
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUreido cytosine and cytosine-containing acrylic copolymers†
Keren
Zhang
 ,
Mingtao
Chen
,
Kevin J.
Drummey
,
Samantha J.
Talley
,
Lindsey J.
Anderson
,
Robert B.
Moore
and
Timothy E.
Long
*
,
Mingtao
Chen
,
Kevin J.
Drummey
,
Samantha J.
Talley
,
Lindsey J.
Anderson
,
Robert B.
Moore
and
Timothy E.
Long
*
Department of Chemistry, Macromolecules Innovation Institute, Virginia Tech, Blacksburg, VA 24061, USA. E-mail: telong@vt.edu
First published on 12th October 2016
Abstract
Regioselective Michael addition afforded a novel N1-substituted cytosine acrylate monomer for the synthesis of acrylic random copolymers with cytosine pendant groups. Quantitative post-functionalization converted cytosine to ureido-cytosine (UCy) with an increased self-association strength due to quadruple hydrogen bond formation. Thermogravimetric analysis (TGA) revealed a lower onset temperature of weight loss (∼200 °C) for UCy-containing copolymers, however, they proved to be more thermally stable at ≤130 °C than the cytosine-containing precursors during isothermal rheological experiments. The incorporation of UCy into random copolymers resulted in higher Tgs, enhanced mechanical performance, and better microphase-separation than the cytosine-containing precursors. Both dynamic mechanical analysis and rheological analysis revealed a plateau regime for each UCy-containing copolymer as well as a tan delta transition that corresponded to hydrogen bond dissociation. In contrast, the viscoelastic behavior of cytosine-containing random copolymers resembled entangled, non-associating polymers with increasing Tg as the cytosine content increased. A solution-cast UCy-containing copolymer film exhibited a more well-defined surface morphology with nano-fibrillar hard domains compared to the cytosine control. Variable temperature FTIR spectroscopy verified the presence of hydrogen bonding, and thermogravimetric sorption analysis (TGA-SA) compared the water uptake of UCy and cytosine-containing copolymers. UCy-containing random copolymers showed various advantages for applications as adhesives and thermoplastic elastomers compared to the cytosine copolymers, including superior cohesive strength, higher thermal stability, wider service temperature window, and lower moisture uptake. Free radical copolymerization of a quadruple hydrogen bond containing acrylic monomers provides a versatile avenue to supramolecular polymers with a tunable composition and improved scalability compared to earlier telechelic oligomers. This report describes the first synthesis of an acrylic monomer family and complementary evidence for tunable association in random copolymers.
Introduction
Supramolecular polymers serve as processable functional materials with reversibility and thermo-responsiveness for many applications including in self-healing materials, adhesives, biomedical materials, templates for controlled polymerization, and optoelectronic materials.1–12 Two main types of supramolecular polymers have received significant attention: step-growth polymerization of noncovalent associating oligomers or monomers, and physically crosslinked polymers with noncovalent associating groups.1 Noncovalent interactions such as hydrogen bonding, ionic interaction, and π–π stacking dominate both solution and solid-state properties for both types of supramolecular polymers. The former type have attracted more academic interest in recent years due to their unique structure and self-healing capability compared to conventional synthetic polymers. However, the latter type of supramolecular polymer offers more potential for the commercialization of new materials due to facile formulation using traditional synthetic methods. In fact, many commercialized polymers fall into the second category of supramolecular polymers, such as Surlyn™ (poly(ethylene-co-methacrylate salt)), Nafion™ (sulfonated polytetrafluoroethylene), and pressure sensitive adhesives (PSA) with hydrogen bonding monomers.10,13–16 The synthesis of pendant functionalized supramolecular copolymers involves the incorporation of one or multiple functional comonomers. Thus, varying the monomer molar ratio and adding/replacing comonomers enable the fine tuning of copolymer compositions, including the molecular weight, associating unit concentration, and sequence distribution. This cost-effective strategy produces new copolymer compositions for diverse commercial impacts, ranging from pressure sensitive adhesives to thermoplastic elastomers. A random copolymerization strategy is readily amenable to commercially viable processes in a range of concentrations in contrast to telechelic systems where the associative group concentration scales with the oligomer molecular weight. In addition, controlled polymerization techniques with associating monomers further extend the tunability of supramolecular polymer architectures for the unique self-assembly of block copolymers.17–21Nature provides many biological supramolecular polymers with pendant associating groups, including proteins and nucleic acids. Cytosine, in combination with the other three nucleobases, encodes DNA using its complementary triple hydrogen bonding to guanine with a higher association constant (>104 M−1) compared to a significantly weaker self-association (ca. 40 M−1).22,23 However, researchers have more extensively investigated the adenine–thymine base pair and the effects on synthetic polymer properties due to the challenging solubility and multiple nucleophilic sites for cytosine and guanine.18,24–29 Hailes and coworkers recently synthesized and investigated the quadruple hydrogen bonding of ureido-cytosine (UCy).23 UCy contains an array of hydrogen bonding AADD (A: acceptor, D: donor) and affords quadruple self-hydrogen bonding, similar to the well-known ureido-pyrimidine (UPy) prominently researched by the Meijer group.3 Ureido-cytosine self-associates into dimers through quadruple hydrogen bonding with a lower binding constant (ca. 2.5 × 105 M−1) than UPy (ca. 107 M−1) in CDCl3.2 The relatively weak hydrogen bonding between the 5-H of cytosine and the urea carbonyl results in an equilibrium between folded and unfolded stereoisomers, and only the unfolded configuration permits the AADD array. Hydrogen bonding at a similar position of UPy drives the equilibrium towards the unfolded conformer, resulting in a higher binding constant that matches the theoretical prediction of quadruple binding arrays of AADD.4,23,30 UPy incorporation afforded both types of supramolecular polymers through step-growth and free radical polymerization.4,31–33 Hailes et al. synthesized UCy-functionalized telechelic supramolecular polymers. Moreover, Cheng and coworkers prepared UCy-functionalized telechelic poly(propylene glycol) (PPG) oligomers, which self-assembled into semicrystalline supramolecular polymers and exhibited self-healing properties.34–36 UCy proved efficient in forming step-growth supramolecular polymers. However, the chain-end functionalization of oligomers largely limits the tunability of supramolecular polymer architectures and scalability. Challenges remain for the impact of telechelic supramolecular polymer synthesis due to more sophisticated polymerization processes at the commercial scale. Thus, conventional radical polymerization of a ureido-cytosine monomer allows a scalable synthetic strategy for pendant group-associating supramolecular polymers simply through tuning comonomer molar ratios in traditional radical polymerization copolymer processes.
This manuscript reports the unprecedented and facile synthesis of acrylic cytosine and ureido-cytosine monomers. This study also details the synthesis and characterization of cytosine and UCy-containing random acrylic copolymers with tunable physical properties, ranging from pressure sensitive adhesives to film-forming films and coatings. Michael addition afforded a regioselective synthesis for the polymerizable cytosine-functionalized acrylate monomer (CyA), which reacted with isocyanates to yield a UCy-functionalized acrylate monomer (UCyA). Free radical polymerization of CyA and n-butyl acrylate (nBA) with varying feed ratios prepared a series of CyA-containing random copolymers with subsequent post-functionalization affording the corresponding UCyA-containing random copolymers. The acrylic backbone formed a low Tg soft matrix, and the flexible spacer from the backbone to cytosine facilitated hydrogen bonding. Two series of copolymers provided direct comparison between weak and strong hydrogen bonding groups to investigate their effects on random copolymer properties. Systematic physical characterization established the structure–morphology–property relationships of CyA and UCyA-containing random copolymers, including thermal, rheological, thermomechanical, and morphological analyses. This study represents the first supramolecular polymers with quadruple hydrogen bonded pendant groups synthesized through post-functionalization of cytosine-containing polyacrylates. UCyA proved more efficient in forming physical crosslinks and the enhancing mechanical strength of random copolymers compared to CyA. UCyA-containing random acrylics enable potential applications as pressure sensitive adhesives and thermoplastic elastomers.
Experimental section
Materials
n-Butyl acrylate (nBA, >99%) was purchased from Aldrich and passed through a neutral alumina column before use. α,α′-Azobis(isobutyronitrile) (AIBN, Fluka, 99%) was recrystallized twice from methanol. 1,4-Butanediol diacrylate (Alfa Aesar, 99%) was used without further purification. Cytosine (C, 99%), triethylamine (TEA, 99%), 4-ethylphenyl isocyanate (98%), and 2,6-di-tert-butyl-4-methylphenol (BHT, 99%) were purchased from Aldrich and used without further purification. Hexane (HPLC grade), chloroform (CHCl3, HPLC), dichloromethane (DCM, ACS grade), dimethylsulfoxide (DMSO, HPLC grade), methanol (MeOH, ACS grade), and N,N-dimethylformamide (DMF, HPLC grade, anhydrous) were purchased from Spectrum Chemicals and used as received.Analytical methods
1H NMR and 13C NMR spectra were collected in CDCl3, or DMSO-d6, or a mixture of CDCl3 and DMSO-d6 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) on an Agilent U4-DD2 spectrometer operating at 400 MHz and 23 °C. High resolution TOF mass spectroscopy (HRMS) for the CyA and UCyA monomers was conducted in the positive ion mode on an Agilent 6220 mass spectrometer with a TOF analyzer. Thermogravimetric analysis (TGA) of UCy and cytosine-containing copolymers was performed on a TA Instruments Q50 TGA with a heating ramp from ambient temperature to 600 °C at a heating rate of 10 °C min−1 under constant nitrogen purging. Td,5 wt% corresponded to the temperature at 5% weight loss of the initial sample weight. The first step weight loss was measured from ambient temperature to the temperature where the first derivative of weight loss versus temperature reached a minimum. Differential scanning calorimetry (DSC) of UCy and cytosine-containing copolymers was conducted on a TA Instruments Q1000 DSC using a heat/cool/heat procedure with a heating rate of 10 °C min−1 and a cooling rate of 10 °C min−1 under a nitrogen flush of 50 mL min−1. The midpoint of the transition in the second heating ramp determined the glass transition temperatures (Tg).
:1, v/v) on an Agilent U4-DD2 spectrometer operating at 400 MHz and 23 °C. High resolution TOF mass spectroscopy (HRMS) for the CyA and UCyA monomers was conducted in the positive ion mode on an Agilent 6220 mass spectrometer with a TOF analyzer. Thermogravimetric analysis (TGA) of UCy and cytosine-containing copolymers was performed on a TA Instruments Q50 TGA with a heating ramp from ambient temperature to 600 °C at a heating rate of 10 °C min−1 under constant nitrogen purging. Td,5 wt% corresponded to the temperature at 5% weight loss of the initial sample weight. The first step weight loss was measured from ambient temperature to the temperature where the first derivative of weight loss versus temperature reached a minimum. Differential scanning calorimetry (DSC) of UCy and cytosine-containing copolymers was conducted on a TA Instruments Q1000 DSC using a heat/cool/heat procedure with a heating rate of 10 °C min−1 and a cooling rate of 10 °C min−1 under a nitrogen flush of 50 mL min−1. The midpoint of the transition in the second heating ramp determined the glass transition temperatures (Tg).
Rheological analyses of UCy and cytosine-containing copolymers were conducted on a TA Instruments Discovery AR-G2 rheometer, using disposable aluminum parallel plates 8 mm in diameter. All measurements were strain-controlled at constant nominal strain values within the linear viscoelastic region, as determined using strain-sweep experiments. Dynamic oscillatory temperature sweep experiments were performed at temperature steps from −40 °C to 130 °C with a 3 °C interval at 1 Hz. The sample was equilibrated for 1 min at each temperature. The maximum of the tan delta curve determined the Tg. Dynamic mechanical analysis (DMA) of annealed UCy and cytosine-containing copolymer films utilized a TA Instruments Q800 dynamic mechanical analyzer in the tension mode at a frequency of 1 Hz, an oscillatory amplitude of 10 μm, and a static force of 0.01 N. Rectangular film samples were cooled to −80 °C, equilibrated for 2 min, their length measured, and subjected to a 3 °C min−1 temperature ramp. Tg values were reported as the maxima of tan delta curves. A Veeco MultiMode scanning probe microscope provided the AFM phase images of UCy and cytosine-containing copolymer films using the tapping-mode. Samples were imaged with Veeco's Nanosensor silicon tips, with a spring constant of 42 N at an approximately 0.5 set-point ratio.
Variable temperature FTIR (VT-FTIR) experiments were performed using a Varian 670-IR spectrometer (DTGS detector) with a Pike Technologies variable temperature GladiATR™ attachment (diamond crystal). The spectra were collected at 4 cm−1 resolution and as an average of 32 scans. The samples were subjected to a temperature ramp of 1 °C min−1, starting from 30 °C to 180 °C and FTIR spectra were collected every 10 °C beginning from 30 °C. A TA Instruments Q5000 thermogravimetric sorption analyzer (TGA-SA) measured the water sorption at relative humidity (RH) steps from 0–95% RH with a 5% increment. Each RH step continued until the sample weight equilibrated (<0.01% change over 10 min). An instrumental pre-drying method at 50 °C and 0% RH was employed for each sample until the weight equilibrated. Water sorption was calculated based on the weight gain of each pre-dried sample weight.
Small angle X-ray scattering (SAXS) experiments were performed using a Rigaku S-Max 3000 3 pinhole SAXS system, equipped with a rotating anode emitting X-rays with a wavelength of 0.154 nm (Cu Kα). The sample-to-detector distance was 1603 mm for SAXS, and the q-range was calibrated using a silver behenate standard. Two-dimensional SAXS patterns were obtained using a 2D multiwire, proportional counting, gas-filled detector, with an exposure time of 1 h. The SAXS data were corrected for sample thickness, and the scattering profiles were vertically shifted to facilitate a comparison of peak positions. All the scattering data were analyzed using the SAXSGUI software package to obtain radially integrated SAXS intensity versus the scattering vector q (SAXS), where q = (4π/λ)sin(θ), θ is one half of the scattering angle and λ is the X-ray wavelength.
Synthesis of the 4-((3-(cytosin-1-yl)propanoyl)oxy)butyl acrylate (cytosine acrylate, CyA) monomer (Scheme 1)
A suspension of cytosine (5.0 g, 45.0 mmol), TEA (1.5 g, 15.0 mmol), BHT (0.3 g), and 1,4-butanediol diacrylate (13.4 g, 68.0 mol) in DMSO (100 mL) was stirred at 50 °C for 24 h. The reaction mixture was poured into water (750 mL) and washed with hexane to remove excess diacrylate. The water layer was extracted with dichloromethane (3 × 100 mL). The combined extracts were dried over MgSO4, filtered, and concentrated in a rotary evaporator to remove all solvents. The evaporation residue was purified using flash column chromatography with CHCl3–MeOH (10:1) on silica gel. The evaporation of the remaining eluent yielded a white solid of 5.2 g (40% yield). The structure and purity of the obtained CyA monomer were confirmed using NMR spectroscopy (Fig. S1†). 1H NMR (400 MHz, DMSO-d6): 7.49 (d, J = 7.2 Hz, 1H, Ha), 6.99 (s, 1H, Hb), 6.91 (s, 1H, Hb′), 6.28 (m, 1H, Hc), 6.13 (m, 1H, Hd), 5.94 (m, 1H, He), 5.60 (d, J = 7.2 Hz, 1H, Hf), 4.07 (m, 4H, Hg), 3.82 (t, J = 6.7 Hz, 2H, Hh), 2.67 (t, J = 6.7 Hz, 2H, Hi), 1.63 (m, 4H, Hj). 13C NMR (100 MHz, DMSO-d6): 171.08, 166.07, 165.50, 155.62, 146.45, 131.50, 128.34, 93.06, 63.68, 45.34, 32.84, 24.74. HRMS (ES+): m/z calculated for [M + H]+ 310.1403 g mol−1; found 310.1389 g mol−1.
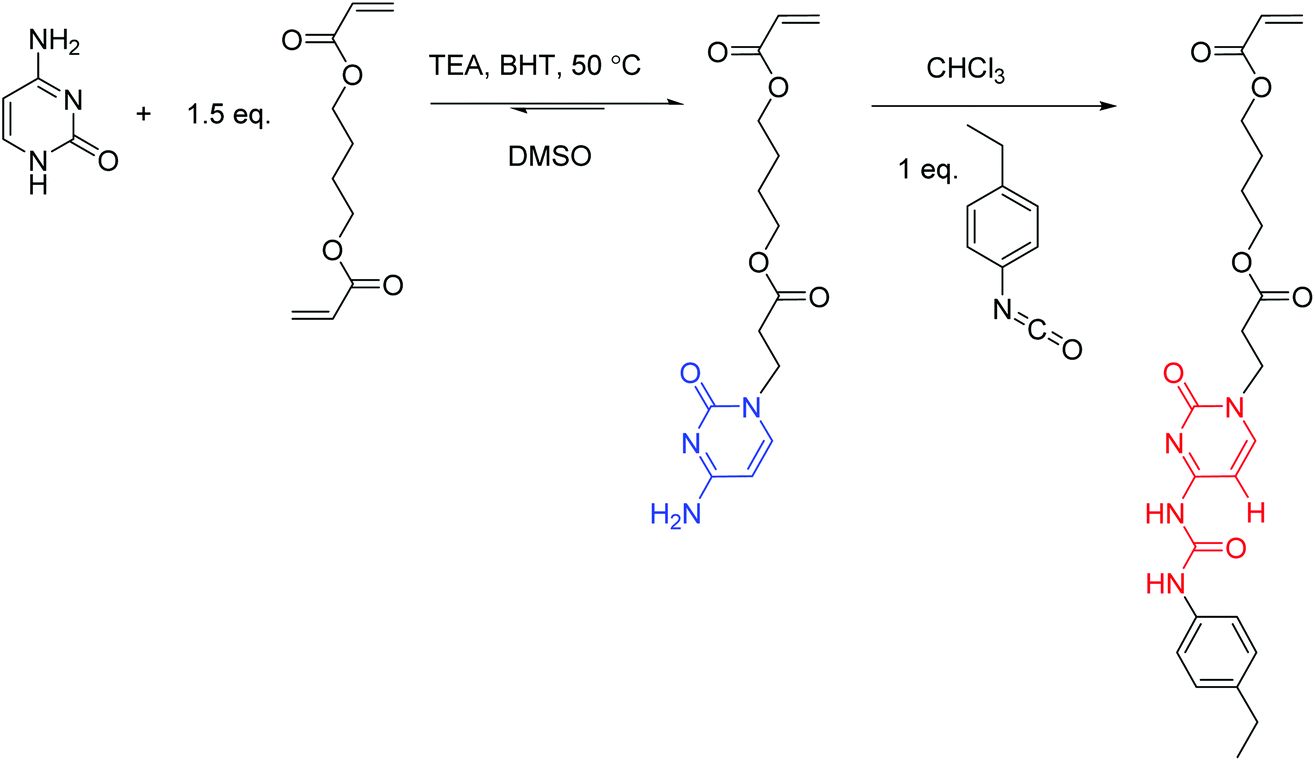 | ||
| Scheme 1 Synthesis of cytosine acrylate (CyA) and ureido-cytosine acrylate (UCyA) monomers. | ||
Synthesis of the 4-((3-(4-(3-(4-ethylphenyl)ureido)-cytosin-1-yl)propanoyl)oxy)butyl acrylate (ureido-cytosine acrylate, UCyA) monomer (Scheme 1)
Cytosine acrylate (66.4 mg, 0.2 mmol), 4-ethylphenyl isocyanate (31.6 mg, 0.2 mmol), and CHCl3 (2 mL) were added to a 25 mL, round-bottom flask equipped with a magnetic stir bar. The reaction mixture was stirred at 0 °C for 2 h and at 25 °C for 12 h, and precipitated into hexane. The resulting suspension was filtered and dried under reduced pressure (20 mmHg) at room temperature to give a white solid of 78.9 mg (81% isolated yield). The structure and purity of the obtained UCyA monomer were confirmed using NMR spectroscopy (Fig. S2†). 1H NMR (400 MHz, DMSO-d6): 11.24 (s, 1H, Ha), 10.06 (s, 1H, Hb), 7.96 (d, J = 7.1 Hz, 1H, Hc), 7.36 (d, J = 8.2 Hz, 2H, Hd), 7.15 (d, J = 8.2 Hz, 2H, He), 6.37–5.88 (m, 4H, Hf+m), 4.25–3.68 (m, 6H, Hg+h), 2.75 (t, J = 6.7 Hz, 2H, Hi), 2.48 (m, 2H, Hl), 1.61 (m, 4H, Hj), 1.14 (t, J = 7.6 Hz, 3H, Hk). 13C NMR (100 MHz, DMSO-d6): 171.28, 165.90, 162.91, 154.28, 151.62, 150.19, 139.24, 136.15, 131.85, 128.73, 128.65, 119.79, 94.63, 64.23, 64.09, 46.54, 32.70, 27.99, 25.16, 16.12. HRMS (ES+): m/z calculated for [M + H]+ 457.2082 g mol−1; found 457.2077 g mol−1.Synthesis of poly(CyA-co-nBA) copolymers (Scheme 2)
Poly(CyA-co-nBA) was prepared using free radical copolymerization in DMF with AIBN as the initiator. A representative example of the polymerization was conducted as follows. A round-bottom flask was charged with CyA (0.4 g, 1.2 mmol), nBA (3.0 g, 23.4 mmol), AIBN (4.0 mg, 24.0 μmol), and DMF (10 mL, 25 wt%) and sparged with N2 for 20 min. The reaction mixture was stirred at 65 °C for 24 h. The resulting solution was precipitated into MeOH–water (5:1, v/v). The final products were collected and dried under reduced pressure (20 mmHg) for 24 h. The product contained 4.5 mol% CyA and 95.5 mol% nBA, with an isolated yield of 80%. The structure and purity of the obtained cytosine-containing copolymers were confirmed using NMR spectroscopy (Fig. S3A†).
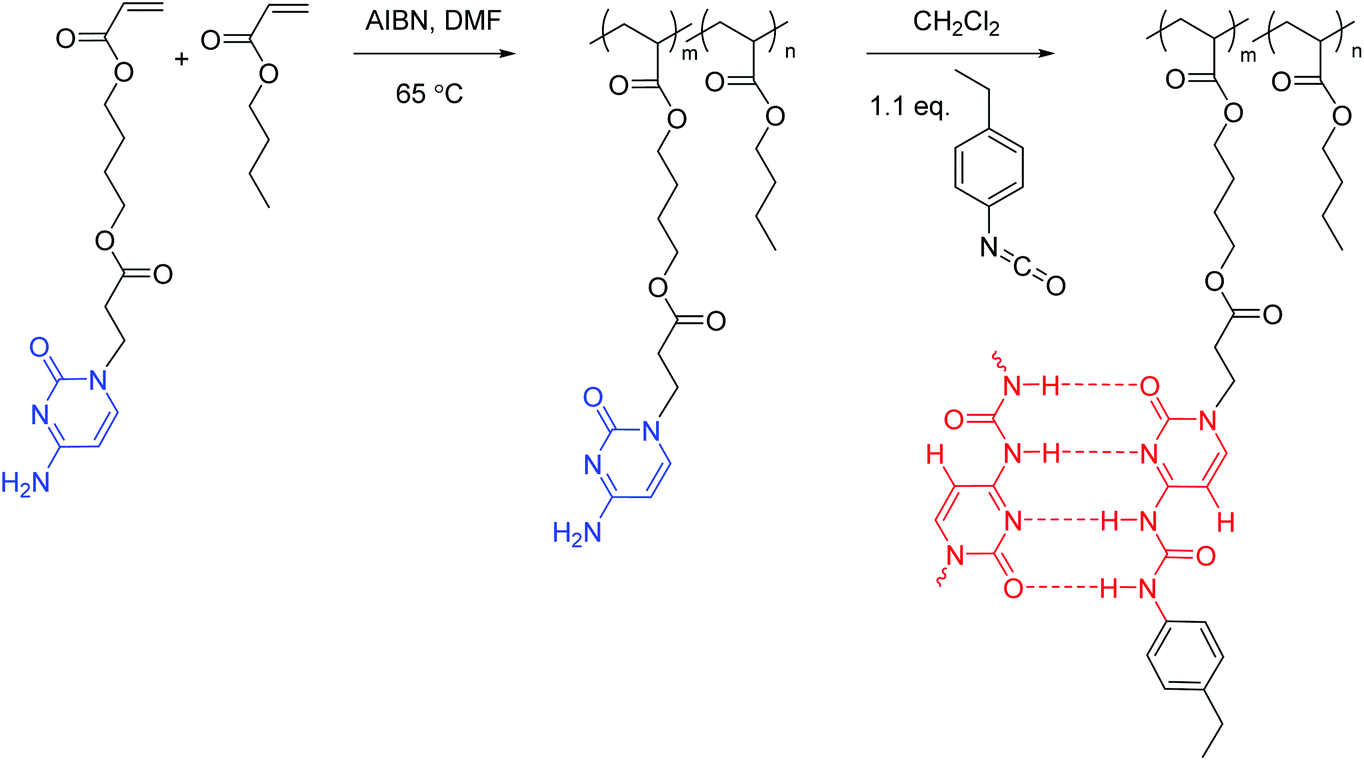 | ||
| Scheme 2 Synthesis of poly(CyA-co-nBA) and poly(UCyA-co-nBA) copolymers, and complementary hydrogen bonding of the ureido-cytosine. Poly(UCyA-co-nBA) and poly(CyA-co-nBA) samples were labeled red and blue, respectively throughout this manuscript for visual guidance. | ||
Synthesis of poly(UCyA-co-nBA) copolymers (Scheme 2)
Poly(UCyA-co-nBA) was prepared using the post-functionalization of poly(CyA-co-nBA). Poly(CyA-co-nBA) (0.45 g) with 4.5 mol% CyA (47.2 mg, 0.15 mmol) and DCM (12 mL) were charged into a 50 mL, round-bottom flask and stirred until the polymer dissolved. A solution of 4-ethylphenyl isocyanate (24.7 mg, 0.17 mmol) in DCM (1 mL) was added into the flask dropwise. The reaction mixture was stirred overnight, concentrated, and precipitated into MeOH–water (6:1, v/v). The final products were collected and dried under reduced pressure (20 mmHg) for 24 h. The product contained 4.5 mol% UCyA and 95.5 mol% nBA, with an isolated yield of 87%. The structure and purity of the obtained UCy-containing copolymers were confirmed using NMR spectroscopy (Fig. S3B†).
Processing and annealing conditions
Poly(CyA-co-nBA) and poly(UCyA-co-nBA) copolymers with relatively high cytosine and UCy contents were dissolved in CHCl3–MeOH (2:1, v/v) and cast into PTFE molds. The molds were covered with glass Petri dishes and maintained at room temperature for 12 h to allow slow evaporation of the solvents. The dried samples were maintained at 50 °C for 12 h, placed under reduced pressure (20 mmHg) for 24 h at room temperature, and finally at 120 °C for 12 h. The resulting films were cooled using a step-wise procedure wherein the oven temperature was reduced by 30 °C and allowed to equilibrate for 3 h, with the process repeated until room temperature was reached. All samples were stored in a desiccator prior to any experiment.
Results and discussion
Synthesis of cytosine acrylate (CyA) and ureido-cytosine acrylate (UCyA) monomers and the corresponding copolymers
This manuscript reports a facile synthetic route for two acrylic monomers (Scheme 1) and the corresponding copolymers that contain cytosine and UCy (Scheme 2), respectively. The base-catalyzed Michael addition of a diacrylate with unprotected cytosine yielded N1-substituted cytosine as the main product due to the higher reactivity of the secondary amine compared to the primary amine.37 This reaction appeared heterogeneous initially due to the challenging solubility of cytosine, and the suspension gradually dissolved as cytosine acrylate formed. Michael addition reached equilibrium when the solution turned completely clear, which served as an indication for reaction completion. Column purification removed the N4-substituted isomer and the difunctionalized byproducts. Derivatization of cytosine and guanine proves challenging compared to adenine and thymine due to the limited solubility and multiple nucleophilic sites, all of which participate in substitution.38 Michael additions of adenine and thymine with acrylates provide a viable route for synthesizing a polymerizable nucleobase with regioselectivity.39 Herein, the reported N1-substituted cytosine synthesis extended the capability of regioselective Michael addition with nucleobases using an optimized catalyst, solvent, and reaction temperature. However, Michael addition of guanine with diacrylate failed in all the tested solvents due to the low solubility of guanine. A reaction of purified cytosine acrylate and 4-ethylphenyl isocyanate converted the primary amine of cytosine to urea quantitatively. UCyA with the 4-ethylphenyl end proved more hydrophobic with an Rf value of 0.72 compared to CyA with an Rf value of 0.19 in CHCl3–MeOH 10:1 (v/v).
The free radical copolymerization of CyA and nBA proceeded homogeneously and afforded a series of random acrylic copolymers with varied cytosine content, controlled through changing the feed ratio of two comonomers. Monomer conversion was higher than 95% based on NMR spectroscopic analysis. Precipitation removed any residual monomer and DMF solvent, affording 80–90% isolated yield. The feed ratios matched polymer compositions as nBA and CyA shared the identical chemical structure of the propagating chain end. NMR spectroscopy assisted in the calculation of cytosine contents in the resulting copolymers using the integration of resonances at 7.4–7.6 ppm and 0.7–1.1 ppm (Fig. S3A†), which corresponded to the chemical shifts of aromatic –CH– (Ha) on cytosine and –CH3 (Hd′) on nBA, respectively. Direct derivatization of cytosine units in poly(CyA-co-nBA) copolymers with isocyanate afforded ureido-cytosine functionalized copolymers with full conversion. This post-functionalization reaction proceeded homogeneously in DCM for poly(CyA-co-nBA) copolymers with less than 12 mol% CyA. The solution viscosity of the reaction mixture for poly(CyA-co-nBA) with 9 mol% CyA increased significantly from 1.3 mPa s to 1.3 Pa s in approximately 70 min after isocyanate addition (Fig. S4†), which was attributed to the strong self-association of UCy compared to cytosine. The reaction of isocyanate and the poly(CyA-co-nBA) copolymer with 21 mol% cytosine turned out to be heterogeneous due to the precipitation of the product in DCM. The resulting poly(UCyA-co-nBA) with 21 mol% UCyA proved insoluble in common organic solvents due to strong physical crosslinking, although UCyA proved less polar than CyA from thin layer chromatography.
The post-functionalization of cytosine copolymers proved more efficient than the direct copolymerization of UCyA and nBA, which led to a lower yield in monomer derivatization and heterogeneous polymerization. The integration of resonances at 7.3–7.4 ppm and 0.7–1.1 ppm (Fig. S3B†) revealed UCy contents in the copolymers, as these resonances represented aromatic –CH– (Hi) on UCy and –CH3 (Hd′) on nBA. The compositions of poly(UCyA-co-nBA) and poly(CyA-co-nBA) matched within the experimental error, according to calculations based on 1H NMR spectroscopy results before and after the post-functionalization. Table 1 summarizes the CyA and UCyA contents for poly(CyA-co-nBA) and poly(UCyA-co-nBA), and the weight fraction of the CyA or UCyA comonomer in each copolymer. All polymerization conditions and purification procedures remained identical for all acrylic copolymers to target comparable molecular weights, which included the reaction temperature, time, initiator/monomer concentration, and precipitation method. Size exclusion chromatography (SEC) results of CyA/UCyA-containing copolymers proved unreliable due to the aggregation and column interaction in all available SEC mobile phases.
| CyA/UCyA in polymer (mol%) | CyA/UCyA in polymer (wt%) | T d,5 wt% (°C) | T g (°C) | T g (°C) predictedb | Calculated first step weight loss (%) | Measured first step weight loss (%) | |
|---|---|---|---|---|---|---|---|
a
Ref. 40.
b Fox equation 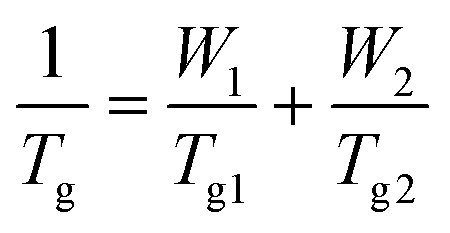 was used to calculate the predicted Tg, where W1 and W2 represent the weight fraction of CyA and nBA respectively, and Tg1 and Tg2 represent the Tg (Kelvin) of poly(CyA) and poly(nBA), respectively. was used to calculate the predicted Tg, where W1 and W2 represent the weight fraction of CyA and nBA respectively, and Tg1 and Tg2 represent the Tg (Kelvin) of poly(CyA) and poly(nBA), respectively.
|
|||||||
| Poly(nBA)a | 0.0 | 0.0 | 343 | −47 | 0 | 0 | |
| CyA | 3.0 | 6.7 | 331 | −42 | −41 | 3 | 4 |
| UCyA | 9.9 | 315 | −38 | 6 | 6 | ||
| CyA | 4.5 | 10.2 | 317 | −38 | −38 | 4 | 5 |
| UCyA | 14.4 | 261 | −36 | 9 | 10 | ||
| CyA | 8.8 | 19.3 | 295 | −26 | −29 | 8 | 8 |
| UCyA | 25.6 | 244 | −18 | 17 | 16 | ||
| CyA | 10.5 | 22.5 | 291 | −28 | −26 | 9 | 10 |
| UCyA | 29.0 | 243 | −16 | 19 | 18 | ||
| CyA | 21.4 | 40.2 | 283 | 14 | −7 | 17 | 17 |
| UCyA | 48.8 | 210 | 28 | 35 | 29 | ||
Thermal analysis
Fig. 1 displays the representative TGA weight loss profiles of poly(CyA-co-nBA) copolymers with poly(UCyA-co-nBA) copolymers with a similar cytosine content. All copolymers exhibited two-step degradation profiles. The first step corresponded to degradation of the CyA/UCyA pendant groups, leaving the acrylic backbone and cytosine solid with a melting point over 300 °C. The measured weight loss in the first step agreed with the theoretical calculation of the linker between cytosine and the polymer backbone (Table 1). The second degradation step mainly involved the degradation of the polymer backbone, which was consistent with previous TGA results of UPy-containing polyacrylates.33 UCyA-containing copolymers exhibited lower Td,5 wt% values than their CyA-containing precursors due to the incorporation of the urea bond, which started to degrade slightly below 200 °C.41,42 The weight loss of cytosine-containing copolymers started at approximately 240 °C, while chemical crosslinking of the primary amines attacking the ester bonds occurred at much lower temperatures. Crosslinking also contributed to the higher Td, 5 wt% of CyA-containing copolymers compared to UCyA copolymers. Isothermal rheology experiments verified the enhanced thermal stability of UCyA-containing copolymers compared to their cytosine precursors at 130 °C and below (Fig. S5 and S6†). The complex viscosity of poly(CyA-co-nBA) increased significantly at 130 °C, while poly(UCyA-co-nBA) remained relatively constant.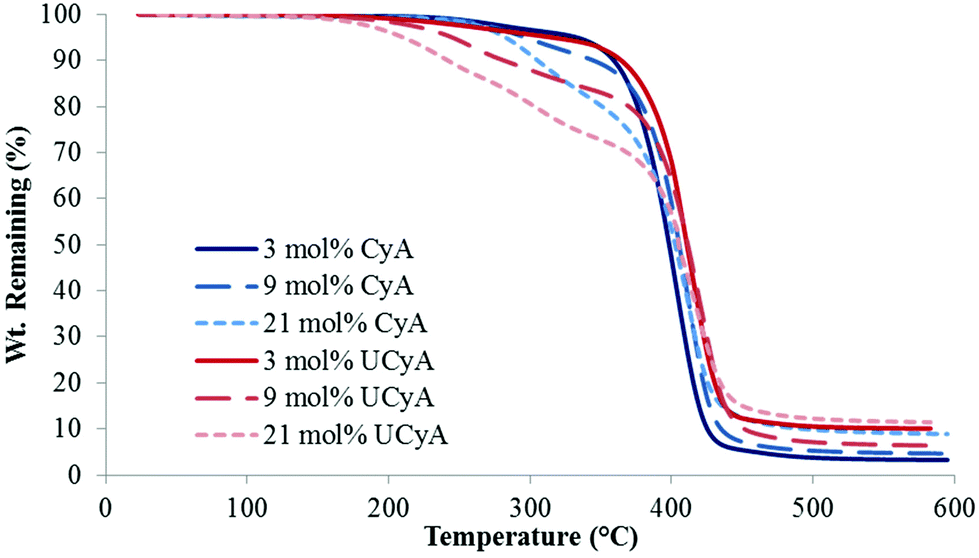 | ||
| Fig. 1 Representative TGA thermograms of poly(CyA-co-nBA) and poly(UCyA-co-nBA) copolymers with varied amounts of CyA and UCyA, respectively. | ||
DSC revealed the thermal transitions of poly(CyA-co-nBA) and poly(UCyA-co-nBA) copolymers with varied cytosine and UCy contents, respectively (Fig. 2, S7 and S8†). Copolymers in each series were amorphous with a single Tg that increased as CyA or UCyA mol% increased. Poly(CyA) exhibited a single Tg at 90 °C, approximately 140 °C higher than the Tg of poly(nBA) with an identical chemical structure near the backbone (Fig. S7†). Both steric hindrance and hydrogen bonding of the pyrimidine ring affected the long range segmental motion of the acrylic backbone and resulted in a higher Tg for poly(CyA).43 The increasing Tg of poly(CyA-co-nBA) copolymers with increasing CyA mol% confirmed the random distribution of two comonomers, which nevertheless deviated from the Fox equation prediction, especially at a relatively high CyA mol% (Fig. 3).44 This phenomenon revealed the presence of an interaction between CyA and nBA, which became more predominant with higher CyA incorporation.24,45 Poly(UCyA-co-nBA) showed higher Tg values compared to the cytosine precursors with a similar polar monomer incorporation (Fig. 2 and 3), resulting from bulkier UCy pendant groups and stronger hydrogen bonding (Scheme 2). This Tg difference between CyA and UCyA-containing copolymers appeared to increase with increasing polar monomer contents. Poly(UCyA-co-nBA) with 21 mol% UCyA exhibited a Tg of 28 °C and an exothermic transition near 150 °C, presumably due to either a phase transition or hydrogen bond dissociation.35
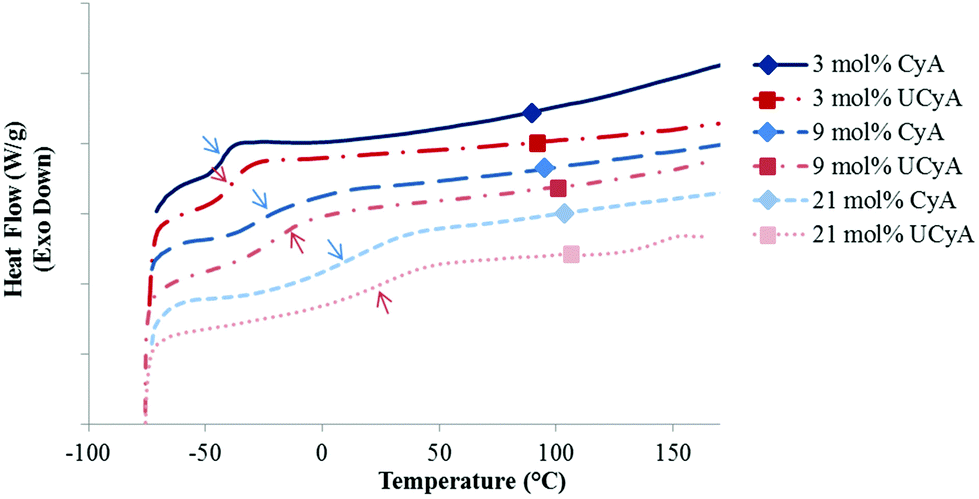 | ||
| Fig. 2 Representative DSC thermograms of poly(CyA-co-nBA) and poly(UCyA-co-nBA) copolymers with varied amounts of CyA and UCyA, respectively. | ||
 | ||
| Fig. 3 Relationship between CyA/UCyA mol% of poly(CyA-co-nBA) or poly(UCyA-co-nBA) and Tg values. | ||
Thermomechanical properties
A rheological temperature sweep experiment investigated the viscoelastic properties of poly(UCyA-co-nBA) and poly(CyA-co-nBA) copolymers in relationship to temperature. Fig. 4 compares the storage and loss moduli (G′ and G′′) of copolymers with 3 mol% of CyA and UCyA. Both samples exhibited similar moduli values before and during their glass transitions from −40 °C to −10 °C, while their viscoelastic behavior differed significantly above −10 °C. A wide plateau of poly(UCyA-co-nBA) started and extended to approximately 40 °C, while the storage modulus exceeded loss modulus. This plateau regime and the following terminal flow regime indicated the formation of a transient network of physical crosslinks from the self-complementary quadruple hydrogen bonding of the ureido-cytosine units. In contrast, the moduli of poly(CyA-co-nBA) continued to decrease after its glass transition and reduced to nearly 20 times lower than poly(UCyA-co-nBA) at 40 °C. The loss modulus curve of poly(UCyA-co-nBA) showed a peak near 50 °C, originating from an increase of the viscous component due to the dissociation of the physically crosslinked network.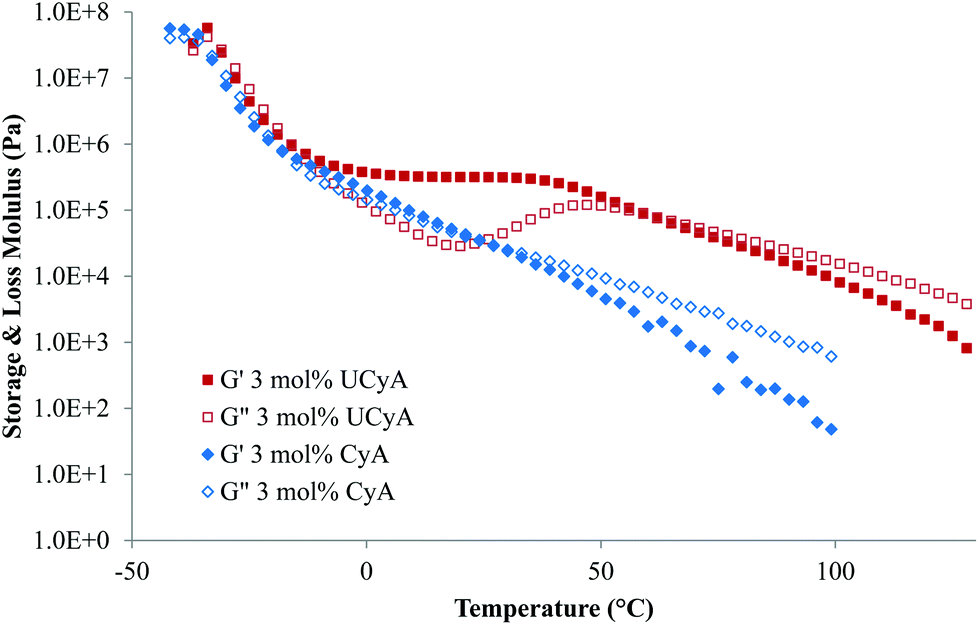 | ||
| Fig. 4 Rheological temperature sweeps of poly(CyA-co-nBA) and poly(UCyA-co-nBA) with 3 mol% CyA and UCyA, respectively. | ||
The plateau modulus of poly(UCyA-co-nBA) with 3 mol% UCyA approached the Dahlquist criterion, which describes an empirical threshold of 3 × 105 Pa at 25 °C and 1 Hz for the storage modulus of pressure sensitive adhesives (PSA).46Fig. 5B shows the decreasing modulus of poly(UCyA-co-nBA) with lower UCyA mol%. As a result, poly(UCyA-co-nBA) copolymers with lower than 3 mol% UCyA serve as potential candidates for PSA with superior cohesive strength than the CyA copolymers. The plateau demonstrated the superior thermal resistance of resulting adhesives with constant cohesive strength at 45 °C. Poly(CyA-co-nBA) with 3 mol% CyA demonstrated sufficient tackiness, however a weaker cohesive strength and significantly worse high temperature performance due to insufficient physical crosslinking.46 The reptation onset of the UCy-containing sample occurred at 59 °C, 32 °C higher than the cytosine precursor. The viscous property dominated after this onset, entering the terminal flow regime, where G′′ exceeded G′. The slopes of the poly(UCyA-co-nBA) modulus curves in the terminal flow region matched closely with those of its CyA-containing precursor, representing an entangled polymer melt behavior.32 The moduli difference between the UCy sample and the cytosine precursor remained at 100 °C, suggesting that not all of the strong hydrogen bonding dissociated at 100 °C. Modulus measurements below 100 Pa exceeded the accuracy limits for the available instrument and geometry.
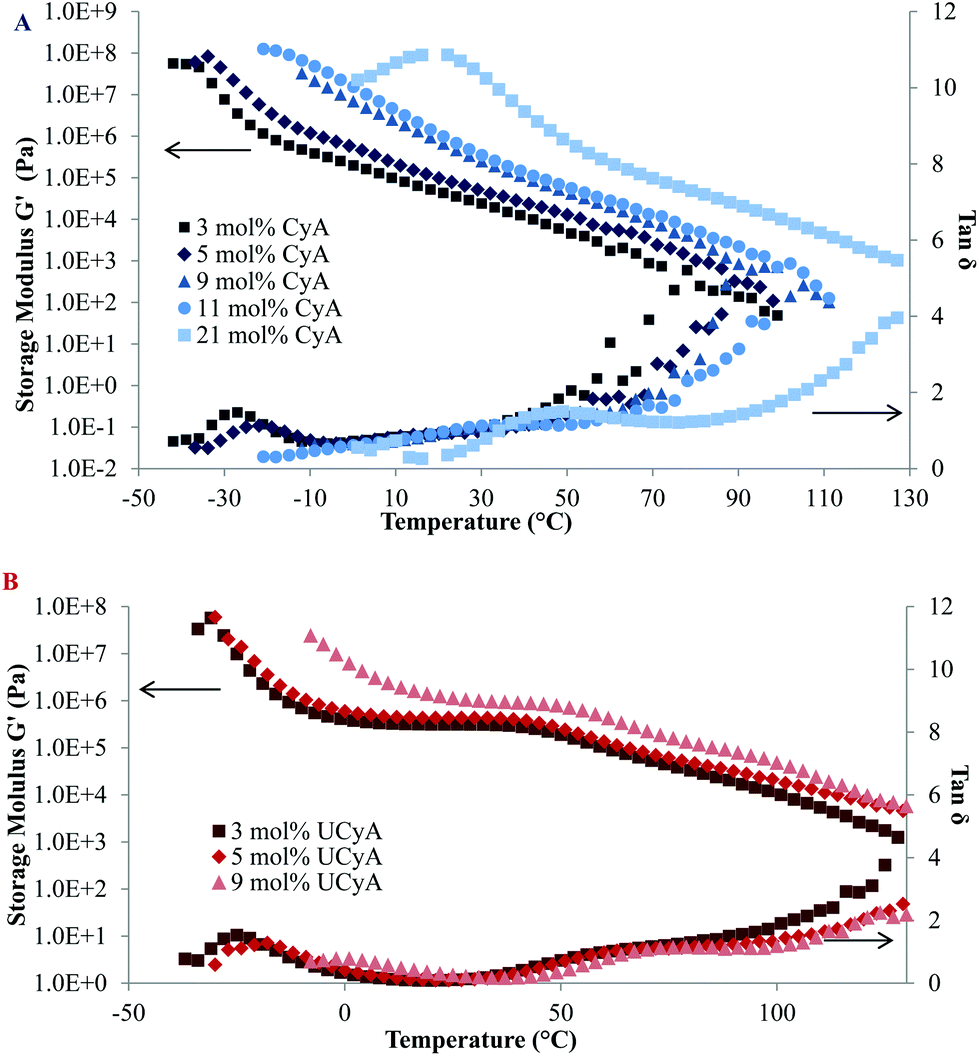 | ||
| Fig. 5 Rheological temperature sweeps of (A) poly(CyA-co-nBA) and (B) poly(UCyA-co-nBA) with varied amounts of CyA and UCyA, respectively. | ||
Fig. 5A and B display both G′ and tan delta curves of the annealed poly(CyA-co-nBA) and poly(UCyA-co-nBA) copolymer films. The sample with higher CyA mol% exhibited higher Tg and higher modulus in the tested temperature range (Fig. 5A), due to a higher density of physical crosslinks. The Tg increase became more significant at higher CyA mol%, which agreed with the thermal analysis results (Fig. 3). Fig. 5B similarly exhibited that both the Tg and modulus increased with increasing UCyA content, attributed to additional hydrogen bonding. However, the tan delta curves of poly(UCyA-co-nBA) copolymers each contained an obvious transition with a peak value near 60–70 °C, which was not shown for poly(CyA-co-nBA). The DMA results of poly(UCyA-co-nBA) films with higher UCyA contents also revealed a second transition near 70 °C (Fig. 6C). Long et al. previously observed similar transitions for block copolymers with functionalized blocks that formed weak noncovalent interactions.40,47 These transitions likely reflected the majority of hydrogen bonding dissociation, which led to a free volume increase with weakened physical crosslinks. The tan delta curves of poly(CyA-co-nBA) films with relatively high CyA contents also showed a secondary transition near 20–30 °C (Fig. 6C), which presumably reflected weaker hydrogen bonding dissociation at lower temperatures than UCyA samples. Thermomechanical analyses proved capable of detecting a transition related to the dissociation of noncovalent interactions, which thermal analysis failed to reveal.
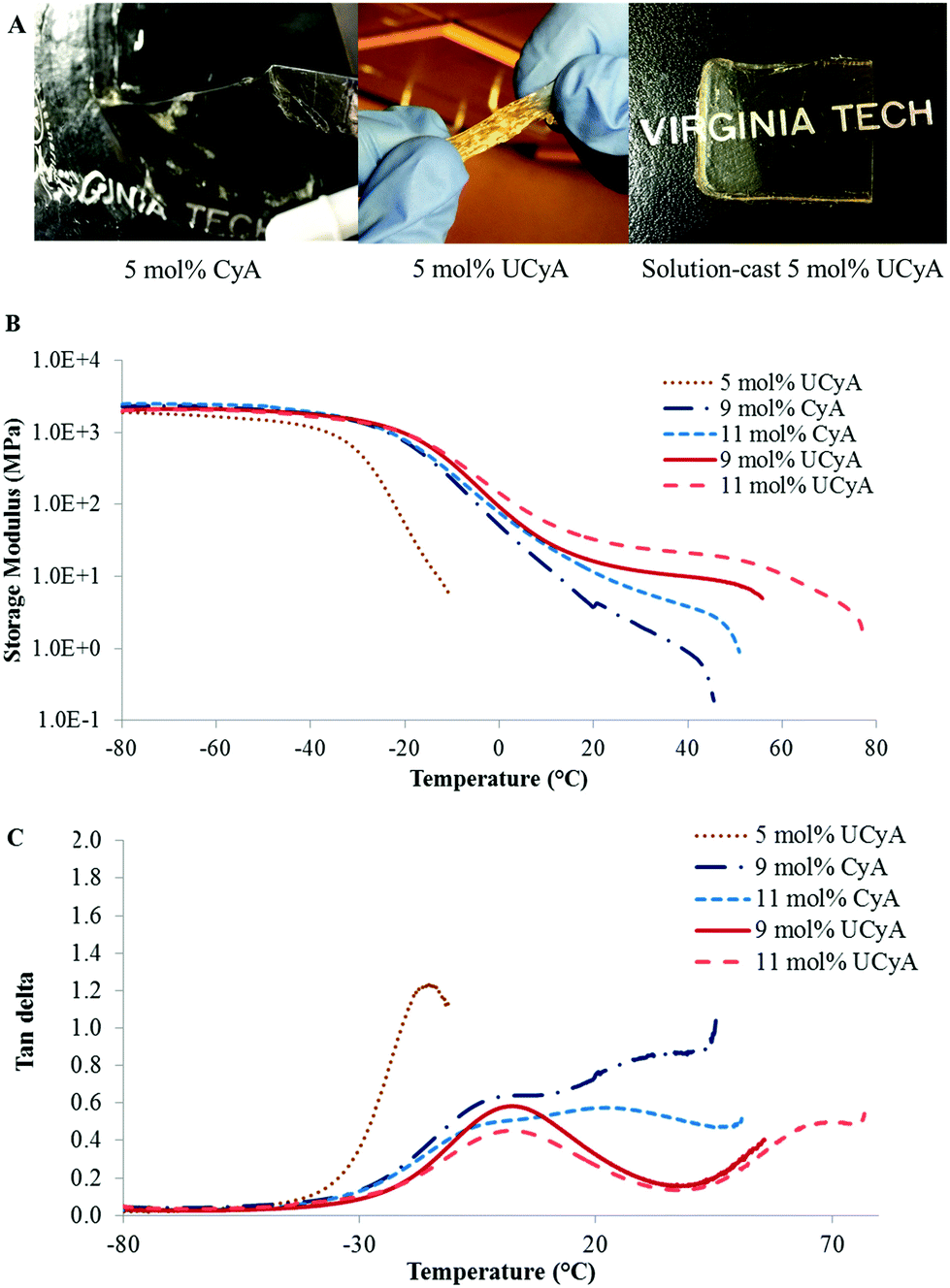 | ||
| Fig. 6 (A) From left to right, pictures of poly(CyA-co-nBA) and poly(UCyA-co-nBA) with 5 mol% cytosine contents, and solution-cast film of the latter. (B) Storage modulus curves and (C) tan delta curves during dynamic mechanical temperature ramps for solution-cast poly(CyA-co-nBA) and poly(UCyA-co-nBA) films with varied mol% of CyA and UCyA, respectively. | ||
Furthermore, DMA closely compared the thermomechanical properties of solution-cast poly(UCyA-co-nBA) copolymer films with their cytosine precursors with uniaxial deformation in the oscillatory mode. The acrylic copolymer with 5 mol% UCyA formed a soft film that showed one-phase thermomechanical behavior with a single modulus drop, while its CyA precursor lacked mechanical integrity for film casting (Fig. 6A). The other samples with a higher mol% of CyA and UCyA shared a similar thermodynamic behavior in the glassy regime and during the glass transition (Fig. 6B). The influence of hydrogen bonding dominated the plateau and terminal flow regimes, agreeing with the rheological analysis for copolymers with lower CyA or UCyA contents. The hydrogen bonding of UCyA contributed to a well-defined plateau, the modulus of which increased with increasing UCyA concentration in the random copolymers. The flow temperature also increased with increasing UCyA content, indicating an enhanced heat resistance of the copolymer mechanical strength. The plateau moduli of UCyA films were in the rubbery range, demonstrating their potential as thermoplastic elastomers. CyA copolymer films lacked a plateau window due to insufficient physical crosslinking from the weaker self-hydrogen bonding of CyA compared to UCyA. The thermomechanical performance of poly(UCyA-co-nBA) films suggested their microphase-separated morphologies.48 The tan delta peak of the UCyA copolymer films, beginning near 50 °C, resulted from the dissociation of the phase-separation enhanced hydrogen bonding of UCy units. The dissociation of noncovalent interactions became visible as a distinguishable transition in thermomechanical analysis because of the reinforcement from phase-separation.1,49
Morphological characterization
Small angle X-ray scattering (SAXS) was used to elucidate the bulk morphology of annealed poly(CyA-co-nBA) and poly(UCyA-co-nBA) films (Fig. 7). Samples were annealed at 120 °C, above the Tg of all copolymers, and step-wise cooling facilitated the formation of noncovalent interactions and phase-separation. The SAXS profiles of all films with >5 mol% cytosine or UCy content exhibited relatively broad scattering maxima, suggesting microphase-separated morphologies, which resulted from hydrogen-bonded cytosine or UCy domains dispersed within the acrylic polymer matrix.50 The scattering peak positions remained constant with varying cytosine or UCy content for the series of poly(CyA-co-nBA) and poly(UCyA-co-nBA), indicating a relatively constant interdomain spacing. This phenomenon is commonly observed and well-studied for random ionomers that contain ionic aggregates.51 The increase in the scattering peak intensity with an overall increase in the mol% of hydrogen bonded groups was attributed to a greater extent of hard domain formation. The absence of a scattering maximum for the film with 5 mol% UCyA revealed its phase-mixed morphology due to insufficient physical crosslinking, consistent with its lower onset temperature of terminal flow below room temperature in DMA (Fig. 6). Converting cytosine to UCy shifted the scattering peak from 1.4 nm−1 to 0.6 nm−1, corresponding to a shift in the interdomain spacing between hydrogen bonded hard domains from 4.5 nm to 10.0 nm, respectively.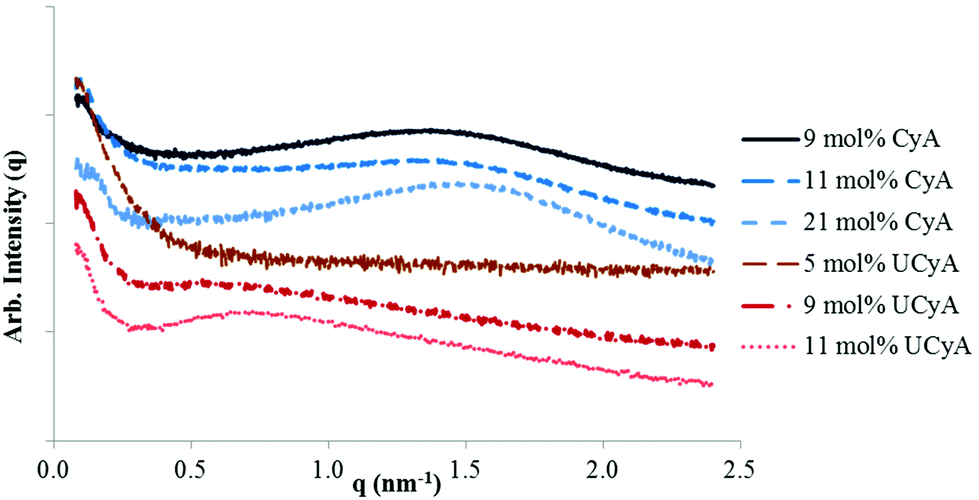 | ||
| Fig. 7 SAXS for solution-cast poly(CyA-co-nBA) and poly(UCyA-co-nBA) films with varied amounts of CyA and UCyA, respectively. For clarity, data were shifted by arbitrary factors vertically. | ||
AFM images confirmed the microphase-separated surface morphology of the annealed poly(CyA-co-nBA) and poly(UCyA-co-nBA) films (Fig. 8). The dark and light areas represented the soft poly(nBA) phase and the hydrogen bonded hard phase, respectively. The poly(CyA-co-nBA) film contained smaller domains with irregular shapes, while the poly(UCyA-co-nBA) film displayed nano-fibrillar hard domains, similar to telechelic urea-UPy supramolecular polymers.52 The π–π stacking of cytosine and phenyl rings presumably assisted the lateral stacking of hydrogen bonded UCyA units, which enabled the formation of nano-fibrillar hard domains. The relatively larger interdomain spacing of the UCy-containing copolymer (Fig. 8B) compared to the cytosine control (Fig. 8A) was consistent with the shift in the scattering peak positions observed in Fig. 7. The physical network containing the nano-fibrillar hard domains likely contributed to the enhanced mechanical properties of the UCy-containing copolymer compared to the cytosine control shown in Fig. 6B. The fibrillar hard domains resembled fillers that reinforced the modulus of the soft matrix, contributing to a rubbery plateau with retained mechanical integrity over a wider temperature range.
 | ||
| Fig. 8 AFM phase images for solution-cast (A) poly(CyA-co-nBA) and (B) poly(UCyA-co-nBA) films with 11 mol% of UCyA and CyA, respectively. | ||
Variable temperature FTIR spectroscopy (VT-FTIR)
VT-FTIR commonly serves as a versatile tool to investigate hydrogen bonding or debonding in response to cooling or heating.4,24,49,53Fig. 9 displays FTIR absorbance spectra in the carbonyl stretching region of copolymers containing CyA or UCyA at varying temperatures, demonstrating hydrogen bonding dissociation with heat. Copolymers with a higher cytosine and UCy content displayed stronger absorbance bands for hydrogen bonded C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H units in reference to the non-hydrogen bonded carbonyls at 1726 cm−1 (Fig. S9†). The wavenumber of this reference band at 1726 cm−1 remained constant with varying temperatures, which indicated that most of the carbonyls on the acrylate backbone were free from hydrogen bonding. In Fig. 9A, both the absorption bands near 1645 cm−1 and 1622 cm−1 at 30 °C blue-shifted to higher wavenumbers with increasing temperature, representing the stretching vibration of CO on the cytosine units. These blue-shifts resulted from the increased frequency of the CO stretching vibration when the hydrogen bond weakened and the CO bond strengthened.54,55 The red-shifts of absorption bands at 1573 cm−1 and 1487 cm−1 from 30 °C to elevated temperatures demonstrated that the frequency of the N–H bond bending vibration decreased with increasing temperature due to the elimination of the anchoring restriction from hydrogen bonding.40 The absorption bands near 1521 cm−1 and 1446 cm−1 corresponded to free N–H bending, which indicated that not all N–H formed hydrogen bonding at room temperature, agreeing with the relatively weak and non-ordered self-association of cytosine. Fig. 9B reveals similar absorption band shifts from the CO stretching and N–H bending for poly(UCyA-co-nBA). The absorption band of the CO stretching near 1710 cm−1 shifted to a lower wavenumber, as the shoulder peak merged with the free CO peak with increasing temperature. The intensity of another absorption band near 1645 cm−1 decreased due to the blue-shift to 1678 cm−1, where a new peak appeared at elevated temperatures. The N–H bending absorption bands for the UCy units near 1550 cm−1 and 1500 cm−1 clearly red-shifted to lower wavenumbers as the temperature increased. The most significant shift occurred at approximately 60 °C to 90 °C for poly(CyA-co-nBA) and 100 °C to 130 °C for poly(UCyA-co-nBA), agreeing with the previous DMA result that the majority of hydrogen bonding from UCyA dissociated at higher temperatures than CyA. However, the heating rate and sample deformation differences of VT-FTIR and DMA led to incomparable temperature values for hydrogen bond dissociation.
O and N–H units in reference to the non-hydrogen bonded carbonyls at 1726 cm−1 (Fig. S9†). The wavenumber of this reference band at 1726 cm−1 remained constant with varying temperatures, which indicated that most of the carbonyls on the acrylate backbone were free from hydrogen bonding. In Fig. 9A, both the absorption bands near 1645 cm−1 and 1622 cm−1 at 30 °C blue-shifted to higher wavenumbers with increasing temperature, representing the stretching vibration of CO on the cytosine units. These blue-shifts resulted from the increased frequency of the CO stretching vibration when the hydrogen bond weakened and the CO bond strengthened.54,55 The red-shifts of absorption bands at 1573 cm−1 and 1487 cm−1 from 30 °C to elevated temperatures demonstrated that the frequency of the N–H bond bending vibration decreased with increasing temperature due to the elimination of the anchoring restriction from hydrogen bonding.40 The absorption bands near 1521 cm−1 and 1446 cm−1 corresponded to free N–H bending, which indicated that not all N–H formed hydrogen bonding at room temperature, agreeing with the relatively weak and non-ordered self-association of cytosine. Fig. 9B reveals similar absorption band shifts from the CO stretching and N–H bending for poly(UCyA-co-nBA). The absorption band of the CO stretching near 1710 cm−1 shifted to a lower wavenumber, as the shoulder peak merged with the free CO peak with increasing temperature. The intensity of another absorption band near 1645 cm−1 decreased due to the blue-shift to 1678 cm−1, where a new peak appeared at elevated temperatures. The N–H bending absorption bands for the UCy units near 1550 cm−1 and 1500 cm−1 clearly red-shifted to lower wavenumbers as the temperature increased. The most significant shift occurred at approximately 60 °C to 90 °C for poly(CyA-co-nBA) and 100 °C to 130 °C for poly(UCyA-co-nBA), agreeing with the previous DMA result that the majority of hydrogen bonding from UCyA dissociated at higher temperatures than CyA. However, the heating rate and sample deformation differences of VT-FTIR and DMA led to incomparable temperature values for hydrogen bond dissociation.
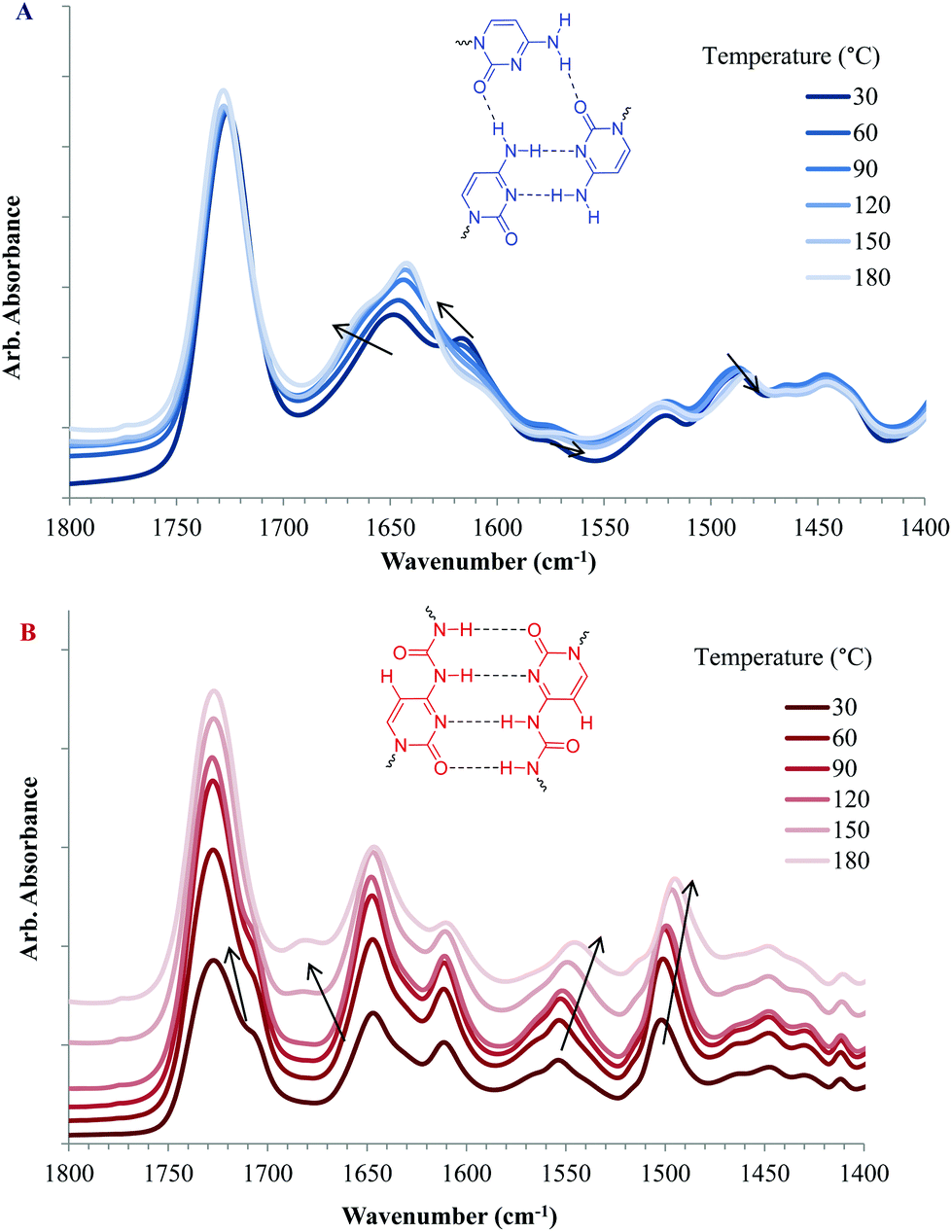 | ||
| Fig. 9 Variable temperature FT-IR spectra of (A) poly(UCyA-co-nBA) and (B) poly(CyA-co-nBA) with 21 mol% of UCyA and CyA, respectively, in the 1500–1700 cm−1 region. Spectra were shifted vertically for visual clarity. | ||
Water sorption
Fig. 10 demonstrates a significantly lower water uptake of poly(UCyA-co-nBA) copolymers as compared to their CyA copolymer precursors. Poly(UCyA-co-nBA) copolymers only exhibited 0.3 wt% moisture uptake at 95% relative humidity with the highest UCyA incorporation, while the corresponding cytosine control gained up to 4.6% weight under 95% relative humidity. The water uptake of poly(CyA-co-nBA) increased with increasing mol% of CyA. The weight changes of poly(UCyA-co-nBA) from the water uptake reached closely to the instrument lower limit. The increased hydrophobicity of UCy relative to cytosine contributed to a lower water uptake for poly(UCyA-co-nBA). Additionally, phase-separation further repelled moisture through packing hydrogen bonding groups into the hard phase and isolating them within the nonpolar soft phase. The low water uptake of poly(UCyA-co-nBA) largely benefits its application in adhesives as moisture generally weakens adhesion and causes degradation during melt processing.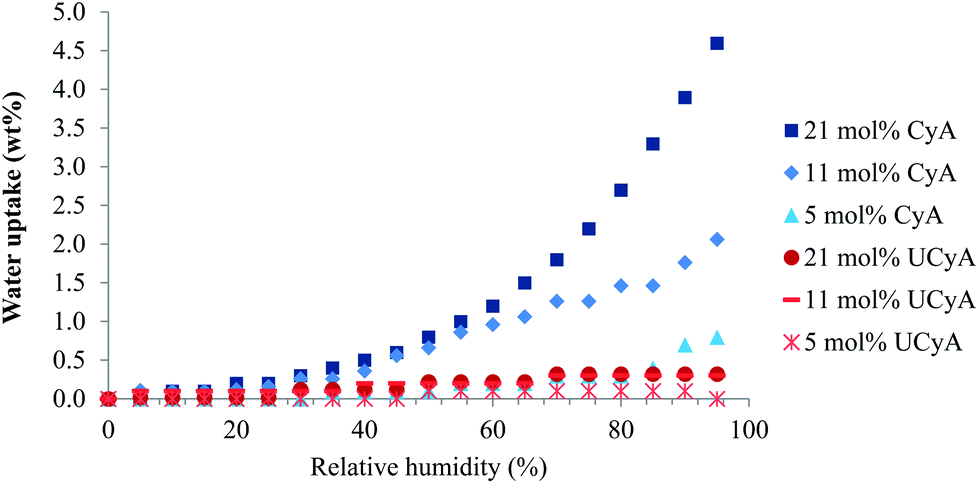 | ||
| Fig. 10 Equilibrium water sorption of poly(UCyA-co-nBA) and poly(UCyA-co-nBA) copolymers with varied amounts of UCyA and CyA in a stepwise relative ramp from 0% to 95%. | ||
Conclusion
This manuscript reports an unprecedented, facile synthetic route for monomer and random copolymers that contain N1-substituted cytosine using the Michael addition reaction of cytosine with diacrylate and free radical polymerization, respectively. Post-functionalization easily converted cytosine pendant groups to ureido-cytosines, which self-associated into quadruple hydrogen-bonded dimers. Physically crosslinked acrylic copolymers formed tacky solids with <5 mol% cytosine contents, while copolymers with ≥9 mol% associating monomers were solution-cast into free-standing films. The physical characterization of UCy and cytosine-containing copolymers provided a direct comparison of weak and strong hydrogen-bonded acrylic copolymer networks.Overall, quadruple hydrogen-bonded UCy units contributed to enhanced thermal stability, higher glass transition temperature, better thermomechanical performance, wider rubbery plateau window, more well-defined morphology, and lower water uptake for a random copolymer compared to its cytosine control. UCyA random copolymers enable potential applications as supramolecular adhesives with ≤3 mol% UCyA and thermoplastic elastomers with 9–11 mol% UCyA. The advantages of using UCyA-containing copolymers over CyA controls for adhesive applications included better melt stability during processing, enhanced cohesive strength, wider functional temperature range, and superior moisture resistance.
Acknowledgements
This research was partially supported by Henkel Corporation. This material is also partially based upon a work supported by the National Science Foundation under Grant No. DMR-0923107 and DMR-1507245.References
- S. Seiffert and J. Sprakel, Chem. Soc. Rev., 2012, 41, 909–930 RSC.
- Analytical Methods in Supramolecular Chemistry, ed. C. Schalley, Wiley-VCH Verlag GmbH & Co. KGaA, 2007 Search PubMed.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS PubMed.
- L. Yang, X. Tan, Z. Wang and X. Zhang, Chem. Rev., 2015, 115, 7196–7239 CrossRef CAS PubMed.
- V. V. Khutoryanskiy, Nat. Mater., 2015, 14, 963–964 CrossRef CAS PubMed.
- R. Deng and X. Liu, Nat. Chem., 2015, 7, 472–473 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS PubMed.
- M. Hutin, E. Burakowska-Meise, W. P. J. Appel, P. Y. W. Dankers and E. W. Meijer, Macromolecules, 2013, 46, 8528–8537 CrossRef CAS.
- K. Zhang, T. E. Long and C. Paul, WO2015058141A1, 2015.
- C. Creton and E. Papon, MRS Bull., 2003, 28, 419–421 CrossRef CAS.
- J. Courtois, I. Baroudi, N. Nouvel, E. Degrandi, S. Pensec, G. Ducouret, C. Chaneac, L. Bouteiller and C. Creton, Adv. Funct. Mater., 2010, 20, 1803–1811 CrossRef CAS.
- R. D. Lundberg, in Structure and Properties of Ionomers, ed. M. Pineri and A. Eisenberg, Springer, Netherlands, 1987, ch. 35, vol. 198, pp. 429–438 Search PubMed.
- H. D. Brooks, J. Y. Kelly, P. H. Madison, C. D. Thatcher and T. E. Long, Proc. Annu. Meet. Adhes. Soc., 2001, 24th, 150–152 CAS.
- W. Griehl and D. Ruestem, Ind. Eng. Chem., 1970, 62, 16–22 CrossRef CAS.
- I. Benedek, Pressure-sensitive adhesives and applications, CRC Press, 2004 Search PubMed.
- S. H. Han, V. Pryamitsyn, D. Bae, J. Kwak, V. Ganesan and J. K. Kim, ACS Nano, 2012, 6, 7966–7972 CrossRef CAS PubMed.
- K. Zhang, G. B. Fahs, M. Aiba, R. B. Moore and T. E. Long, Chem. Commun., 2014, 50, 9145–9148 RSC.
- H. S. Bazzi, J. Bouffard and H. F. Sleiman, Macromolecules, 2003, 36, 7899–7902 CrossRef CAS.
- H. J. Spijker, A. J. Dirks and H. J. C. M. Van, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4242–4250 CrossRef CAS.
- N. Hosono, M. A. J. Gillissen, Y. Li, S. S. Sheiko, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501–510 CrossRef CAS PubMed.
- J. Sartorius and H.-J. Schhneider, Chem. – Eur. J., 1996, 2, 1446–1452 CrossRef CAS.
- E. Greco, A. E. Aliev, V. G. H. Lafitte, K. Bala, D. Duncan, L. Pilon, P. Golding and H. C. Hailes, New J. Chem., 2010, 34, 2634–2642 RSC.
- M. Tamami, K. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromol. Chem. Phys., 2014, 215, 2337–2344 CrossRef CAS.
- R. McHale, J. P. Patterson, P. B. Zetterlund and R. K. O'Reilly, Nat. Chem., 2012, 4, 491–497 CrossRef CAS PubMed.
- R. McHale and R. K. O'Reilly, Macromolecules, 2012, 45, 7665–7675 CrossRef CAS.
- P. K. Lo and H. F. Sleiman, J. Am. Chem. Soc., 2009, 131, 4182–4183 CrossRef CAS PubMed.
- J.-F. Lutz, A. F. Thuenemann and K. Rurack, Macromolecules, 2005, 38, 8124–8126 CrossRef CAS.
- J. C. Kim, J. Jung, Y. Rho, M. Kim, W. Kwon, H. Kim, I. J. Kim, J. R. Kim and M. Ree, Biomacromolecules, 2011, 12, 2822–2833 CrossRef CAS PubMed.
- R. P. Sijbesma and E. W. Meijer, Chem. Commun., 2003, 5–16 RSC.
- G. T. F. A. de, G. B. W. L. Ligthart, M. Lutz, A. L. Spek, E. W. Meijer and R. P. Sijbesma, J. Am. Chem. Soc., 2008, 130, 5479–5486 CrossRef PubMed.
- K. E. Feldman, M. J. Kade, E. W. Meijer, C. J. Hawker and E. J. Kramer, Macromolecules, 2009, 42, 9072–9081 CrossRef CAS.
- K. Yamauchi, J. R. Lizotte and T. E. Long, Macromolecules, 2003, 36, 1083–1088 CrossRef CAS.
- V. G. H. Lafitte, A. E. Aliev, E. Greco, K. Bala, P. Golding and H. C. Hailes, New J. Chem., 2011, 35, 1522–1527 RSC.
- C.-C. Cheng, F.-C. Chang, J.-H. Wang, Y.-L. Chu, Y.-S. Wang, D.-J. Lee, W.-T. Chuang and Z. Xin, RSC Adv., 2015, 5, 76451–76457 RSC.
- C.-C. Cheng, F.-C. Chang, J.-K. Chen, T.-Y. Wang and D.-J. Lee, RSC Adv., 2015, 5, 101148–101154 RSC.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- S. Sivakova and S. J. Rowan, Chem. Soc. Rev., 2005, 34, 9–21 RSC.
- S. Cheng, M. Zhang, N. Dixit, R. B. Moore and T. E. Long, Macromolecules, 2012, 45, 805–812 CrossRef CAS.
- K. Zhang, M. Aiba, G. B. Fahs, A. G. Hudson, W. D. Chiang, R. B. Moore, M. Ueda and T. E. Long, Polym. Chem., 2015, 6, 2434–2444 RSC.
- G. Armstrong and M. Buggy, Mater. Sci. Eng., C, 2001, 18, 45–49 CrossRef.
- M. Ravey and E. M. Pearce, J. Appl. Polym. Sci., 1997, 63, 47–74 CrossRef CAS.
- G. G. Odian, Principles of polymerization, Wiley-Interscience, Hoboken, NJ, 2004 Search PubMed.
- P. C. Hiemenz and T. P. Lodge, Polymer Chemistry, Taylor & Francis, 2nd edn, 2007 Search PubMed.
- M. Tamami, S. T. Hemp, K. Zhang, M. Zhang, R. B. Moore and T. E. Long, Polymer, 2013, 54, 1588–1595 CrossRef CAS.
- E. P. Chang, J. Adhes., 1991, 34, 189–200 CrossRef CAS.
- K. Zhang, S. J. Talley, Y. P. Yu, R. B. Moore, M. Murayama and T. E. Long, Chem. Commun., 2016, 52, 7564–7567 RSC.
- K. Zhang, K. J. Drummey, N. G. Moon, W. D. Chiang and T. E. Long, Polym. Chem., 2016, 7, 3370–3374 RSC.
- S. Sivakova, D. A. Bohnsack, M. E. Mackay, P. Suwanmala and S. J. Rowan, J. Am. Chem. Soc., 2005, 127, 18202–18211 CrossRef CAS PubMed.
- A. Feula, A. Pethybridge, I. Giannakopoulos, X. Tang, A. Chippindale, C. R. Siviour, C. P. Buckley, I. W. Hamley and W. Hayes, Macromolecules, 2015, 48, 6132–6141 CrossRef CAS.
- A. Eisenberg, B. Hird and R. B. Moore, Macromolecules, 1990, 23, 4098–4107 CrossRef CAS.
- W. P. J. Appel, G. Portale, E. Wisse, P. Y. W. Dankers and E. W. Meijer, Macromolecules, 2011, 44, 6776–6784 CrossRef CAS.
- K. Zhang, A. M. Nelson, S. J. Talley, M. Chen, E. Margaretta, A. G. Hudson, R. B. Moore and T. E. Long, Green Chem., 2016, 18, 4667–4681 RSC.
- E. Arunan, R. Desiraju Gautam, A. Klein Roger, J. Sadlej, S. Scheiner, I. Alkorta, C. Clary David, H. Crabtree Robert, J. Dannenberg Joseph, P. Hobza, G. Kjaergaard Henrik, C. Legon Anthony, B. Mennucci and J. Nesbitt David, Pure Appl. Chem., 2011, 83, 1637 CAS.
- M. M. Coleman, K. H. Lee, D. J. Skrovanek and P. C. Painter, Macromolecules, 1986, 19, 2149–2157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01519k |
| This journal is © The Royal Society of Chemistry 2016 |