016, 16, 50 CrossRef CAS PubMed.
- G. Morstyn, L. Souza, J. Keech, W. Sheridan, L. Campbell, N. Alton, M. Green, D. Metcalf and R. Fox, Lancet, 1988, 331, 667 CrossRef.
- O. Kinstler, G. Molineux, M. Treuheit, D. Ladd and C. Gegg, Adv. Drug Delivery Rev., 2002, 54, 477 CrossRef CAS PubMed.
- J. Hoggatt, T. A. Tate and L. M. Pelus, Int. J. Nanomed., 2015, 10, 2647 CAS.
- A. Mero, B. Spolaore, F. M. Veronese and A. Fontana, Bioconjugate Chem., 2009, 20, 384 CrossRef CAS PubMed.
- C. Maullu, D. Raimondo, F. Caboi, A. Giorgetti, M. Sergi, M. Valentini, G. Tonon and A. Tramontano, FEBS J., 2009, 276, 6741 CrossRef CAS PubMed.
- A. Fontana, B. Spolaore, A. Mero and F. M. Veronese, Adv. Drug Delivery Rev., 2008, 60, 13 CrossRef CAS PubMed.
- P. J. Coussons, N. C. Price, S. M. Kelly, B. Smith and L. Sawyer, Biochem. J., 1992, 282, 929 CrossRef CAS PubMed.
- S. L. Snyder and P. Z. Sobocinski, Anal. Biochem., 1975, 64, 284 CrossRef CAS PubMed.
- A. Grigoletto, Ph.D. Thesis, University of Padova, 2015.
- A. Mero, Z. Fang, G. Pasut, F. M. Veronese and T. X. Viegas, J. Controlled Release, 2012, 159, 353 CrossRef CAS PubMed.
- Y. Zhang, M. Huo, J. Zhou and S. Xie, Comput. Methods Programs Biomed., 2010, 99, 306 CrossRef PubMed.
- A. Besheer, T. C. Hertel, J. Kressler, K. Mäder and M. Pietzsch, J. Pharm. Sci., 2009, 98, 4420 CrossRef CAS PubMed.
- G. Gregoriadis, S. Jain, I. Papaioannou and P. Laing, Int. J. Pharm., 2005, 300, 125 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |