
Glutathione responsive polymers and their application in drug delivery systems
John F.
Quinn
*a,
Michael R.
Whittaker
a and
Thomas P.
Davis
*ab
aARC Centre of Excellence in Convergent Bio-Nano Science & Technology, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, Melbourne, Victoria 3052, Australia
bDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: john.f.quinn@monash.edu; thomas.p.davis@monash.edu
First published on 3rd October 2016
Abstract
Materials which respond to biological cues are the subject of intense research interest due to their possible application in smart drug delivery vehicles. In particular, novel polymers which respond to biochemical differences between the extra-and intracellular environments may be useful for preparing particles which can chaperone a therapeutic agent in the extracellular environment, and release said agent only when the particle is internalised by a target cell. To that end, polymers that exploit the elevated glutathione (GSH) concentration in the intracellular compartment are attracting substantial research effort. In this review we describe a number of different strategies for the preparation of glutathione responsive materials. In particular, we examine the use of GSH responsive linkers to prepare polymers that degrade upon exposure to millimolar concentrations of GSH, and the use of these polymers to prepare particles that disassemble at these concentrations. We also describe the use of such GSH responsive polymers in the controlled delivery of both chemotherapeutic agents and genetic material, and highlight a number of strategies employed to trigger release of an encapsulated drug using GSH. Additionally, we highlight some of the more novel GSH responsive systems which have recently been reported, and suggest further areas where GSH responsive materials are likely to see continued and highly focused research effort.
 John F. Quinn | Dr John Quinn received his PhD from UNSW in 2003. His thesis, jointly supervised by Professor Tom Davis and Dr Ezio Rizzardo (CSIRO), examined the application of ionizing radiation as an initiation source in reversible addition fragmentation chain transfer (RAFT) polymerization. He then undertook postdoctoral work at the University of Melbourne with Professor Frank Caruso (2003–2004), and was an ARC Australian Postdoctoral Fellow in the same group from 2005–2007. After a number of years outside the university sector, Dr Quinn returned to academic research in 2014 and is currently a Senior Research Fellow at Monash Institute of Pharmaceutical Sciences. His research is principally concerned with the use of novel macromolecules in drug delivery applications. |
 Michael R. Whittaker | Dr Michael Whittaker undertook his doctoral studies at the University of Queensland with Prof. Andrew Whittaker and Dr David J. T. Hill. In 2001 he joined Polymerat Pty Ltd as a senior research scientist, and then from 2004 to 2008 was a Senior Research Officer in the Department of Chemistry at the University of Queensland. In 2008, Dr Whittaker moved to the Centre for Advanced Macromolecular Design (CAMD) at UNSW where he was Research Manager until 2012. Since 2013 has been a Senior Research Fellow at the Monash Institute of Pharmaceutical Sciences (MIPS). His current work examines the use of stimuli-responsive materials for pharmaceutical applications. |
 Tom P. Davis | Professor Tom Davis is the Monash-Warwick Professor of Medical Nanotechnology at Monash University (Australia) and the Director of the Australian Research Council (ARC) Centre of Excellence in Convergent Bio-Nano Science and Technology. He also holds an appointment as Professor of Polymer Nanotechnology at the Department of Chemistry at the University of Warwick in the United Kingdom. Prof. Davis previously spent 21 years as a senior academic at the University of New South Wales, where he founded and directed two research centres: the Centre for Advanced Macromolecular Design (CAMD) and the Australian Centre for Nanomedicine (ACN). Prof. Davis’ research focuses on the application of polymer science and nanotechnology to therapeutic problems, and enhancing the fundamental understanding of how nanomaterials interact with biological systems. He is an author on over 400 peer-reviewed papers, and his work has received more than 25 |
1. Introduction
During the last decade there has been intensifying research interest in the preparation of particulate drug delivery systems.1,2 For example, using a passively or actively targeted particle to deliver a chemotherapeutic agent may be useful for reducing the side effects associated with systemic exposure to the agent.3,4 Additionally, particle based delivery also holds some promise for materials that are otherwise degraded before they can reach the site of action. As such, particle based delivery systems are of considerable interest for the delivery of genetic material such as small interfering RNA (siRNA) or plasmid DNA (pDNA).5 The breadth of materials being explored for these applications is dazzling, and of course it is inevitable that only a tiny fraction, if any, will ultimately reach the clinic. Nevertheless, there are important lessons to be learned from the preparation of smart drug delivery vehicles, and particularly in the chemistries that are employed to sequester and release the drug or genetic cargo.In many cases, drugs are encapsulated within polymeric particles by non-covalent associations such as van der Waals forces, hydrogen bonding, π–π stacking or electrostatic associations. The precise interaction (or combination thereof) which maintains a drug in a particle is not commonly elucidated, and researchers tend to rely on empirical evidence for encapsulation to support successful drug loading. In addition to physical association of therapeutic cargo, there are also examples in the literature in which drugs are covalently attached to the carrier through some type of labile linkage. However, successful drug loading into a particle is only half of the story: the cargo must also be released in order to exert a therapeutic effect. Release from a polymeric particle is normally facilitated by inducing some change to the polymer in the biological milieu of the targeted tissue. To this end, changes in intratumoral or intracellular pH, exposure to certain enzymes or temperature gradients have been proposed as potential release triggers.6,7 Changes in the redox environment between intra- and extracellular environments can also be harnessed as a possible stimulus for drug release,8 and the development of novel redox responsive systems is clearly receiving a great deal of research attention.9–11 One advantage of employing redox responsive materials is that there are countless redox processes which occur in the normal physiology of the intracellular environment: the oxygen/superoxide (O2/O2˙−) system, the nicotinamide adenine dinucleotide phosphate (NADP+/NADPH) system, the glutathione (GSH/GSSG) system (Fig. 1) and the thioredoxin TrxSS/Trx(SH)2 system, to name only four as examples. Of these, the GSH/GSSG system piques the interest of drug delivery researchers due to the high concentrations of GSH encountered in the intracellular compartment (1–10 mM).12 Interestingly, even within the intracellular compartment there are significant gradients of GSH concentration.13 Researchers have postulated that the high GSH concentration in the intracellular pool should be adequate to facilitate degradation of certain chemical linkages.14 Moreover, concentrations of GSH in the extracellular environment are typically 100–1000 times lower (1–10 μM), and as such materials can be engineered to have stability outside the cell and to release their cargo only when internalised. One of the more studied linkages for achieving this purpose is the disulphide bridge, for which the intracellular GSH concentration can be exploited to trigger thiol–disulfide exchange. In other words, exposure to the intracellular pool of GSH can be employed to “decrosslink” an otherwise disulphide stabilised material. A simple search on the terms “glutathione”, “responsive” and “polymer” yields 97 hits in the last two full calendar years (2014–2015), demonstrating that there is intense research activity in this area (Fig. 2). It should be noted that this is by no means an exhaustive search for materials exhibiting GSH responsive behaviour: not all papers describing GSH-responsive polymers would necessarily use the word “responsive”, or even “polymer”, for that matter. Nevertheless, we would contend that the trend observed over the last nine years is broadly indicative of the growth in this field (Fig. 2). Moreover, the ease with which in vitro assays can be performed has no doubt stimulated this intense interest in glutathione as a potential trigger for drug release: reduced glutathione is readily available and relatively inexpensive. That said, there is an increasing amount of literature which explores the application of these materials in vivo, and there are promising results that suggest GSH-responsive materials may be useful in pharmaceutical applications. We have an ongoing interest in exploiting thiol chemistry in tandem with reversible-addition fragmentation chain transfer (RAFT) polymerization to prepare next-generation nanomedicines15–19 and, as will be seen throughout, RAFT polymerization is frequently used in the preparation of GSH-responsive polymers due to the availability of thiocarbonylthio functionality at the chain end.
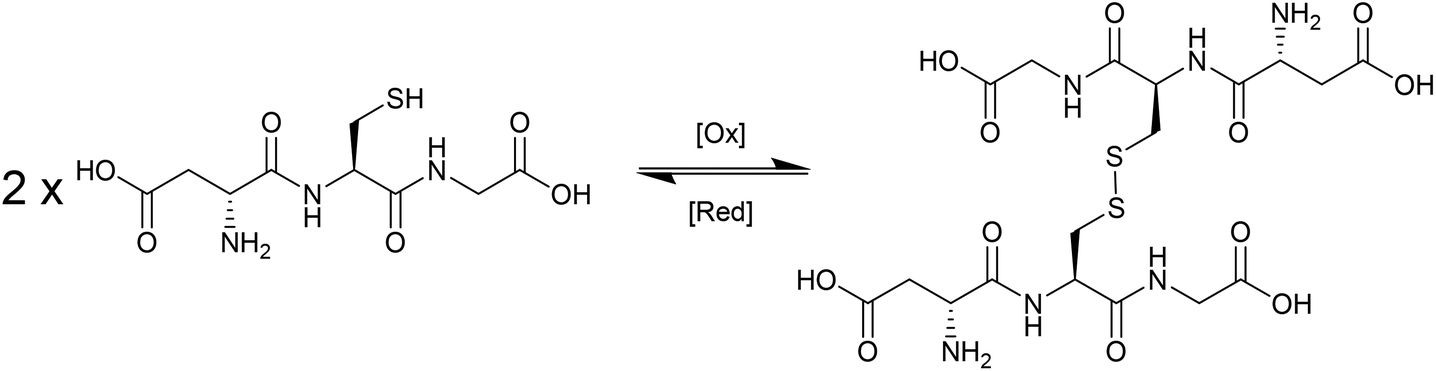 | ||
| Fig. 1 Reduced and oxidised forms of glutathione. | ||
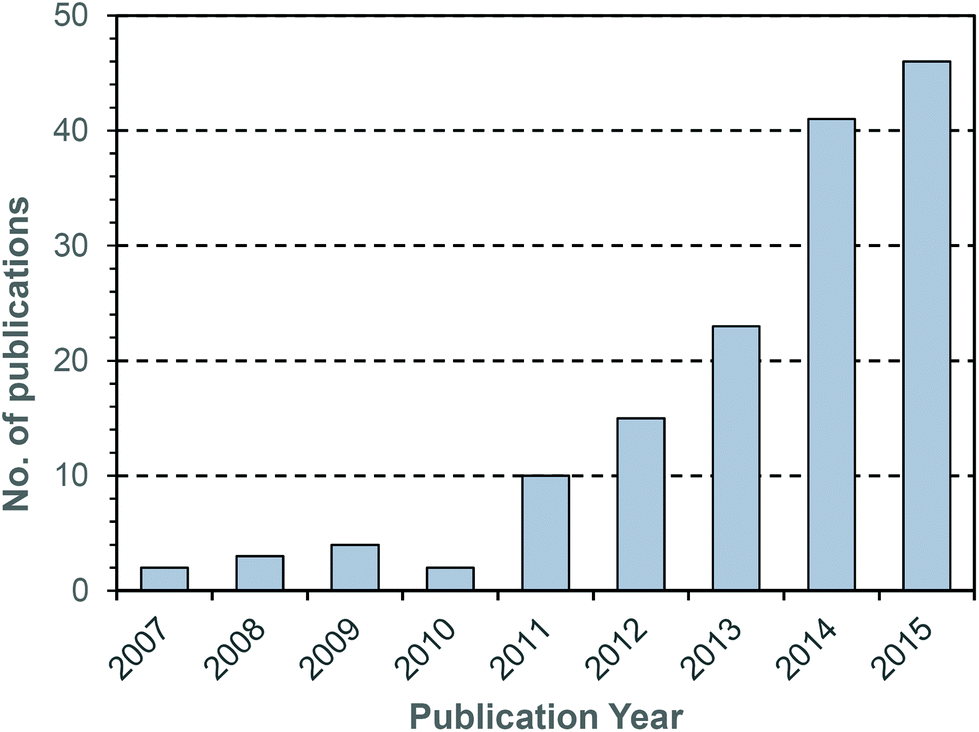 | ||
| Fig. 2 Number of publications per year obtained using the search terms “glutathione” and “responsive” and “polymer”. Data obtained from Thomson Reuters’ Web of Science® as at 29th July 2016. | ||
In this review, we will examine recent progress in the development of GSH-responsive polymer systems. We will briefly discuss the application of such materials in hydrogels and novel polymeric materials, and then explore recent examples in which glutathione responsive groups have been used in the preparation of responsive particles and drug delivery systems. In particular, we will examine the contrasting scenarios of drug release facilitated by degradation of the polymeric carrier versus cleavage of a GSH-responsive linker. We will provide some recent examples where GSH-responsive polymers been deployed in both siRNA and chemotherapeutic delivery, as well as mention some recent studies which have reported co-delivery of genetic material and drug for next generation cancer therapies. The use of GSH-responsive materials in imaging applications will also be described, as well as the development of new H2S releasing materials which release is triggered by a thiol stimulus. We will conclude with a brief discussion of the outlook for this important class of materials.
2. Glutathione reactive materials
(a) Reactive to GSH via thiol–disulphide exchange
In 2003 Bulmus et al. reported the synthesis of polymers containing pyridyl disulphide pendants, and demonstrated that these were highly reactive to thiol-containing compounds such as glutathione (Fig. 3).20 Specifically, the authors reported the synthesis of pyridyl disulphide acrylate (PDSA) by reacting acryloyl chloride with 3-hydroxypropyl pyridyl disulphide, and the subsequent terpolymerization of this monomer with butyl acrylate and methacrylic acid (MAA) in DMF at 65 °C. The resulting polymers were shown to react with GSH at concentrations of 0.01, 1 and 10 mM. The authors proposed that such materials may be useful in preparing disulphide-linked therapeutics wherein the active molecule could be released due to the elevated levels of GSH in the intracellular environment. Aside from the use of PDSA, pyridyl disulphide functionalised materials have been synthesized by numerous other approaches. For instance, pyridyl disulphide functionalised poly(vinyl pyrrolidone) has been prepared by modifying a carboxylated PVPON precursor.21 Pyridyl disulphide chemistry has become a mainstay in the preparation of GSH responsive materials, and there are numerous examples in the recent literature where the pyridyl disulphide group has been used to excellent effect in the preparation of novel GSH responsive polymers.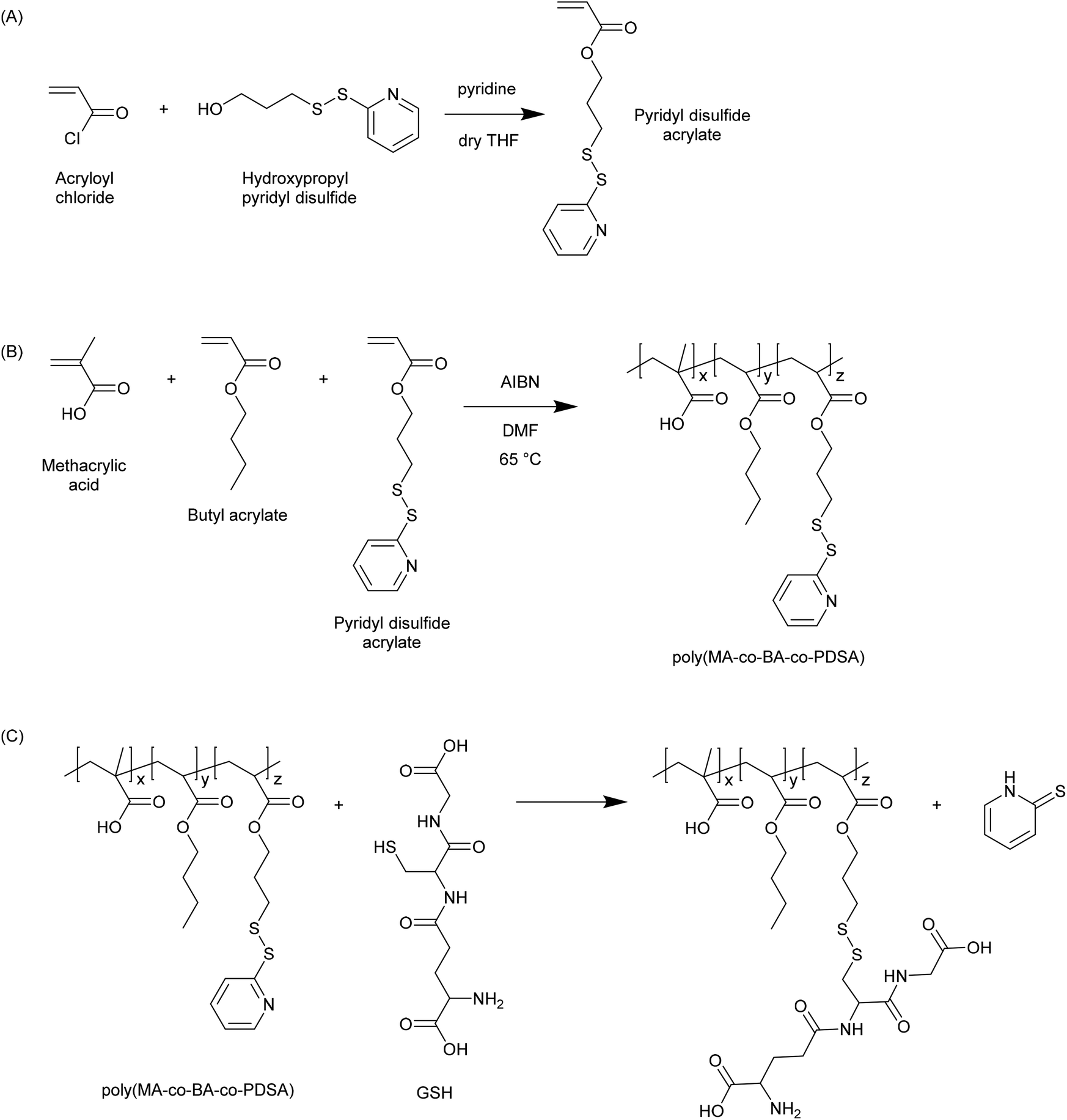 | ||
| Fig. 3 Synthesis of pyridyl disulphide functional polymers as reported by Bulmus et al.20 (A) Synthesis of pyridyl disulphide acrylate (PDSA) monomer. (B) Synthesis of poly(methacrylic acid-co-butyl acrylate-co-pyridyl disulphide acrylate) (poly(MA-co-BA-co-PDSA)). (C) Reaction of poly(MA-co-BA-co-PDSA) with glutathione. | ||
Armes et al. also reported a prototypical GSH-responsive polymer system comprised of an ABA triblock copolymer with a disulphide linkage in the middle of the central block.22 This was achieved by employing bis[2-(2-bromoisobutyryloxy)ethyl] disulfide as a difunctional atom transfer radical polymerization (ATRP) initiator to synthesize an ABA triblock copolymer with a central block of poly(2-(methacryloyloxy)ethylphosphorylcholine) (PMPC) and surrounding segments of PNIPAM. By exploiting the thermoresponsive nature of PNIPAM, the authors were able to prepare a free standing gel at physiological conditions (pH 7.4 and 37 °C) using aqueous solutions with a concentration above 8% (w/v). Importantly, exposure to GSH at the same conditions led to the gels to revert to free flowing solutions. These results demonstrate that GSH can be employed to trigger macroscopic changes to appropriately designed polymer materials.
There has also been some interest in the preparation of hybrid organic–inorganic nanomaterials with GSH responsive behaviour. You et al. prepared multiwalled carbon nanotubes (MWNT) with pyridyl disufide functionality, and then used these to conjugate a thiol-terminated PNIPAM to the surface of the MWNT.23 The polymer coating could be removed by exposure to 15 mM GSH, while exposure to 5 μM had no impact on the PNIPAM coating. Importantly, the concentrations explored correspond to the intra- and extracellular concentrations, indicating that the polymer coating could be engineered to mask the surface prior to internalisation, and then reveal the underlying material once internalised.
(b) Reactive to GSH via Michael addition
In addition to reaction via thiol–disulfide exchange, the thiol of GSH is also potentially reactive via Michael addition. Wang et al. have exploited this reactivity to prepare GSH–polymer conjugates (Fig. 4).24 In this case, a copolymer of a protected glycomonomer (3-O-methacryloyl-1,2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5,6-di-O-isopropylidene-D-glucofuranose, MAIpGlc) and 2-hydroxyethyl methacrylate (HEMA) was first prepared. The pendant hydroxyl functionality afforded by the HEMA units was then reacted with acryloyl chloride to give polymers with pendant acrylate functionality. Following deprotection of the glycopolymer units, the acrylate groups were reacted with GSH via Michael addition. The resulting thioether linkages are, of course, far less labile than the disulfides formed by thiol–disulfide exchange, and so such glutathione reactive materials are unlikely to be applicable in a drug delivery scenario. Nevertheless, the use of maleimide functionalised polymer in an inverse opal templated using a colloidal silica crystal has been used to provide a self-reporting sensing platform for sensitively detecting the reduction of oxidized glutathione by certain enzymes.25 As such, so called thiol–ene GSH reactive materials may have some applications in sensing, if not drug delivery.
:5,6-di-O-isopropylidene-D-glucofuranose, MAIpGlc) and 2-hydroxyethyl methacrylate (HEMA) was first prepared. The pendant hydroxyl functionality afforded by the HEMA units was then reacted with acryloyl chloride to give polymers with pendant acrylate functionality. Following deprotection of the glycopolymer units, the acrylate groups were reacted with GSH via Michael addition. The resulting thioether linkages are, of course, far less labile than the disulfides formed by thiol–disulfide exchange, and so such glutathione reactive materials are unlikely to be applicable in a drug delivery scenario. Nevertheless, the use of maleimide functionalised polymer in an inverse opal templated using a colloidal silica crystal has been used to provide a self-reporting sensing platform for sensitively detecting the reduction of oxidized glutathione by certain enzymes.25 As such, so called thiol–ene GSH reactive materials may have some applications in sensing, if not drug delivery.
 | ||
| Fig. 4 Preparation of acrylate-functionalised glycopolymers reactive to GSH by Michael addition, as reported by Wang et al.24 | ||
(c) Reactive to GSH via thiol–thioester exchange
An alternative approach in the preparation of GSH responsive polymers is to prepare materials with pendant thioester linkages. To this end, Liu and coworkers have prepared copolymers of N,N-dimethylacrylamide (DMA) and pyridyldisulfide ethylacrylamide (PDSEA) via RAFT polymerization (Fig. 5).26 By sequentially reacting the pyridyl disulfide functional polymers with TCEP and thymine thioester, the authors were able to prepare so-called thymine-conjugated biodynamer. In the presence of relatively high concentrations of GSH (0.1 M) these materials underwent a thiol–thioester exchange to reveal thiol functionality on the polymer and form a GSH–thymine thioester adduct. This reaction can potentially be used to trigger a solubility switch or crosslinking event within the polymer, although further optimisation is required for the materials to respond to physiological concentrations of GSH.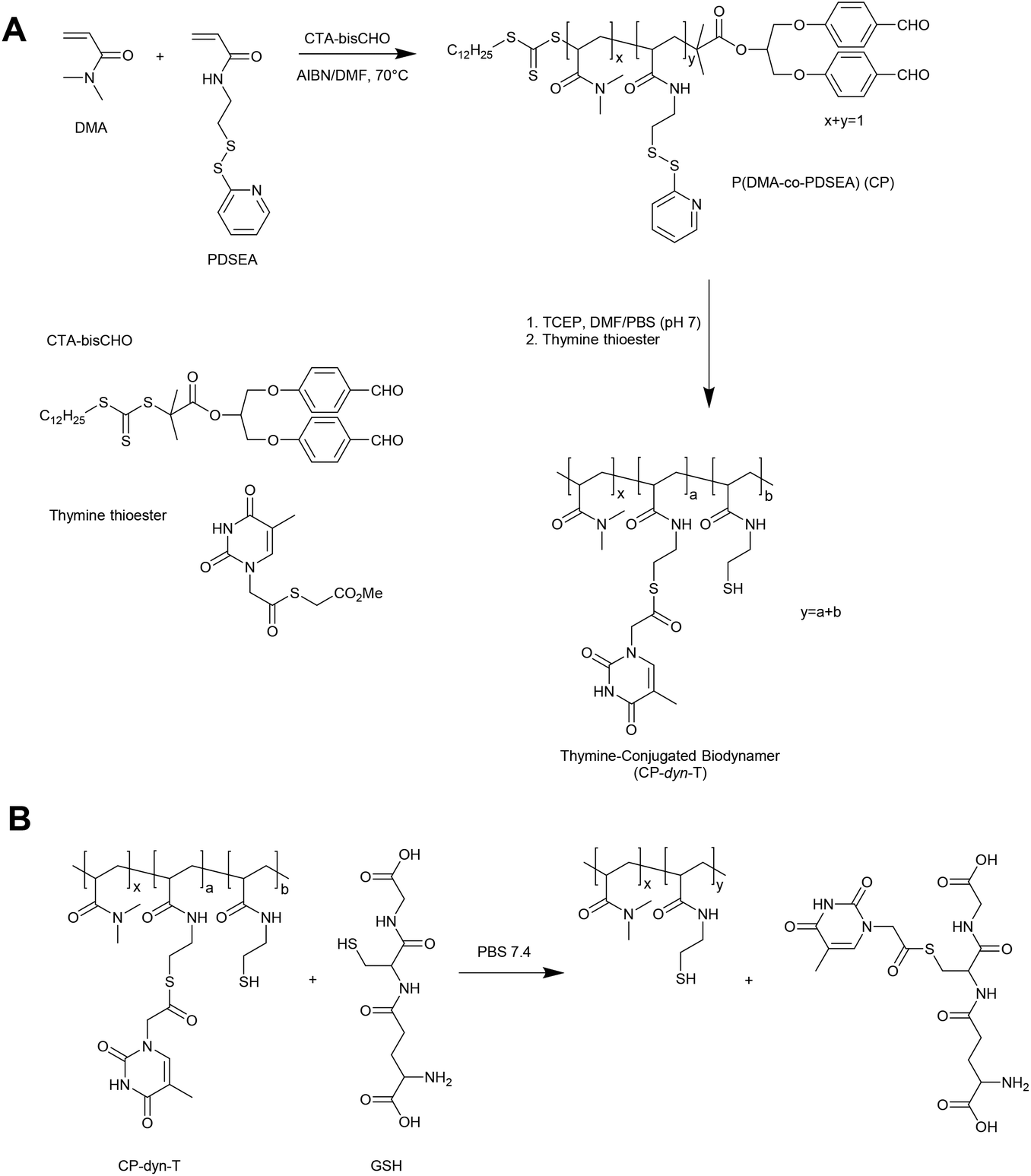 | ||
| Fig. 5 Synthesis of the pyridyl disulfide-functionalized copolymer P(DMA-co-PDSEA) and subsequent preparation thymine-conjugated biodynamer (CP-dyn-T) via thiol–thioester exchange. Adapted from L. Liu et al., Polym. Chem., 2015, 6, 3934–3941 © 2015 Royal Society of Chemistry. | ||
3. Glutathione triggered responsive materials
(a) Polymer disassembly
Materials which are degradable in response to a glutathione stimulus have attracted considerable attention. In almost all cases, the degradation is triggered by a thiol–disulfide exchange reaction, wherein a crosslink is effectively broken and one side of the crosslink is replaced with a small molecule thiol. Through this reaction the crosslinked polymer is essentially fragmented into soluble units which may themselves be either polymeric or monomeric, depending on the specific system employed.Caruso and coworkers developed PMAA films which were stabilised with disulphide linkages using a layer-by-layer methodology.27 In this work, the PMAA was functionalised with cysteine and then assembled into a thin film via sequential alternate adsorption with poly(vinyl pyrrolidone) (PVPON). The film buildup was facilitated by hydrogen bonding between the protonated carboxylic acid groups of the PMAA and the pyrrolidone units of the PVPON. The cysteine units enabled formation of disulphide crosslinks between the PMAA chains, and then when the pH was elevated (deprotonating the PMAA and eliminating the hydrogen bonds with PVPON) the disulphide links effectively stabilised the PMAA film. These crosslinks were subsequently broken by exposure to dithiothreitol (DTT), although subsequent reports from the same group have employed glutathione as the stimulus for breaking the crosslinks and facilitating film erosion.28–30 Importantly, this system has been effectively applied to both planar and particulate substrates. A similar approach has been employed by Haynie and Li,31 although in that case the film assembly was performed using polypeptides and the crosslinking was thus effected by the direct inclusion of cysteine in the polypeptide units.
Grayson and coworkers have reported hydrogels crosslinked via disulphide linkages and degradable via exposure to GSH.32 In this case the materials were prepared by the free radical copolymerization of HEMA with the difunctional crosslinker bis(2-methacryloxyethyl) disulfide (DSDMA). Inclusion of a disulphide linkage in the crosslinker enables the hydrogels prepared to be readily degraded by exposure to GSH. Importantly, under non-reducing conditions the materials exhibit comparable stability to hydrogels prepared with a crosslinker having an ether link in place of the disulphide. These materials were shown to elute a dye at a rate which could be altered by exposure to millimolar concentrations of GSH, indicating a role for crosslink density in the release mechanism.
Du Prez and coworkers have prepared hydrogel materials stabilised by disulfides using Michael addition chemistry.33 Specifically, amine terminated trifunctional polyethylene glycol was reacted with a difunctional maleimide incorporating a disulphide linkage (dithiobis(maleimido)ethane). Subsequent exposure of the resulting hydrogels to GSH yielded soluble products, and the materials were shown to be suitable for fibroblast culture.
Phillips and Gibson reported an elegant methodology for the preparation of polydisulfides using prepolymers prepared via RAFT polymerization.34 In this case, a RAFT agent (trithiocarbonate) with a pyridyl disulphide group on the R group was employed to prepare poly(N-isopropylacrylamide) (PNIPAM), poly(dimethyl acrylamide) (PDMA) and poly(oligoethylene glycol methyl ether methacrylate) (POEGMA). The resulting polymers included a trithiocarbonate at one end of the chain and a pyridyl disulphide at the other end. Subsequent exposure of these materials to ethanolamine led to conversion of the thiocarbonylthio endgroup to thiol, which rapidly reacts with the pyridyl disulphide endgroups to facilate a condensation between the polymer chains. Exposure to triphenyl phosphine or GSH was shown to reverse the polycondensation, and in the case of GSH this was demonstrated to be concentration dependent. In an interesting extension of this work, the same group employed pyridyl disulphide-terminated PNIPAM to demonstrate that LCST could be modified by exposure to GSH.35 Further, pyridyl disulphide-terminated PNIPAM was successfully used to prepare nanoaggregates at physiological temperature.36 When the nanoaggregates were exposed to GSH under these same conditions, an endgroup modification occurs which increases the LCST and therefore facilitates disassembly of the nanoaggregate. This so-called isothermal transition is an elegant way to trigger release of an encapsulated cargo.
An alternative method for the formation of GSH-responsive hydrogels was reported by Yang et al.37 In this case the authors first prepared a polyfunctional (4 arm) PEG in which each arm was terminated with a thiol group. Exposure of the thiol terminated PEG to hydrogen peroxide facilitated oxidation of the thiols to form disulphide linked hydrogels. The materials were shown to be degradable in the presence of GSH, with the time frame highly dependent on the GSH concentration. Millimolar concentrations led to degradation in a matter of hours, while micromolar concentrations facilitated degradation over a number of days. These materials could be loaded with an osteoinductive factor (recombinant human bone morphogenetic protein-2 (rhbMP-2)), and their utility in bone regeneration was demonstrated.
Chitosan based hydrogels have also been prepared with GSH-responsive linkages.38 This was achieved by reacting chitosan with dimethyl 3,3′-dithiobispropionimidate (DTBP) to form crosslinks incorporating a disulphide bridge. Interestingly, the reaction between dimethyl 3,3′-dithiobispropionimidate and chitosan results in the formation of imidoamide bonds, and it was demonstrated (using ethylenediamine) that these links are also labile to nucleophilic attack by amines. As GSH includes both a thiol and an amine group, it was hypothesized that the degradation of this particular hydrogel occurred through a hybrid mechanism incorporating both thiol–disulfide exchange and nucleophilic attack on the imidoamide.
Redox-responsive hydrogels have also been synthesized using poly(vinyl alcohol) (PVA) and 4-mercaptophenylboronic acid (MPBA).39 The interaction between boronic acid and PVA is well-known and has been widely employed in hydrogel synthesis. By use of the thiol containing boronic acid, these authors were able to assembled a hydrogel by mixing the components in an oxidative environment (hydrogen peroxide or atmospheric oxygen). Interestingly, the gels formed could be dissembled not only in GSH (1–7 mM), but also with exposure to D-glucose (10–250 mM).
Liang and Kiick have recently reported an interesting system in which maleimide-functional liposomes are used with thiol-terminated multifunctional PEG to form a crosslinked hydrogel network.40 Two different thiol endgroups were employed: alkyl thiol and aryl thiol. The authors demonstrate that exposure of the resulting networks to 10 mM GSH results in appreciable mass loss only when aryl thiols are used for the gel formation. Moreover, the materials show good stability when exposed to much lower concentrations of GSH (10 μM). The use of succinimide thioether exchange in the presence of thiols represents a novel approach for rendering materials responsive to elevated levels of GSH.
GSH-degradable polyurethanes have been successfully synthesized using diamines derived from cysteine and bischloroformates derived from alditols having L-arabino or xylo configuration.41 These step growth polymers had molecular weights typical for such materials (5–25 K), and were shown to be reduced by both hydrolysis and exposure to GSH. Importantly, the degradation induced by glutathione was faster and more substantial than that induced purely by hydrolysis.
Kim and coworkers have recently reported on the preparation of redox-degradable hyperbranched polyglycerols (Fig. 6).42 These materials were prepared by polymerizing a novel disulphide-containing glycerol monomer (2-((2-(oxiran-2-ylmethoxy)ethyl)disulfanyl)ethan-1-ol) via anionic ring-opening polymerization from a trimethylolpropane initiator. Both homopolymer and copolymers with glycidol were prepared, and the polymerization kinetics followed using 13C NMR. The resulting hyperbranched materials were examined for their stability against a reducing agent (DTT), and were shown to degrade into smaller segments after only 1 hour. The materials were also tested for cytoxicity against WI-38 (human diploid cells) and HeLa (human epithelial carcinoma cells), and were well tolerated up to 100 μg mL−1.
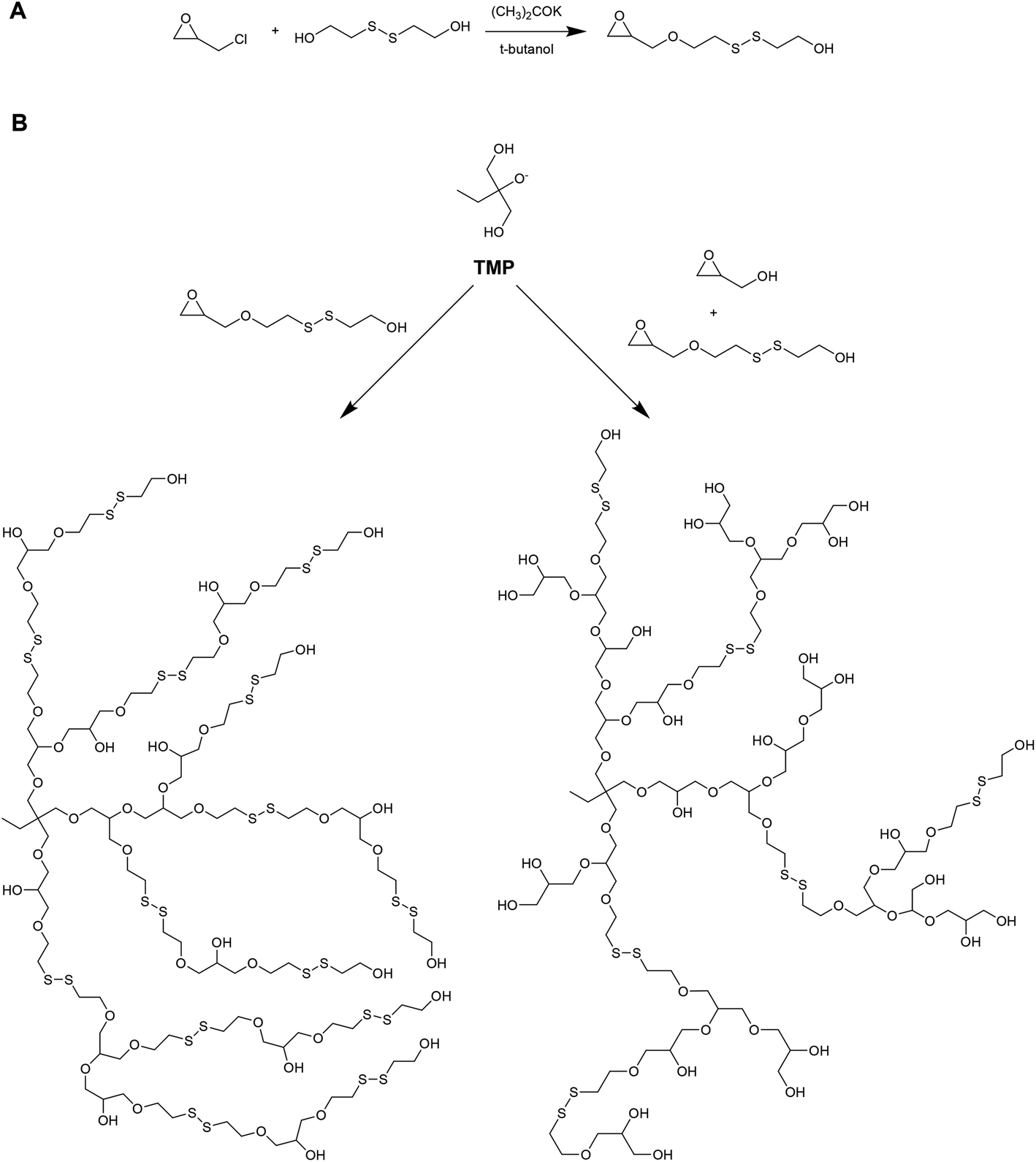 | ||
| Fig. 6 (A) Synthesis of the redox-active disulfide monomer 2-((2-(oxiran-2-ylmethoxy)ethyl)-disulfanyl)ethan-1-ol. (B) Synthesis of (i) homopolymers of 2-((2-(oxiran-2-ylmethoxy)ethyl)-disulfanyl)ethan-1-ol, and (ii) copolymers of 2-((2-(oxiran-2-ylmethoxy)ethyl)-disulfanyl)ethan-1-ol and glycidol. Adapted from Son et al., Macromolecules, 2015, 48, 600–609. © 2015 American Chemical Society. | ||
Glutathione responsive moieties have also been successfully incorporated into poly(ester triazoles).43 In this case, 3,3′-dithiobispropionic acid was first esterified with propargyl alcohol to provide a difunctional alkyne. A corresponding trifunctional azide (tris(hydroxymethyl)ethane tri(4-azidobutanoate)) was prepared by reacting 1,1,1-tris(hydroxymethyl)ethane sequentially with 4-chlorobutyryl chloride and sodium azide. Copper catalyzed azide–alkyne cycloaddition (CuAAC) was then employed to facilitate polymerization. Exposure of the resulting materials to DTT demonstrated that thiol–disulfide exchange was effective for degrading the materials in a matter of hours. Although GSH was not employed in this case, these materials are highly likely to respond similarly to GSH as they do to DTT, albeit more slowly due to the slightly poorer reducing ability of GSH compared to DTT.
Gan et al. have recently reported on the preparation of biodegradable, thermoresponsive PNIPAM-based hydrogel scaffolds prepared by free radical polymerization.44 In this case, the authors employ a terpolymerization of poly(ε-caprolactone dimethacrylate) (PCLDMA), N,N′-bis(acryloyl)cystamine and NIPAM using 1,4-dioxane as solvent and AIBN as the initiator to prepare the GSH responsive hydrogel. Subsequent exposure of the hydrogels to 3 mM GSH leads to dramatic changes in the pore morphology, although it must be noted that hydrolytic degradation of the caprolactone linkages could also contribute to these observations.
(b) Particle disassembly
In addition to degrading polymeric materials into smaller molecular units, GSH can also be used to trigger the disassembly of particles formed from certain polymers. For instance, Liu et al. have recently reported on the synthesis of ABA triblock copolymers incorporating a GSH-responsive aliphatic middle segment between two PEG blocks.45 1,6-Hexanethiol was first reacted with 2,2′-dithiodipyridine to give pyridyl disulphide functionalised 1,6-hexanethiol. This could then be readily reacted with thiol terminated PEG to yield the target material. Subsequent exposure of the ABA triblock to DTT was shown to destabilise micelles formed from the polymer, as demonstrated by loss of fluorescence due to encapsulated Nile Red.Zhang and coworkers have reported on use of diselenide containing materials as GSH-responsive materials.46 In this case a polyurethane was prepared using toluene diisocyanate and a diselenide containing diol, and this was subsequently incorporated into the middle block of an ABA triblock copolymer with PEG. The materials formed micelles due to the mismatch in hydrophilicity between the respective domains, and these micelles were shown to successfully encapsulate Rhodamine B. Moreover, the micelles were sensitive to both oxidant (hydrogen peroxide) and reductant (glutathione), and either material could be used to trigger release of the dye. A similar diselenide functionalised material has been reported by Xu and co-workers,47 although in this case cationic units were incorporated via inclusion of 2,2′-(piperazine-1,4-diyl) diethanol in the polyurethane synthesis, and the micelles were coassembled with commercially available 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy-(poly(ethylene glycol))-2000]. Disruption to the micelles was observed when exposed to relatively low levels of GSH (0.05 mM), although the impact was not as dramatic as exposure to hydrogen peroxide. Diselenide linkages have also been incorporated into a supramolecular polymer assembled from pillar[5]arene dimer and a neutral guest.48 In this particular example the diselenide is built in to the pillar[5]arene dimer, and so the supramolecular assembly can be disrupted by exposure to thiols such as GSH, leading to a loss of macroscopic structure. For a comprehensive treatment of selenium-containing materials as redox responsive materials more generally, the reader is referred to an excellent review on the subject by Zhang and coworkers.49
Cajot et al. have also reported on the preparation of micelles from ABA triblock copolymers and the incorporation of GSH sensitive linkages to stabilise these materials.50 In this case the core was formed using the central (B) domain of polycaprolactone which incorporated azide functionality, while the corona was provided through the PEG (A) blocks. The polycaprolactone core was crosslinked using copper catalyzed azide–alkyne cycloaddition with a suitable di-alkyne having a disulphide bridge. The resulting micelles were effectively stabilised by the disulphide and were shown to disassemble upon exposure to GSH at 10 mM. Interestingly, the authors were able to precisely control the locus of the azide groups within the PCL block, and were thus able to examine whether the location of the crosslinks with the hydrophobic core substantially affects the micelle stability.
Cellulose-based nanogels with GSH reactive crosslinks have been prepared by grafting polymeric pendants via ATRP.51 Specifically, hydroxypropylcellulose was successfully modified by reaction with bromoisobutyryl bromide to provide initiation sites for ATRP. Copolymerization of oligo(ethylene glycol) monomethyl ether methacrylate (OEGMA) and a monomer incorporating a disulphide linkage (HMssEt) provided the final cellulose graft copolymer. Reduction of the disulphide linkages with DTT and subsequent reoxidation of the generated thiols with time provided disulphide crosslinked nanogels. These disulphide crosslinked nanogels were shown to disintegrate upon exposure to 10 mM GSH, as characterised using DLS.
Tang and co-workers have explored GSH as a trigger for the release of encapsulated protein from within nanogels formed by free radical polymerization.52 In this example the particle was formed by first dispersing the protein (caspase-3, bovine serum albumin or enhanced green fluorescent protein) in buffer, after which acrylamide, N-(3-aminopropyl) methacrylamide (APMAAm) and a difunctional crosslinker (either N,N′-methylenebisacrylamide or N,N′-bis(acryloyl)cystamine) were added. N,N′-Bis(acryloyl)cystamine was employed to yield particles having glutathione responsive crosslinks, whereas N,N′-methylenebisacrylamide afforded stable crosslinked materials. Formation of the encapsulated proteins was achieved by initiating the free radical polymerization of the monomers with ammonium persulfate. Protein release was observed from the disulphide crosslinked particle when exposed to GSH, and this was observed to proceed in a concentration dependent manner (i.e., faster for 2 mM than for 0.5 mM). Moreover, delivery of caspase-3 using the glutathione responsive particles was effective in inducing apoptosis in HeLa, MCF-7 and U-87 MG cells, thus demonstrating the potential utility of this system.
Random copolymers have also been used in the production of glutathione responsive systems. For example, copolymerization of either ethyl acrylate (EA) or methyl methacrylate (MMA) with tert-butyl methacrylate, followed by removal of the tert-butyl groups with HCl, has been used to prepare the carboxylic acid functionalised polymers poly(EA-co-MAA) and poly(MMA-co-MAA).53 After assembling into particles, the carboxylic acid groups were used to conjugate cystamine as a means of incorporating disulphide crosslinks. The resulting particles were stable (although swollen) when challenged by an elevation of pH, but could be disassembled by exposure to GSH at 5 mM. These results demonstrate that more sophisticated molecular architectures (e.g., block copolymers) are not necessarily essential in the preparation of GSH-responsive materials.
Caruso and co-workers have prepared disulphide stabilised particles based on poly[(N,N-diisopropylamino)ethyl methacrylate] (PDPA).54 In this case N,N′-(diisopropylamino)ethyl methacrylate was first copolymerized with 2-(2-(2-(3-(trimethylsilyl)prop-2-ynyloxy)ethoxy)ethoxy)ethyl methacrylate to prepare a polycation having pendant alkyne groups. This could then be assembled on a planar or colloidal surface via the LbL methodology using PMAA as a polyanion. The pendant alkyne functionalities were then used to crosslink the film via introduction of a disulphide-containing bisazide crosslinker, which reacts through CuAAC. Elevation of pH leads to deprotonation of the PDPA and loss of the PMAA due to abolition of the electrostatic interactions, yielding stable particles incorporating essentially PDPA and disulphide-containing crosslinks. The particles formed could be degraded in the presence of GSH (5 mM). Moreover, by varying the amount of crosslinker used the degradation kinetics could be effectively tuned, demonstrating the important contribution of crosslink density to particle stability.
Harada and coworkers have incorporated GSH-responsive properties into vesicles assembled from poly(amidoamine) dendron-poly(lysine) block copolymer (PAMAM dendron-PLL).55 The vesicles were first assembled in a mixture of water and methanol, and then stabilised by crosslinking with iminothiolane. Dialysis against water yielded well-defined vesicles stabilised through disulphide moieties. Exposure to GSH at concentrations from as little as 5 μM were shown to impact on vesicle stability, with concentrations above 50 μM effective for releasing an encapsulated dye.
Guan and coworkers have reported on polymer nanogels prepared using copolymers of dicysteine pentafluorophenolate with either oligoethyelenimine or tetra-L-lysine.56 These polymers were assembled into particles using a coacervation method, whereby a non-polar solvent such as diethyl ether or methyl tert-butyl ether was added to a solution of the polymer in a polar solvent such as DMF or DMSO. The coacevated droplets were subsequently crosslinked by addition of a suitable crosslinker (such as homobifunctional PEG N-hydroxysuccinimide or dimethyl suberimidate) and a base. The resulting particles were stable at pH 7.4, but completely disintegrated within 90 min when exposed to 10 mM GSH. Moreover, the materials were well tolerated by 3T3 fibroblasts. This approach highlights an alternative methodology whereby the GSH sensitive unit is incorporated into a prepolymer rather than through the crosslinker applied in particle formation.
Tardy et al. have examined the use of polyrotaxane assemblies incorporating a disulphide linkage as a route to prepare GSH-responsive particles.57 In this case the authors start by threading α-cyclodextrins onto bis(o-pyridyldisulfide) poly(ethylene glycol) using literature methods, followed by capping with an tri-alkyne functionalised aromatic thiol. CuAAC is then used to incorporate additional PEG pendants into the material. The functionalised polyrotaxanes are shown to form assemblies with hydrodynamic diameter ∼200 nm, and to be readily degraded by exposure to GSH (5 mM). These results demonstrate the potential complexity which can be employed when assembling GSH responsive particles.
You and coworkers have prepared silica nanoparticles coated with a GSH-responsive polymer shell using a novel approach involving surface precipitation and crosslinking.58 Initially, a polymer incorporating disulphide linkages was synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization of 2-(2-methoxyethoxy)ethyl methacrylate (MEO2MA) and OEGMA. A difunctional monomer, N,N′-bis(acryoyl)cystamine, was added to facilitate branching of the polymer. The obtained material exhibited an LCST of approximately 45 °C, and so heating to 50 °C led to the polymer precipitating on the surface of the particle where intermolecular disulphide exchange facilitated the formation of a crosslinked polymer shell. Importantly, this approach was not limited to polymers prepared using free radical approaches such as RAFT: a similar approach was employed with a polymer synthesized through Michael addition polymerization of N,N′-dimethyldipropylenetriamine (DMDPTA) and N,N′-bis(acryloyl)cystamine. Importantly, exposure to GSH was effective for removal of the shell, indicating the potentially bioresponsive nature of these materials.
Whittaker and coworkers have reported on the preparation of core-crosslinked star (CCS) polymers incorporating a glutathione responsive linkage in the polymer core.59 Initially, linear polymers were synthesized by polymerizing an initial block of poly(ethylene glycol)methyl ether methacrylate (PPEGMA) followed by a second copolymeric block of 2-(dimethylamino)ethyl methacrylate (DMAEMA) and 2,2,2-trifluoroethyl methacrylate (TFEMA). These linear “arm” polymers were then formed into a GSH-responsive CCS polymer by chain extension with bis(2-methacryloyl)oxyethyl disulfide (DSDMA). A control material was synthesized without a GSH responsive linker by extending the arm with ethylene glycol dimethacrylate (EGDMA) and butyl methacrylate (BMA). The authors demonstrated that exposure to 10 mM GSH lead to some degradation of the CCS polymers, noting that faster and more complete disintegration is observed with the stronger reducing agent tris(2-carboxyethyl)phosphine (TCEP).
Cunningham et al. have explored block copolymers of poly(L-lactide) (PLA) and PEG with a central disulphide linkage as redox responsive systems.60 The polymers were prepared by ring opening polymerization of L-lactide initiated with 2-hydroxyethyl disulphide, followed by carboxylation of the PLA with succinic anhydride. PEG chains were then coupled to the PLA via esterification. The resulting polymers formed micellar structures which could be degraded in the presence of DTT. Moreover, after loading with doxorubicin the authors demonstrated that exposure to 10 mM GSH could lead to triggered release of the loaded drug. These results attest to the broad spectrum of materials that can be used to prepare glutathione responsive systems.
Ghosh and coworkers have recently reported the synthesis of ABA triblock copolymers incorporating central poly(disulphide) domain bracketed by segments of poly(triethylene glycol monomethyl ether)methacrylate (PTEGMA).61 The poly(disulphide) domain was synthesized by step polymerization of 2,2′-dithiodipyridine with either 2,2′-(ethane-1,2-diylbis(oxy))-diethanethiol or 1,6-hexanedithiol. The resulting pyridyl disulphide capped prepolymer was then reacted with mercaptoethanol to give hydroxyl terminated poly(disulphide). The terminal hydroxyls were then reacted with 2-bromoisobutyrylbromide to give a macromolecular initiator for ATRP. These initiators were used to polymerize 2-(2-(2-methoxyethoxy) ethoxy)ethyl methacrylate, thus yielding the target ABA triblock copolymer. The resulting polymers were shown to assemble into micelles, encapsulate a dye (Nile Red) and to degrade in the presence of GSH with concomitant release of the dye. Importantly, the choice of comonomer in the initial synthesis of the poly(disulphide) was shown to significantly impact the degradation kinetics in the presence of GSH. Specifically, the use of a more hydrophobic comonomer (1,6-hexanedithiol) led to slower degradation of the micelles in the presence of GSH.
Monodisperse GSH-responsive polymer capsules have been prepared by precipitation polymerization of vinylcaprolactam (VCL) or VCL/MAA onto dimethyldiethoxysilane (DMDES) emulsion droplets, followed by removal of the DMDES by dissolution with ethanol.62 Inclusion of a disulphide containing crosslinker, N,N′-bis(acryloyl)cystamine, in the polymerization system enabled the preparation of capsules which could be degraded in the presence of GSH. In this case little difference in degradation kinetics was observed between 10 mM and 20 mM GSH. Moreover, the capsules could be effectively loaded with doxorubicin via the strong affinity of doxorubicin for MAA units, and the cargo released through disintegration of the particle in the presence of GSH.
Glutathione-responsive block copolymer vesicles have been successfully prepared using a block copolymer having segments of PEG and poly(cholesteryl acryloyloxy ethyl carbonate) (PAChol).63 The block copolymer is synthesised such that a disulphide linkage is included between the PEG and PAChol domains. The PEG–PAChol is shown to form vesicles via nanoprecipitation, with these vesicles capable of encapsulating calcein. Exposure to GSH led to vesicle degradation, as characterised by calcein release and TEM observations of the partially degraded vesicles. However, in this case the concentrations of GSH necessary to degrade the vesicles were quite high (∼35 mM), and such concentrations unlikely to be encountered physiologically. The authors attribute this observation to reduced diffusion of GSH into the vesicle, and as such there is some scope for reengineering the system to such that it is responsive to lower concentrations of GSH.
Kempe et al. have reported the preparation of particles based on poly(2-ethyl-2-oxazoline) brushes with pendant thiol groups (PEtOxMASH).64 The brushes were synthesized by ATRP of oligo(2-ethyl-2-oxazoline)methacrylate and glycidyl methacrylate (GMA). Ring opening of the oxirane afforded by the GMA enabled incorporation of pyridyl disulphide groups. The PEtOxMASH brushes were assembled into thin films on either planar or colloidal substrates by sequential adsorption with PMAA, a complementary hydrogen bonding donor. After crosslinking by overnight heating, subsequent exposure to 5 mM GSH resulted in disintegration of the particles in a matter of hours. These materials are interesting insofar that the use of poly(2-ethyl-2-oxazoline) represents an alternative antifouling polymer to the commonly employed PEG.
(c) GSH-responsive gates for porous materials
Porous inorganic materials are attracting an increasing amount of attention in the preparation of drug delivery systems. Nanoporous or mesoporous silica is particularly interesting in that the pores can be employed to sequester therapeutic agent to reasonably high loadings. In order to retain the loaded drug, stimuli responsive “gates” are often employed. Given the elevated concentrations of GSH found intracellularly, GSH responsive polymers are ideal candidates for the formation of gates on porous inorganic materials. This is perhaps one of the more innovative areas in which GSH responsive materials have been deployed.Feng and co-workers first reported the preparation of mesoporous silica with disulphide functionalised polymeric gates in 2008.65 Specifically, these authors conjugated poly(N-acryloxysuccinimide) (PNAS) to the pore entrance on MCM-41 mesoporous silica particles. The use of PNAS enables relatively straightforward crosslinking with cystamine. To examine whether this procedure was effective for encapsulation and release of a model drug, the PNAS functionalised MCM-41 was first loaded with the dye Rhodamine B prior to crosslinking. It was demonstrated that the dye remained encapsulated until the addition of the reducing agent DTT, which led to cleavage of the disulphide and concomitant release of the dye. Importantly, the rate of release was shown to be dependent on the concentration of the DTT, suggesting that the release may be able to be tuned between the micromolar concentrations of thiol in the bloodstream and the millimolar concentrations found intracellularly. The same authors have extended this concept to incorporate host–guest interaction gates into MCM-41 mesoporous silica.66 In this case, β-cyclodextrin is tethered to the PNAS via a disulphide bridge, and a difunctional azolinker is used to close the molecular gates by interacting with the β-cyclodextrins. These pores may then be opened in three ways: (i) addition of α-cyclodextrin which dissociates the β-cyclodextrin-diazo linker by preferential formation of an inclusion complex; (ii) application of light to trigger a trans-to-cis isomerization of the azobenzene and accompanying dissociation from the β-cyclodextrin inclusion complex; and (iii) reductive cleavage of the disulphide with DTT to remove the gate altogether.
Giménez et al. have employed similar chemistry to coat MCM-41 mesoporous silica with PEG chains tethered via a disulphide linkage.67 In this case, the dye safranin-O was first loaded into the particles and the surface modified using (3-mercaptopropyl)trimethoxysilane (to give thiol groups on the particle surface). Reaction with 2,2′-dipyridyl disulphide was used to yield pyridyl disulphide functionalised mesoporous silica particles. A GSH-responsive PEG coating was then afforded by reacting the pyridyl disulphide with O-(2-mercaptoethyl)-O′-methyl-hexa(ethylene glycol). Release of safranin-O (or preloaded doxorubicin) was able to be triggered by exposure to a physiologically relevant concentration of GSH (10 mM). A similar approach has been employed by Xie et al.68 and by Wang et al.,69 with the key difference being the methods used to prepare both the thiol-functionalised particles and the thiol terminated PEG polymer. Similar release profiles are observed in the presence GSH. Taken together, these reports demonstrate the applicability of the gated approach for releasing chemotherapeutics from mesoporous materials.
Kim and coworkers have also employed disulphide linked cyclodextrin (CD) as a suitable GSH-responsive gate on mesoporous silica.70 These authors demonstrated effective retention of the commonly employed chemotherapeutic agent doxorubicin within the CD gated pores, and that exposure to millimolar concentrations of GSH could effectively release the agent through cleavage of the disulphide and “ungating” of the pores. Moreover, the CD units could be imparted with “stealth” properties via reaction with isocyanate terminated PEG.
Using a slightly different approach, doxorubicin loaded MCM-41 particles have been modified with glutathione responsive molecular gates by tethering C18 alkyl chains to the particle surface via disulphide linkages.71 The alkyl chain was then used to facilitate association of an amphiphilic peptide containing an RGD targeting sequence through hydrophobic interactions between the surface-tethered C18 and the hydrophobic portion of the amphiphilic peptide (also C18). Exposure to DTT or GSH led to shedding of the gates and release of the encapsulated drug. In an extension of this work, the same group conjugated a α,ω-functionalised peptide sequence including a targeting sequence (RGD) and hydrophobic segment (4 phenylalanine units) to the surface of doxorubicin-loaded MCM-41 via a disulphide linkage.72 The other end of the peptide was functionalised with an azide to enable click conjugation of an alkyne-functionalised PEG chain incorporating a pH labile benzoic–imine bond. In this case the hydrophobic phenylalanine units were proposed to provide the molecular gates preventing release of the encapsulated drug. The researchers demonstrated that doxorubicin release could be effectively triggered by exposure to a reducing agent (DTT, 0.1–10 mM).
Yang and coworkers have recently reported the development of dual-responsive mesoporous silica particles with pH- and GSH-gated release of doxorubicin (Fig. 7).73 These materials were prepared by first functionalising the surface of mesoporous silica particles with disulphide-linked carboxylic acid groups, and then conjugating hydroxyl-terminated poly(glycidyl methacrylate) (PGMA) via formation of an ester linkage. The pendant epoxide groups on the PGMA were then reacted with ethylenediamine to give an amine-functional polymer. The protonated pendant amines were used to form pH-dependent complexes with cucurbit[7]urils, and these served to gate the mesoporous silica and prevent release of encapsulated doxorubicin. Moreover, the ammonium–cucurbit[7]urils complexes could be dissociated by decreasing the pH (due to competing complexation by hydronium ions), and this facilitated release of the doxorubicin. Moreover, release could also be triggered by exposure to millimolar concentrations of GSH, which effectively shed the polymeric shell by cleavage of the disulfide.
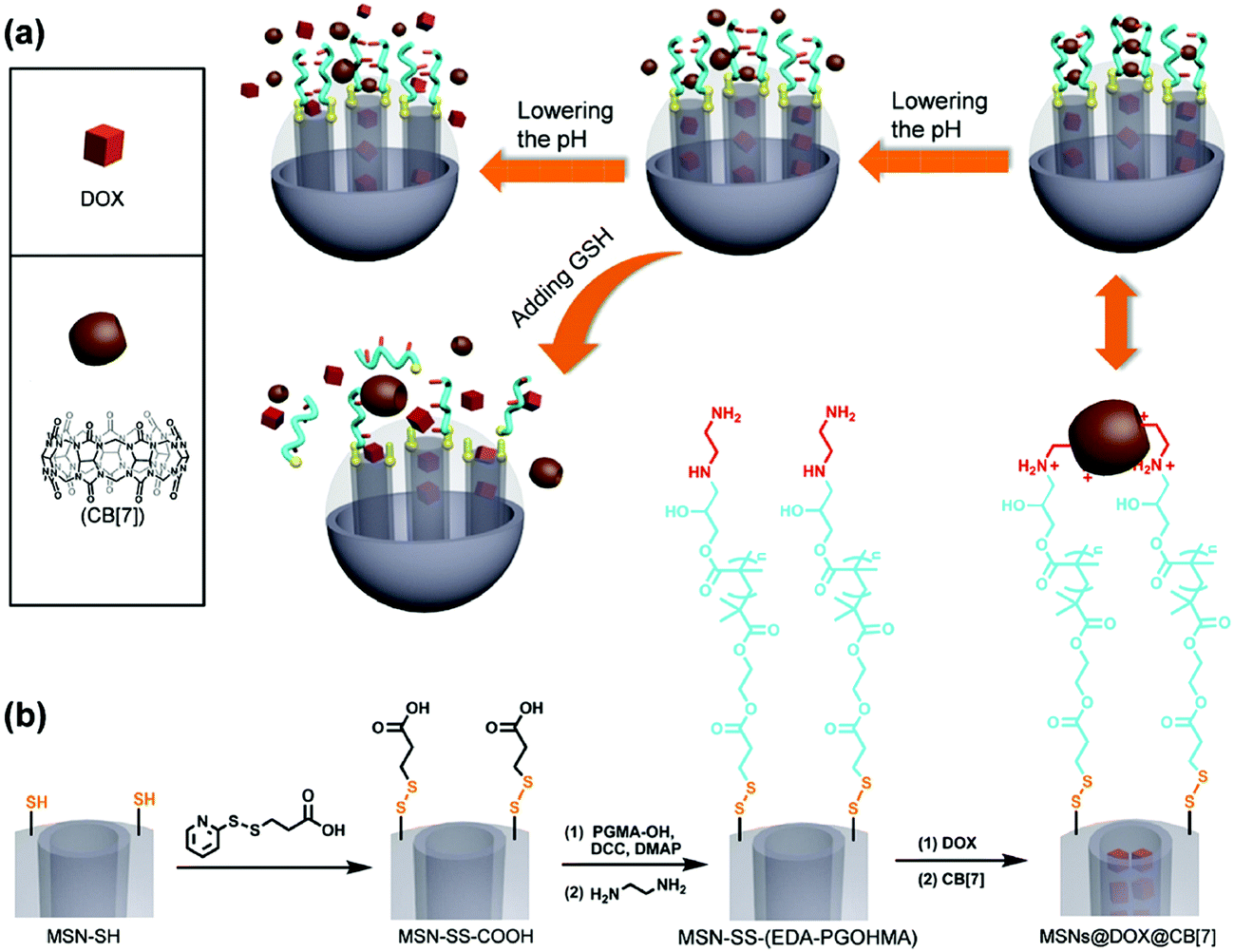 | ||
| Fig. 7 (a) Preparation of drug-loaded mesoporous silica nanoparticles with a dual pH- and GSH-responsive cross-linked supramolecular coating: (b) synthetic approach for the attachment of disulphide-linked hydroxyl-terminated poly(glycidyl methacrylate) (PGMA-OH) and subsequent assembly with cucurbit[7]uril (CB[7]). Adapted from Li et al., ACS Appl. Mater. Interfaces, 2015, 7, 28656–28664. © 2015 American Chemical Society. | ||
Zhang et al. have also reported a pH- and GSH-gated mesoporous silica system.74 In this case the particle surface was initially endowed with thiol groups which were converted to disulphide linked amines by reaction with S-(2-aminoethylthio)-2-thiopyridine hydrochloride. These were then used to conjugate alkynes via reaction with propargyl bromide. The alkynes provided a functional handle for coating with azide-functionalised chitosan via CuAAC. Finally, the amine functionality inherent to chitosan was used to form a pH labile imine bond with methoxy poly(ethylene glycol) benzaldehyde. The authors demonstrated pH and GSH dependent release for doxorubicin in pH and concentration ranges relevant to the tumour microenvironment.
A further example of pH- and GSH-gated mesoporous silica has been reported by Chang et al.75 In this case, mesoporous silica particles were coated with disulfide-cross-linked poly(N-vinylcaprolactam-co-MAA) (poly(VCL-s-s-MAA)) by precipitation polymerization of N-vinyl caprolactam, N,N′-bis(acryoyl)cystamine and MAA in the presence of modified mesoporous silica. The resulting particles could be effectively loaded with doxorubicin, and exhibited minimal release at pH 7.4 and 37 °C. However, release could be triggered by lowering the pH to the range 6.5–5.0 (to collapse the polymer coating and reduce electrostatic binding of the drug) or by exposure to 10 mM GSH (to disrupt the polymer coating through cleavage of the disulphide linkages afforded by the N,N′-bis(acryoyl)cystamine). A similar approach has also been employed by using precipitation polymerization of acrylic acid (AA) on the surface of mesoporous silica, followed by crosslinking with cystamine hydrochloride.76
Zhang and coworkers have reported the application of mesoporous silica nanoparticles for pH and GSH stimulated codelivery of topotecan and Tpep (a mitochondria-targeted therapeutic agent).77 In this case, the silica is first modified with 3-(mercaptopropyl)trimethoxysilane to provide a thiol, and then loaded with topotecan. The surface thiol is then converted to a pyridyl disulphide intermediate using 2,2′-dipyridyl disulphide, and the Tpep conjugated via thiol–disulfide exchange. The Tpep decorated particles are then masked using poly(ethylene glycol)-blocked-2,3-dimethylmaleic anhydride-modified poly(L-lysine) (PEG-PLL(DMA)). The authors propose that the Tpep-coated particles can be unmasked by degradation of the of the DMA–PLL linkages due to tumoral acidity, and that Tpep and topotecan are then released due to cleavage of the disulphide bonds by intracellular GSH. These results demonstrate the sophisticated materials which can be designed to exploit multiple biological cues, including elevated GSH concentrations in the intracellular compartment.
In an interesting twist on the formation of GSH responsive organic–inorganic hybrids, Botella and coworkers have prepared poly(lactic-co-glycolic acid) (PLGA) particles with an outer shell of thiol-responsive amorphous organosilica.78 The PLGA particles are first assembled by adding a solution of PLGA and pyrene in chloroform dropwise to an aqueous solution of cetyltrimethylammonium bromide. After removal of the chloroform and washing by repeated centrifugation cycles, the particles were resuspended in 1:9 isopropanol:water, after which tetraethyl orthosilicate (TEOS), bis[3-(triethoxysilyl)-propyl]disulfide (TESPDS) and ammonium were added. The amount of added TESPDS was varied to provide particles with different densities of disulphide gates. It was demonstrated that the pyrene release was increased in the presence of DTT (100 mM), although the concentration of thiol molecule required is far in excess of what would be encountered physiologically. Nevertheless, these materials provide an interesting platform from which further GSH-responsive hybrid inorganic–organic systems might be developed.
4. Glutathione responsive delivery systems
(a) Delivery of chemotherapeutic agents
An enormous array of different materials have been employed for the delivery of chemotherapeutics. For instance, chemotherapeutic agents have been physically encapsulated into GSH-responsive block copolymers incorporating linear PEG and polypeptide,89–91 linear PEG and polyurethane,92 linear PEG and polyester,93–95 linear PEG, polyester and polyurethane,96 linear PEG and polycarbonate,97 linear PEG and polymethacrylate,98 linear PEG, polyacrylate and polyester,99,100 and brush PEG and polyester.101,102 These materials typically incorporate at least one reducible disulphide or diselenide linkage at either the junction of the two blocks, or within the core103 or corona of the micellar structure. As such, exposure to GSH at millimolar levels triggers destabilization of the micelle and release of the encapsulated material. In addition to block copolymers, random copolymers have also been employed such as brush PEG copolymers,104 and hyperbranched polymethacrylates.105 Polysaccharide based materials, such as poly(ε-caprolactone)-graft-chitosan,106 and succinic anhydride/cystamine modified dextran107 have also been used to load chemotherapeutic agents and release them in response to millimolar concentrations of GSH. Other novel materials, such as PEGylated hyperbranched polyphosphoesters108 have also been employed.
Aside from self-assembled polymers, crosslinked nanogels have also received considerable attention as candidates for particle based chemotherapeutic delivery. These materials offer considerable advantages in that they are potentially able to be loaded with higher amounts of therapeutic agent. In particular, nanogels formed through precipitation polymerization in water or precipitation–distillation polymerization in acetonitrile have been successfully prepared using N,N′-bis(acryloyl)cystamine as a disulphide-incorporating crosslinker. For instance, N,N′-bis(acryoyl)cystamine has been used to prepare nanogels with N-(2-hydroxypropyl)methacrylamide and MAA,109 VCL, MAA and PEGMA,110 and 2-(2-methoxyethoxy)ethyl methacrylate and OEGMA.111 In each case, the prepared nanogels were successfully loaded with doxorubicin and GSH-triggered release was demonstrated. In a slight variation, Jin et al. prepared nanogels using reflux-precipitation polymerization of N,N′-bis(acryoyl)cystamine and MAA, and subsequently PEGylated these by conjugating PEG–NH2 using carbodiimide chemistry.112 As in other nanogel systems, the release of loaded doxorubicin was again shown to be dependent on pH and GSH concentration.
Nanogels for GSH triggered release have also been prepared by two-stage precipitation distillation polymerization.113 MAA and N,N′-bis(acryloyl)cystamine were initially copolymerized in boiling acetonitrile, and the formed nanogels employed as seeds for the subsequent terpolymerization of N,N′-bis(acryloyl)cystamine, NIPAM and GMA. The oxirane groups on the GMA units were ring-opened with ethylendiamine, and folic acid conjugated to the pendant amines via amidation. Once again, the resulting nanogels could be loaded with doxorubicin, and release triggered by exposure to millimolar concentrations of GSH. A two stage distillation–precipitation polymerization process has also been used to prepare multistimuli-responsive nanoparticles incorporating superparamagnetic iron oxide nanoparticles.114 In this case, methacrylate-functionalised Fe3O4 particles were incorporated into the first polymerization with N,N′-bis(acryloyl)cystamine and MAA. A thermoresponsive shell was subsequently formed around the particle through the second-stage polymerization of N,N′-methylenebis(acrylamide) and NIPAM. When loaded with doxorubicin, the resulting particles were shown to release the drug in a temperature, pH and GSH dependent manner. Liu and coworkers have also examined the use of Fe3O4 particles in the preparation of nanogels with GSH sensitivity.115 In this case the surface of the particle was functionalised with a trithiocarbonate chain transfer agent, after which nanogel coatings could be formed by the RAFT copolymerization of acrylamide, NIPAM and N,N′-bis(acryloyl)cystamine on the particle surface. The resulting particles were shown to load both doxorubicin and curcumin. Moreover, the authors demonstrated that particles loaded with curcumin were able to release the cargo in the presence of 5 or 10 mM GSH.
Polymeric hollow nanocapsules comprising a crosslinked hydrogel have been prepared by Liu and coworkers.116 To prepare these materials, the authors first prepared a polymeric stabilizer of mPEG-block-poly(tert-butyl acrylate) via ATRP, and then employed this as the stabilizer in the miniemulsion interfacial polymerization of N,N′-bis(acryoyl)cystamine and tert-butyl acrylate via activator generated electron transfer ATRP (AGET ATRP). The resulting PtBA particles were hydrolysed using trifluoroacetic acid to yield essentially PAA particles crosslinked via disulphide linkages. As with the other nanogel systems studied, the particles were successfully loaded with doxorubicin, and release triggered by exposure to millimolar concentrations of GSH and/or decreased pH. Interestingly, the authors also employed a folic acid terminated stabilizer as a convenient route for incorporating folic acid targeting moieties into the final polymer particles.
An alternative approach to the preparation of drug-loaded nanogels involves the formation of disulfide crosslinked hydrogel coatings on the surface of cubical and spherical mesoporous manganese oxide (Mn2O3) particles.117 This is achieved through hydrogen-bonded layer-by-layer assembly of PMAA and PVPON on Mn2O3 particles, followed by crosslinking of the PMAA network with cystamine. Removal of the PVPON (by elevating the pH to 8) and the sacrificial mesoporous templates (with EDTA) provides both cubical and spherical disulphide crosslinked PMAA particles. The particles were successfully loaded with doxorubicin, and release of the drug was facilitated by exposure to millimolar levels of GSH.
Xu's group have described a novel methodology to develop GSH responsive materials for drug delivery by using a co-ordination responsive approach (Fig. 8).118 Specifically, selenide containing polyurethanes were first synthesized by reaction of di(1-hydroxylundecyl)selenide with toluene diisocyanate. The isocyanate endgroups formed were then reacted with mPEG to form an ABA triblock copolymer. These materials formed stable aggregates in water when dispersed with platinum(II) chloride, in part due to formation of a ca. 3:1 complex between the Se atoms in the polyurethane domain and Pt2+. Importantly, this interaction stabilised the particles to elevated ionic strengths. However, exposure to millimolar concentrations of GSH led to competitive complexation with the Pt2+ and a concomitant destabilisation of the aggregates. The authors exploited this interesting feature in order to release doxorubicin incorporated into the aggregates, with sustained release over 20 hours demonstrated in response to 10 mM GSH. A similar approach has been employed for the delivery of cisplatin from selenide-containing ABA block copolymers of mPEG and polylactide.119 In this case, the coordination-induced stabilization is afforded by the drug itself, rather than exogenously applied platinum.
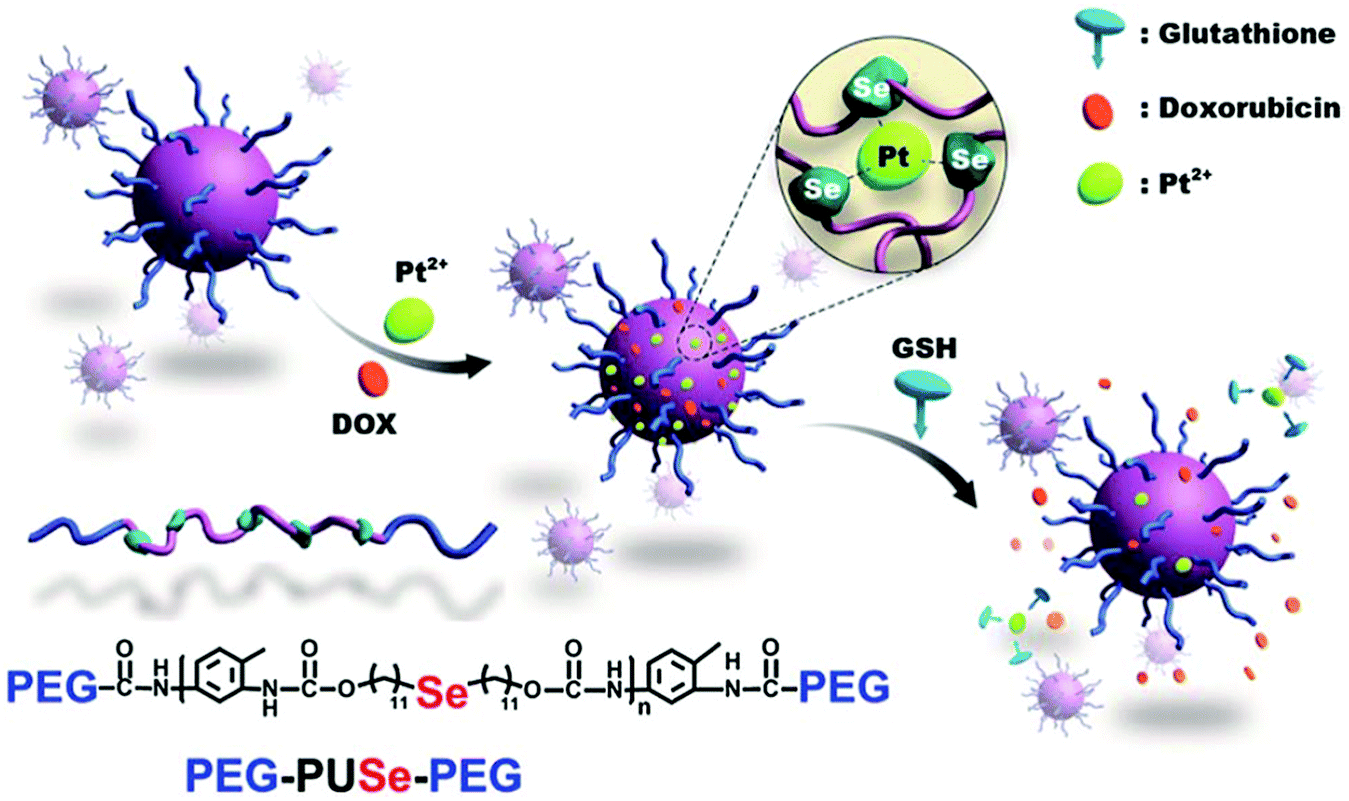 | ||
| Fig. 8 Schematic illustration of GSH-responsive micelles based on the coordination of Pt2+ to the selenium-containing polymer PEG–PUSe–PEG, and triggered release of doxorubicin through competitive coordination with glutathione. Adapted from Cao et al., Chem. Sci., 2012, 3, 3403–3408. © 2012 Royal Society of Chemistry. | ||
Dou and coworkers have reported an interesting GSH-responsive micelle system based on thiol-modified Pluronic P123 and F127.120 In this example, Pluronic P123 and F127 were first coupled with cystamine via a N,N′-carbonyldiimidazole-mediated approach, and then the disulfide reduced to give thiol-terminated copolymer. The copolymer was then used to form paclitaxel loaded micelles by a thin-film hydration method. Specifically, the copolymer and paclitaxel were dissolved in a common solvent (acetonitrile) and then dried into a film in a round bottomed flask. After removal of residual acetonitrile with high vacuum, the micelles were formed by hydrating the film with water. Introduction of gold nanoparticles resulted in crosslinking of the micelle shell due to the well-known Au–SH interaction. The authors noted that release could be triggered by exposure to GSH (due to competitive binding with the gold nanoparticles), although there was significant leakage of the paclitaxel even without the GSH stimulus.
Sun and coworkers have reported a novel system in which glutathione has been used to trigger the production of reactive oxygen species.121 In this example, the authors conjugated the copper chelator D-penicillamine (D-pen) to chitosan via the formation of a disulphide linkage. This was then used to prepare self-assembled nanoparticles via ionotropic gelation with tripolyphosphate, either with or without the presence of doxorubicin or Cu(II)-doxorubicin complex. The particles were then coated with hyaluronic acid (HA). Exposure to 10 mM GSH resulted in release of free D-pen, which reduces Cu2+ to Cu+, forms a stable complex with the Cu+, and initiates production of ROS. Moreover, reduction of Cu(II) to Cu(I) also accelerates the release of doxorubicin. Studies with HT-29 cells demonstrated the utility of this system for sensitizing the cells to doxorubicin cytotoxicity, leading to significantly reduced cell proliferation.
Departing from the general theme of chemotherapeutic delivery, Kim et al. have reported the application of thiol-responsive micelles formed from gemini surfactants to encapsulate the nonsteroidal anti-inflammatory drug indomethacin.122 In this case trityl protected cysteine-mPEG (H-Cys(Trt)-mPEG) was first synthesized by condensing Fmoc-Cys(Trt)-OH with mPEG–NH2 using DCC, and subsequently deprotecting the Fmoc group. The H-Cys(Trt)-mPEG was then coupled with Fmoc-Lys(Fmoc)-OH using DCC, and the Fmoc group removed to provide H-Lys-Cys(Trt)-mPEG. The H-Cys(Trt)-mPEG and H-Lys-Cys(Trt)-mPEG were then coupled with stearic acid, after which the trityl groups were removed by reacting with trifluoroacetic acid and the exposed cysteines oxidised to form the gemini surfactants ((C18)2-Lys-Cys-mPEG)2 and ((C18)2-Lys-Cys-mPEG)2. The surfactants were then used to encapsulate indomethacin by an oil-in-water emulsion method, adding a solution of the drug in methylene chloride to a stirred aqueous solution of the surfactant, followed by evaporation of the methylene chloride. Exposure to GSH at 5 or 10 mM triggered release of the indomethacin, while exposure to oxidised GSH (GSSG) did not stimulate any release above and beyond that occurring in the absence of any stimulus.
Stenzel, Zetterlund and coworkers have reported the development of GSH responsive materials for encapsulating water soluble drugs using an inverse miniemulsion periphery RAFT polymerization (IMEPP) approach.123 These authors first synthesized poly(N-(2-hydroxypropyl methacrylamide))-b-poly(pentafluorophenyl methacrylate) (PHPMA-b-PPFPMA) as a macromolecular chain transfer agent and stabiliser. The PHPMA-b-PPFPMA was then added to toluene with a monomer (pentafluorophenyl methacrylate), crosslinker (ethylene glycol dimethacrylate or bis(2-methacryloyloxyethyl) disulfide), and initiator (azobisisobutyronitrile) to form the organic phase. Meanwhile, an aqueous phase was prepared separately using deionized water, a lipophobe (sodium chloride) and any drug to be encapsulated (gemcitabine). The aqueous phase was added to the organic phase and the mixture sonicated for 5 min before polymerizing at 70 °C for 7 hours. Nanocapsules were collected from the emulsion by de-emulsifying the system using hexane, and subsequent centrifugation. The pentafluorophenyl ester side groups were subsequently reacted with glucosamine to provide glycopolymer nanocapsules. The materials were shown to release encapsulated gemcitabine rapidly upon exposure to 10 mM GSH.
Liu and coworkers have recently reported a novel drug delivery system in which biocompatible PEGylated alginate (ALG-PEG) brushes are grafted onto graphene oxide nanoparticles (GONs) via GSH responsive disulfide bridges.124 To achieve this, the alginate is first modified using amine-terminated PEG to yield biocompatible PEG brushes. These were further modified by conjugation with cystamine to provide a disulphide-containing amine linker. The amine was then used to conjugate the brushes to graphene oxide nanoparticles prepared by the Hummers method. The resulting particles were able to load doxorubicin to very high loadings (97.64% ± 3.36), which the authors attribute to π–π stacking with the highly conjugated GON surface. Doxorubicin release from the particles could be facilitated by exposure to GSH (5 mM or 10 mM), and further accelerated by decreasing the pH (from 7.4 to 5.0).
Huang et al. have reported the preparation of so-called pseudo-poly(aminoacid)s via click polymerization, and examined their utility as a potential carrier for doxorubicin.125 To achieve this, the authors first synthesised a bisalkyne including a disulphide linkage 2,2′-dithiobis[1-(prop-2-ynylcarbamoyl)-ethyl-carbamic acid tert-butyl ester, and two bisazides: 2,2-bis(azidomethyl)propane 1,3-diol (BAP) and 2,2-bisazidohexane (BAH). To conduct the polymerization, the bisalkyne was combined in DMF with a slight excess of BAP or BAH (to provide azide endgroups on the resulting polymer), ascorbic acid and CuSO4·5H2O. The polymer was precipitated in diethyl ether, and then converted to an ABA triblock copolymer by reaction with alkyne-terminated mPEG. After removing the excess PEG by dialysis, the resulting polymer was lyophilized. The resulting triblock copolymer was loaded with doxorubicin by dissolving both polymer and drug in organic solvent, and then adding slowly to distilled water. The resulting particle dispersion was dialysed against water to remove residual organic solvent. Doxorubicin could be released from the particles by exposure to 10 mM GSH, or by lowering the pH from 7.4 to 5.0. Further, empty particles were tested for cytotoxicity, and it was demonstrated that these materials were generally well tolerated by L929 mouse fibroblasts up to 1 mg mL−1.
Mallik's group have reported a novel application of GSH responsive linkers in their development of polymer coated echogenic lipid nanoparticles with dual release triggers (Fig. 9).126 In this example, liposomes were formed using 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycerol-3-phosphoethanolamine derivative with three pendant propargyl groups (POPE-G). The outer leaflet of the liposome was successfully crosslinked using CuAAC with a disulphide-containing bisazide. After encapsulating doxorubicin in the nanoparticles via a pH gradient approach, release could be triggered by exposure to millimolar concentrations of GSH. Importantly, GSH exposure at micromolar concentrations triggered only minimal leakage from the liposomes. Moreover, the authors demonstrated that application of ultrasound together with GSH led to enhanced release.
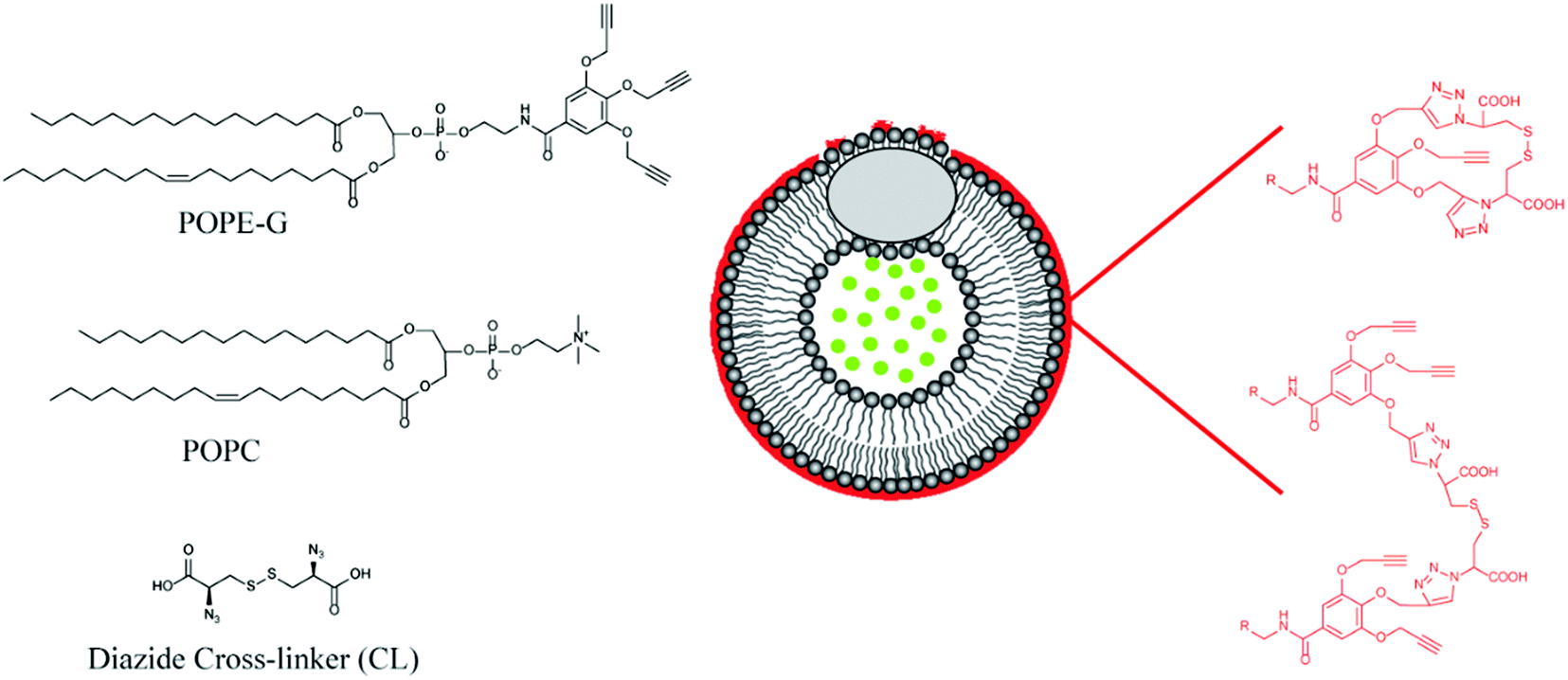 | ||
| Fig. 9 Lipid nanoparticles with a GSH-responsive outer leaflet prepared by polymerizing the tri-alkyne-functionalized lipid with a diazide cross-linker. Adapted from Nahire et al., Biomacromolecules, 2013, 14, 841–853. © 2013 American Chemical Society. | ||
The same group have prepared GSH sensitive polymersomes through the assembly of PEG-polyester block copolymers with a disulphide linkage incorporated at the chain junction.127 This was achieved by reacting mPEG sequentially with succinic anhydride (to yield acid terminated PEG), and then cystamine (to provide amine terminated PEG with a disulphide linkage). This material was then used to initiate the ring opening polymerization of lactide to provide the final block copolymers. Polymersomes were formed by a solvent exchange methodology by adding a solution of the block copolymer in THF to a solution of calcein (10 μM) in HEPES buffer. After sonication and extrusion through polycarbonate membrane polymersomes of ca. 150–200 nm were obtained. The polymersomes could be successfully loaded with doxorubicin or gemcitabine through a pH gradient approach, and release the cargo in response to intracellular levels of GSH and/or ultrasound. These materials were shown to be useful for decreasing the viability of breast and pancreatic cancer cells in monolayer and spheroid cultures.
Feijen, Zhong and coworkers have recently reported reversibly crosslinked pH-responsive biodegradable micelles based on poly(ethylene glycol)-poly(2,4,6-trimethoxybenzylidene-pentaerythritol carbonate-co-pyridyl disulfide carbonate) [PEG-P(TMBPEC-co-PDSC)].128 PTMBPEC-containing materials have advantages over traditional aliphatic polyesters such as PCL, PLA, and PLGA in that the PTMBPEC domain can be employed to cause rapid swelling or hydrolytic dissociation of the acetal pendants at only mildly acidic conditions. The polymer was prepared by ring opening copolymerization of mono-2,4,6-trimethoxybenzylidene-pentaerythritolcarbonate (TMBPEC) and pyridyl disulfide cyclic carbonate (PDSC) using methoxy PEG as an initiator and zinc bis[bis(trimethylsilyl)amide] as a catalyst. Doxorubicin-loaded particles were prepared by slowly adding phosphate buffer to an organic solution of the polymer and doxorubicin, with crosslinking subsequently induced by addition of DTT. An irreversibly crosslinked control was prepared by substituting acryloyl carbonate for the PDSC in the polymerization, and crosslinking with a free radical initiator. The organic solvents were removed by dialysis against phosphate buffer. The authors demonstrated that doxorubicin release from the reversibly crosslinked micelles could be triggered by exposure to 10 mM GSH, and that this was accelerated at pH 5.0 (compared to pH 7.4). Further, the polymers were effective for delivery of doxorubicin to RAW264.7 cells, and the authors noted that doxorubicin accumulated more quickly in the nucleus when the GSH responsive linkages were employed. This was attributed to the more rapid release facilitated by intracellular GSH, as opposed to the irreversibly crosslinked system. The polymer particles themselves (i.e., without loaded cytotoxic agent) were well tolerated by RAW264.7 and HeLa cells, indicating the potential utility of the system for drug delivery applications.
In an alternative approach, Zhu and coworkers prepared multithiol branched polymers by copolymerizing PEGMA with the polymerizable chain transfer agent S-(4-vinyl) benzyl S′-propyltrithiocarbonate (VBPT).130 The thiocarbonylthio groups in the resulting branched poly(VBPT-co-PEGMA) were converted to pyridyl disulphide functionality by reacting with propylamine in the presence of PDS. A thiol containing drug (6-mercaptopurine monohydrate) was then conjugated by thiol–disulfide exchange. The resulting particles released the drug over a matter of hours in the presence of 10 mM GSH, while exhibiting minimal release without the addition of a GSH stimulus.
Doxorubicin has also been attached to polymer particles through GSH responsive disulphide linkages.131 In one approach, doxorubicin was conjugated with dithiobis(succinimidyl propionate) to yield doxorubicin with a disulphide containing reactive handle (succinimidyl ester). This was then used to conjugate the doxorubicin to stearic acid grafted chitosan. The authors demonstrated that release was dependent on the concentration of DTT, with millimolar concentrations giving much faster release than micromolar. Derivatives of dithio(bispropionic acid) have also been used to conjugate paclitaxel to hyaluronic acid,132 and curcumin to mPEG-PLA,133 while bis(2-hydroxyethyl) disulphide has been used in the conjugation of mPEG to camptothecin via carbonate linkages.134 In this final example there is an interesting dependence of the particle shape on the molecular weight of the conjugated mPEG, with mPEG350 yielding filamentous particles as opposed to the spherical aggregates formed by mPEG1900.
Conjugates of mPEG and paclitaxel have also been explored as nanocarriers for controlled delivery applications.135 In this case, the authors first modified 3-mercaptopropionic acid with 2,2′-dithiodipyridine to form 2-pyridyl-2-carboxyethyl disulfide, and then reacted this with thiol-terminated PEG to form carboxylic acid-terminated PEG incorporating a disulphide linkage. The carboxylic acid group was then exploited to form a conjugate with paclitaxel via one of the secondary alcohols. The resulting conjugates were used to form nanoaggregates with free paclitaxel (Fig. 10). Paclitaxel conjugates have also been formed with polymers incorporating PEG brushes.136 In this case, the authors copolymerized 2-((3-(pyridin-2-yldisulfanyl)propanoyl)oxy)ethyl methacrylate with PEGMA, and then reacted the pendant pyridyl disulphide with 3-mercaptopropionic acid to give a carboxylic acid group linked to the polymer via a disulphide linkage. Carbodiimide chemistry was then used to conjugate the paclitaxel to the carboxylic acid through ester formation.
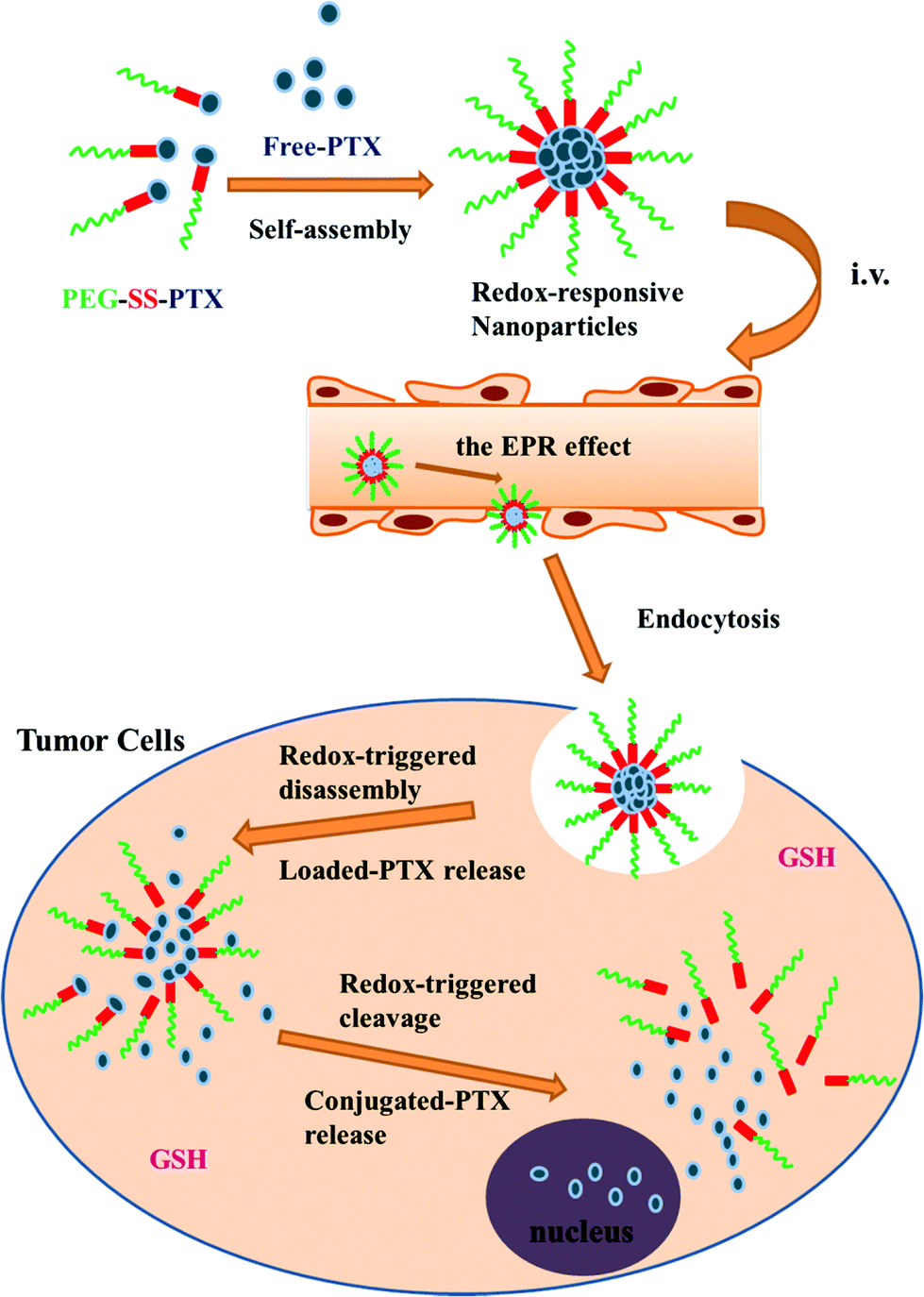 | ||
| Fig. 10 Preparation, tumour accumulation, and GSH-stimulated intracellular drug release of redox-responsive PEG-SS-PTX/PTX nanoparticles as prepared by Chuan et al. Release of PTX occurs via two modalities: release of physically encapsulated PTX and liberation of conjugated PTX via thiol–disulfide exchange. Reproduced with permission from Chuan et al., Mol. Pharmaceutics, 2014, 11, 3656–3670. © 2014 American Chemical Society. | ||
Camptothecin has been conjugated to a biodegradable diblock polyphosphoester poly(butynyl phospholane)-block-poly(ethylethylene phosphate) (PBYP-b-PEEP).137 In this example, 2,2′-disulfanediylbis(ethan-1-ol) was first reacted with bromoisobutyrylbromide to give 2-((2-hydroxyethyl)disulfanyl)ethyl 2-bromo-2-methylpropanoate, and the bromine was then substituted with azide to yield 2-((2-hydroxyethyl)disulfanyl)ethyl 2-azido-2-methylpropanoate. This material was then conjugated to camptothecin via a carbonate linkage, and the azide used to connect the resulting adduct to the pendant alkyne in the PBYP-b-PEEP. Release of the camptothecin from the polymer was highly dependent on the GSH concentration, with minimal release triggered by micromolar concentrations and much higher release afforded in the presence of 5–10 mM GSH.
Polymeric carriers for the GSH-stimulated release of phenylarsine oxide have also been developed.138 These materials were prepared by reacting the free carboxylic acid groups of poly(ethylene oxide)–block-poly(α-carboxylate-ε-caprolactone) (PEO-b-PCCL) with 6-mercaptohexylamine to provide a block copolymer with pendant thiol functionality. The pendant thiols facilitated association of the phenylarsine oxide through the well-known affinity of thiol and trivalent arsenicals. Moreover, the authors demonstrated that the phenylarsine oxide could be liberated through exposure to GSH concentrations of between 1 and 10 mM. These strategies are of considerable interest given the usefulness of trivalent arsenic compounds such as arsenic trioxide for the treatment of conditions such as acute promyelocytic leukemia (APL).
In a number of the examples given above, the drug (doxorubicin, paclitaxel or camptothecin) is necessarily modified in order to conjugate it via a disulphide linkage. As such, the chemical entity that is ultimately delivered from the conjugate is not the pristine drug. Indeed, only thiol-containing drugs can ordinarily be used for direct delivery of an unmodified therapeutic via thiol–disulfide exchange. To address this severe limitation, Zelikin and co-workers have recently reported the use of disulphide containing self-immolative linkers (SILs) as an elegant solution to this problem.139 In their first example, the antiretroviral drug azidothymidine (AZT) was incorporated into a poly[N-(2-hydroxypropyl)methacrylamide (PHPMA) macromolecular prodrug (MP) via copolymerization of a monomer having AZT attached via an SIL (Fig. 11).140 The AZT-containing monomer was prepared by first reacting the oxidised, dimerized form of 2-mercaptoethanol with methacryloyl chloride to afford a monomeric precursor with a hydroxyl pendant linked through a disulphide to a methacrylate. Simultaneously, AZT was modified with 4-nitrophenyl chloroformate to give an activated carbonate precursor. Reacting the monomeric precursor with the activated carbonate afforded the desired AZT-containing monomer. The authors demonstrated that this monomer could be readily copolymerized with HPMA without compromising the drug or SIL. Moreover, the resulting MPs were shown to release the pristine AZT in the presence of 5 mM GSH or lysate from TXM-bI cells. This reaction occurred via thiol-induced cleavage of the disulphide, which in turn initiated a cyclization of the linker to reveal the AZT molecule. A similar methodology has been employed to prepare MPs of ribavirin, although in this case it was also necessary to protect the secondary hydroxyls on the ribavirin prior to activation with the 4-nitrophenyl chloroformate.141 A slightly different approach has been employed by the same authors to conjugate a histone deacetylase inhibitor (Panobinostat) to HPMA copolymers for the development of MPs incorporating HIV anti-latency agents.142 In this case, the authors prepared an activated monomer by reacting oxidised 2-mercaptoethanol with methacryloyl chloride, and then activating the pendant hydroxyl with 4-nitrophenyl chloroformate. This activated monomer could be readily copolymerized with HPMA, and the resulting polymers conjugated to Panobinostat via reaction with the drug's secondary amine group. Release of the pristine drug could be triggered using 5 mM DTT. In each of these examples the authors demonstrated that the MPs were suitable for drug delivery in cell culture studies, providing solid evidence for the utility of SILs to deliver a therapeutic agent. Given that these SILs are not dependent on the drug having a particular functionality (i.e., thiol), it is likely that they will attract increased research attention for delivering a range of therapeutic agents.
 | ||
| Fig. 11 (A) Synthesis of AZT-containing monomer incorporating a self-immolative linker. (B) Synthesis of macromolecular prodrugs of AZT from HPMA and AZT-containing monomer. (C) Release of AZT via cleavage of the disulphide and linker cyclization. Reagents and conditions: (a) methacryloyl chloride, TEA, DCM (anhydrous), 76%; (b) 4-nitrophenyl chloroformate, TEA, DCM, 82%; (c) DIEA, DMAP, DCM, 80%; (d) 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid, 4,4′-azobis(4-cyanovaleric acid), DMSO. Adapted from A. Kock et al., Chem. Commun., 2014, 50, 14498–14500. © 2014 Royal Society of Chemistry. | ||
Koul and coworkers have recently reported a triblock copolymer system (poly(ethylene glycol)-polylactic acid-poly(ethylene glycol)) incorporating multiple disulphide linkages.146 The polymers were assembled into doxorubicin-loaded polymersomes via nanoprecipitation in the presence of the drug, and then labelled with (i) folate, (ii) trastuzumab or (iii) folate and trastuzumab via carbodiimide coupling. After studying the GSH response and targeting efficiency in vitro, the dual folate/trastuzumab targeted polymersomes were administered to Ehrlich ascites tumour (EAT) bearing Swiss albino mice every three days for 15 days. A significant reduction in tumour volume was observed over the course of administration. Additionally, no damage to vital organs was observed upon histopathological assessment, in contrast to free doxorubicin which caused significant liver fibrosis and tubular damage in the kidney. Moreover, blood serum analysis indicated that there was no significant elevation to markers associated with liver or kidney damage in the case of the nanoparticles, whereas free doxorubicin resulted in elevation of several key markers of liver and kidney toxicity. Again, the absence of a control group without disulphide linkages makes it difficult to judge whether the in vivo results are particular to a GSH sensitive system, or whether comparable results would be achieved without GSH responsive linkers.
Jin et al. have explored the use of GSH-responsive PMA nanogel systems as potential vehicles for the codelivery of doxorubicin and paclitaxel.147 In this study, the authors confined their in vivo investigations to a preliminary assessment of biodistribution and toxicity, with particular note given to the impact of conjugating PEG chains to the nanogel. Using intravital real-time fluorescence imaging, the authors noted that naked PAA hydrogels quickly accumulated in the kidney, with significant accumulation also occurring in the liver, spleen, heart and lung. In contrast, PEGylated nanogels were distributed throughout the body over the first two hours, with only limited accumulation in the kidney and minimal liver accumulation. The authors also determined an LD50 value for these materials, with a value of ca. 140 mg kg−1 determined for the MAA hydrogel as opposed to ca. 500 mg kg−1 for the PEGylated variant. These results support the important role of PEG in improving the tolerance of various materials in vivo.
A number of GSH-responsive polysaccharide based systems have also been investigated in vivo. For instance, Chen and co-workers have recently reported on the use of PLGA–hyaluronic acid (HA) conjugates linked via a cystamine bridge (HA-SS-PLGA).148 These materials were employed to encapsulate both doxorubicin and cyclopamine (a primary inhibitor of the hedgehog signaling pathway of cancer stem cells) using a double emulsion method. Weekly administration of particles loaded with doxorubicin and cyclopamine to MDA-MB-231 tumour bearing nude mice demonstrated a remarkable inhibition of tumour growth, to the extent that no tumours were evident over 40 days. Moreover, tumour inhibition was maintained for a further 35 days after cessation of treatment. However, the precise role of the GSH responsive linkage in these results is somewhat unclear as the various controls examined did not include a conjugate of HA and PLGA without a GSH-sensitive linkage.
Hu and co-workers have recently reported on stearic acid grafted chitosan oligosaccharides with doxorubicin conjugated through a GSH-responsive linker.131 These materials were administered to BEL-7402 tumour-bearing nude mice. Maximal tumour accumulation was observed after 36 h, and this was attributed to the enhanced permeation and retention effect of nanoscale delivery vehicles. Significant accumulation was also observed in the liver, spleen, and lungs. The authors observed comparable inhibition of tumour growth over 24 days to that observed with free doxorubicin, with the added benefit of reduced cardiac injury as determined by histopathological assessment. A similar system in which the stearoyl groups are conjugated through a disulphide-containing linker and the drug (paclitaxel) is encapsulated via physical interactions has also been investigated.149 These materials also accumulated in tumour tissue, and led to inhibition of tumour growth. Unfortunately, due to difficulties in loading paclitaxel into a non-GSH responsive analogue, control experiments without the GSH-responsive property could not be conducted. As such, it is difficult to elucidate the precise role of the GSH-responsive groups in the observed tumour inhibition.
A further polysaccharide system investigated in vivo involves paclitaxel conjugated to hyaluronic acid via a GSH responsive disulphide linkage.132 In this case, nude mice bearing a murine hepatic carcinoma (Heps) were administered HA-SS-PTX every second day for 30 days. Five groups were investigated: HA9.5-SS-PTX and HA35-SS-PTX, (corresponding to HA molecular weights of 9.5 kDa and 35 kDa respectively), an mPEG-SS-PTX conjugate, free PTX and saline. The authors observed the best inhibition of tumour growth and survival rate with HA9.5-SS-PTX, followed by HA35-SS-PTX, mPEG-SS-PTX, PTX and saline. Importantly, mice administered with the conjugates also exhibited less wasting compared to those administered free PTX, indicating improved safety over the free drug. To further investigate the safety profile of the conjugates, histopathological assessment was also conducted on major organs, with little evidence of inflammatory response, cell degeneration or necrosis noted.
A similar system to the mPEG-SS-PTX control described above has been reported by Wang and coworkers.135 In this case, the material is self-assembled into particles and loaded with additional unconjugated paclitaxel. Both free paclitaxel and a non-GSH responsive analogue (PEG-PTX) are employed as controls. The particles incorporating the GSH-sensitive bonds provide the best tumour inhibition of the materials studied, and also lead to less weight loss than the dose of free PTX giving similar tumour inhibition. This study provides some of the clearest evidence to date for the utility of GSH responsive linkers in nanoparticle delivery vehicles for chemotherapeutic agents.
In a slightly different approach, Mallik and coworkers have examined liposomes with PEG chains attached via a GSH-responsive disulphide linkage.150 The PEG chains are included in this system in order to shield an incorporated lipopeptide from degradation by matrix metalloproteinase-9 (MMP-9), thus ensuring that the liposomes remain stable. However, upon contacting elevated levels of GSH in the tumour environment the protective PEG layer is shed, enabling MMP-9 to degrade the lipopeptide and thereby disrupt the liposome, releasing the encapsulated material. The authors demonstrate that the system can be employed for the delivery of gemcitabine by administering the particles (both with and without the lipopeptide component) to tumour bearing Nude-Foxn1 mice. The liposomes incorporating the lipopetide component demonstrated a more significant reduction in tumour volume relative to the control, thus demonstrating that the PEG protected system may offer some promise for the smart delivery of chemotherapeutic agents.
In another recent and highly creative approach, Guo et al. have reported the preparation of GSH-responsive block copolymer micelles incorporating a chemosensitizer as part of the polymer structure.151 Specifically, curcumin is reacted with dithiodipropionic acid to prepare a disulphide-containing polyester, and the resulting material is then converted into a block copolymer by reaction with either mPEG or biotin-terminated PEG. The block copolymers are then used to form particles with doxorubicin. Administration of the biotinylated particles to MCF-7/ADR tumour bearing mice leads to a significant reduction in tumour volume compared to administration of doxorubicin alone. The authors attribute this improved efficacy to release of the curcumin being triggered by GSH in the intracellular environment.
While in the majority of studies where GSH-responsive materials have been applied in vivo the materials seem to be generally well tolerated with good haemocompatibility, there is at least one study which suggests that disulphide crosslinks may lead to increased glutathione reductase activity in the spleen as a general indicator of oxidative stress.152 Importantly, though, the same authors did not observe any tissue damage upon histological investigation of spleen, kidney or liver tissue.
(b) Delivery of genetic material
As noted in the Introduction, one of the primary motivations for the use of particle based delivery is the protection of sensitive molecules from degradation in the bloodstream. Indeed, much of this endeavour has been concentrated in the field of RNA and DNA delivery. In many cases this does not involve the development of particles per se, rather the development of materials which, upon complexation with a polynucleic acid, form particles which can be applied in therapeutic delivery. Of course, a necessary element of any such system is that the complex can be disrupted to release the genetic material in the cytosol. As such, there has been considerable interest in the development of materials that are both bioreducible and endosomolytic.For example, Wagner and coworkers have reported on siRNA conjugates formed by complexing siRNA with a novel polymer incorporating polylysine (PLL) (to bind and protect the RNA), PEG (to solubilize and shield the complex), and a lytic peptide (melittin).153 The melittin component is masked by protecting the lysine residues with dimethylmaleic anhydride (DMMAn) which is removable at endosomal pH, thus ensuring that the membrane-lytic activity is confined to the endosomal compartment. In this example the siRNA is attached to the PLL via a bioreducible disulfide bond, in order to prevent unpacking of the complex by heparin. While the synthesized materials showed excellent gene knockdown in vitro, significant levels of toxicity were observed when the materials were administered to mice, with the authors noting macroscopic liver damage and intestinal bleeding. The authors suggested that the observed toxicity may be due to lack of targeting, incomplete PEG shielding or conjugate aggregation.
Cai et al. have reported on polymers for plasmid DNA (pDNA) delivery in which PEG chains are conjugated to a PLL backbone via disulphide containing linkers.154 In this case, mPEG is first modified by reaction with succinic anhydride to give carboxylic acid terminated PEG. This was then reacted with cysteamine to give amine-terminated PEG incorporating a disulphide linkage. This material was then used to initiate polymerization of ε-benzyloxycarbonyl-L-lysine N-carboxyanhydride, thus obtaining block copolymers of ε-benzyloxycarbonyl-protected PLL and PEG with a disulphide at the junction between the two blocks. Deprotection yielded disulphide linked block copolymers of PLL and PEG, with the PEG block shown to be cleavable in the presence of 10 mM GSH. The materials exhibited comparable transfection activity to PEI 25 kDa in both 293 T and Hela cells, with the additional benefit of lower cytotoxicity. The same group employed a similar system with the added feature of pH-sensitive crosslinking of the PLL domain using glutaraldehyde, and again found good transfection efficiency with reduced cytotoxicity compared to PEI.155 Exposure to GSH at 10 mM was again found to unpack the complex, enabling release of the genetic material for transfection.
Kataoka and coworkers have reported on similar blocks in which a poly{N-[N-(2-aminoethyl)-2-aminoethyl]aspartamide} (PAsp(DET)) segment was used instead of a PLL domain.156 Moreover, these authors introduced stearoyl functionality by reacting a proportion of the pendant amino groups with N-succinimidyl octadecanoate. The resulting materials were shown to effectively complex siRNA and lead to significant VEGF silencing compared to an analogue without a disulphide linkage. Moreover, cell viability was maintained at the levels required for silencing, and haemolytic activity could be minimized by selecting the N/P ratio appropriately. In vivo investigations showed that the delivery of the siRNA reduced tumor growth for between 11 and 15 days, after which the tumour growth returned to a similar trajectory to an untreated tumour.
Poly(amidoamines) (PAMAMs) with disulphide linkages in the backbone have also been explored for the delivery of genetic material.157 To prepare such materials, 1-(2-aminoethyl)piperazine (AEPZ) and N,N′-bis(acryloyl)cystamine (BAC) were stirred together in a ratio of 1:2 at 50 °C for five days, after which a further 2 equivalents of amine (either AEPZ, 3-amino-1,2-propanediol, PEG-amine or a 1:1 mixture of AEPZ and PEG-amine) was added to cap the terminal unsaturation and provide a suite of materials for further investigation (amine terminated, diol terminated, PEG terminated and 50/50 amine and PEG terminated). All materials synthesized showed reduced cytotoxicity compared to PEI, and this was attributed to the degradability of the disulphide linkages in the PAMAM core. The addition of PEG chains to the PAMAM somewhat reduced the transfection efficiency, which was attributed to reduced internalisation of the particles due to the shielding effect of the PEG chains. Altogether, these results indicate that disulfide containing PAMAM holds considerable promise as a delivery vehicle for genetic material.
Lin and coworkers have reported a GSH-responsive four-armed PEG star capped with poly(disulfide histamine) oligomers, and employed this to deliver DNA both in vitro and in vivo.158 The polymer was prepared by first synthesizing the poly(disulfide histamine) oligomers through the reaction of histamine with bis(acryloylcystamine), and then conjugating these oligomers to vinylsulfone capped four armed PEG stars via Michael addition. The resulting polymers were shown to effectively complex DNA into approx. 150 nm particles. Although these materials had lower transfection efficiency compared to PEI, they also exhibited substantially lower cytotoxicity, as demonstrated both in cell viability assays and by administering the particles to nude mice. Moreover, the particles induced gene expression in tumour to a greater extent than in other organs, as opposed to PEI which led to significantly higher levels of expression in the lung and spleen, compared to the tumour.
An et al. have recently reported “Lipopolymer 49” as a suitable siRNA carrier for gene silencing in glioma cells.159 This material is a solid-phase synthesized T-shaped peptide-like oligoamide with two central oleic acids, 20 aminoethane units, and two terminal cysteine units. The authors demonstrate that these materials form polyplexes of approx. 200 nm with siRNA, and that they efficiently release siRNA upon exposure to GSH. The authors demonstrate excellent gene downregulation in glioma cells, and after intravenous delivery to glioma model nude mice. This is one of an increasing number of papers showing good in vivo data for gene knockdown with appropriately designed systems.
Functionalised oligosaccharides incorporating disulphide linkages have also been employed in delivery of genetic materials. In one example chitosan was functionalised with (i) PEI and (ii) PEG through disulphide linkages by first reacting native chitosan with 3,3′-dithiodipropionic anhydride.160 The resulting acid functional chitosan was then reacted sequentially with amine terminated mPEG and PEI (1800 Da). The materials were shown to effectively bind pDNA into particles with diameter from 100–150 nm, depending on the N/P ratio. Exposure to 10 mM GSH led to an unravelling of the complex and concomitant increase in particle size to ca. 800 nm. The materials showed transfection efficiency comparable to PEI, but with significantly diminished cytotoxicity.
Zentel and coworkers have developed a novel nanogel system for complexing siRNA using block copolymers of tri(ethylene glycol methyl ether) methacrylate (TEGMA) and pentafluorophenyl methacrylate.161 In this system, the reactive pentafluorophenyl ester groups were used to prepare nanogels through reaction with a novel diamine crosslinker prepared from spermine and cystamine. The resulting crosslinks included secondary amine links that were suitable for complexing the siRNA. Importantly, the authors were able to demonstrate release of the siRNA from the nanogel in the presence of a suitable reducing agent (DTT) at 20 mM.
Zhang and coworkers have explored three-armed peptides as an alternative vector for the efficient delivery of pDNA.162 In this report two peptides were employed: one comprised of lysine units with a terminal cysteine on each arm, and the other comprising primarily lysine residues with a single histidine in each arm adjacent to the terminal cysteine. Formation of disulphide linkages between the terminal cysteines enables the formation of highly branched polypeptides which are susceptible to disassembly via reduction by intracellular GSH. The authors demonstrated that the peptides efficiently bound pDNA, forming polyplexes of 150–300 nm. The peptides were used to transfect HeLa and COS7 cells, and it was demonstrated that both polypeptides exhibited improved transfection efficiency and reduced cytotoxicity compared to linear polylysine. Moreover, of the three armed polypeptides those incorporating histidine units showed slightly higher transfection efficiency. This observation was attributed to enhanced endosomal escape.
Inorganic materials functionalised with disulfides have also been employed as a basis for the delivery of pDNA. For instance, molybdenum(IV) sulphide (MoS2) sheets have been decorated with PEI and PEG chains by exploiting surface interactions with thiols.163 In this case PEI was first modified by amidation with lipoic acid to yield disulphide-functionalised PEI. Exposure of the MoS2 sheets to the modified PEI and PEG thiol with sonication, followed by removing the excess polymer with repeated centrifugation/re-dispersion cycles provided MoS2–PEI–PEG nanocomposites. Interestingly, irradiation of MoS2 with light in the near infrared led to a localised heating which could be employed to facilitate particle escape from the endosome. Once in the reducing environment of the cytosol, exposure to millimolar concentrations of GSH leads to cleavage of the PEI and PEG from the MoS2, resulting in release of the DNA and efficient transfection.
An alternative hybrid organic–inorganic system for oligonucleotide delivery has been proposed by Rosenholm and coworkers.164 In this example, amine functional mesoporous silica was first reacted with succinic anhydride to provide carboxylic acid functionalised particles. These were then reacted with cystamine to provide particles with disulphide linked amine groups. The resulting particles were then used for two purposes: (i) to sequester oligonucleotides via electrostatic associations; and (ii) to provide a handle through which to conjugate N-hydroxysuccinimidyl ester activated PEG. The authors demonstrated that oligonucleotide sequences could be efficiently encapsulated in the mesoporous silica, and that exposure to 1 or 10 mM GSH could be used to trigger release. Importantly, lower concentrations of GSH (10 μM) led to only minimal release of the genetic material.
Materials with diselenide bonds have also been incorporated into gene delivery systems. Cheng et al. prepared oligoethyleneimine (OEI) incorporating (i) diselenide; (ii) monoselenide and (iii) disulphide linkages using the disuccinimidyl esters of 3,3′-diselanediyldipropanoic acid, 3,3′-selenodipropanoic acid and 3,3′-disulfanediyldipropanoic acid, respectively.165,166 Both diselenide and disulphide materials were susceptible to reduction using GSH or DTT, although the diselenide-functionalised PEI was somewhat more stable than the disulphide containing OEI. Nevertheless, the authors noted that the cytoxicity and transfection efficiency of the diselenide linked OEI was comparable to that observed for the disulfide linked OEI, and that both exhibited lower cytotoxicity than non-degradable 25 K PEI. As such, diselenide containing materials may have some utility in delivery of genetic material.
(c) Co-delivery of drugs and genetic material
In some therapeutic situations it is desirable to deliver not just one, but two or more therapeutic agents. In particular, there are certain conditions for which the delivery of a cytotoxic agent together with gene therapy may lead to better clinical outcomes. As such there has been some interest in the development of particle based systems for the dual delivery of drugs together with pDNA or siRNA. One such example has recently been reported by Wang et al.167 In this example, the authors modified branched PEG with cystamine via 1,1′-carbonyldiimidazole mediated coupling. The terminal amines introduced through the cystamine were then reacted with succinimidyl ester terminated linear PEG. Finally, the terminal hydroxyl groups were conjugated with the amine groups on G2 PAMAM dendrimers, again using 1,1′-carbonyldiimidazole mediated coupling. The resulting materials were shown to bind siRNA (or DNA) and drug (doxorubicin) to a high loading in particle form, which the authors attributed to structure inversion. Particles incorporating B-cell lymphoma 2 (Bcl-2) siRNA and doxorubicin were then administered to BALB/c nude mice with SMMC-7721 xenografts. It was observed that co-delivery of both drug and siRNA led to the greatest inhibition of tumour growth, compared to administration of the particles with doxorubicin or siRNA alone. Altogether, these branched PEG-dendrimer conjugates with GSH responsive linkages offer considerable potential as efficient co-delivery vectors for next generation cancer medications.An alternative co-delivery vector has been reported by Ma et al.168 In this case mesoporous silica nanoparticles were first prepared by condensing tetraethylorthosilicate (TEOS) and 3-mercaptopyltrimethoxysilane (MPTMS) in the presence of the structure-directing agent cetyltrimethylammonium bromide (CTAB). After removal of the CTAB, the surface thiols were reacted with S-(2-aminoethylthio)-2-thiopyridine hydrochloride to yield amine surface functionality linked to the silica through a GSH responsive disulphide bond. These were then used to conjugate adamantane-1-carboxylic acid via the formation of an amide linkage. The particles were then loaded with doxorubicin, after which the pendant adamantane groups were used to form stable host–guest complexes with ethylenediamine modified β-cyclodextrin (CD-2NH2). The CD-2NH2 served the dual purpose of capping the pores of the mesoporous silica, thus retaining the drug, and also providing positive charge for binding the negatively charged siRNA. The authors demonstrated controlled release of drug and siRNA in response to elevated levels of GSH in vitro. Moreover, successful delivery of siRNA in vivo was demonstrated using delivery of GFP siRNA to transgenic zebrafish larvae, while anticancer activity of the drug loaded particles was demonstrated in a zebrafish mifepristone-inducible liver tumor model.
A third system for potential co-delivery of plasmid DNA and chemotherapeutic based on thiolated gelatin nanoparticles has been reported by Amiji's group.169 In this case, type B gelatin is first thiolated by reacting the primary amine groups with 2-iminothiolane. Particles are then formed from an aqueous 1% (w/v) thiolated gelatin solution at 37 °C with the pH adjusted to 7 using sodium hydroxide. After dispersing 1 mg of plasmid DNA in the solution, chilled ethanol is slowly added with continuous stirring at 600 rpm. The gelatin nanoparticles form when the solvent composition reaches 75% water, at which point the thiol groups are crosslinked by dropwise addition of 8% (v/v) glyoxal solution. The particles can be PEGylated by addition of methoxy-PEG-succinimidylcarbosyl methyl ester or maleimide PEG-succinimidylcarbosyl methyl ester. Maleimide functionality enables subsequent functionalisation with cysteine terminated EGFR binding peptide. Gemcitabine functionalised particles were prepared by reacting the thiolated gelatin particles with succinimidyl 3-[2-pyridyldithio]propionate-modified gemcitabine, thus providing particles wherein the drug is conjugated through a GSH responsive disulphide linker. Panc-1-tumour bearing mice were administered with plasmid DNA-only particles, gemicitabine-only particles or a combination of both particles. The authors demonstrated that the combination treatment groups showed significantly greater anti-tumor activity compared to those administered only the pDNA or drug, and that targeted nanoparticles exhibited the best efficacy. Although this system did not involve co-delivery simultaneously from the same particle, the underlying platform is amenable to modification toward that end, and as such it represents an important development in the preparation of next generation co-delivery vectors for combination gene/drug therapy.
5. Other applications of GSH-responsive materials
There have been several reports of using GSH responsive materials in fluorescence and MRI imaging. For instance, Ang et al. have reported polymer nanoparticles incorporating a fluorescence probe for sensing biological thiol molecules.170 This system involves incorporating a weakly fluorescent molecule containing a disulphide linkage (PySSCou) into nanoparticles formed through supramolecular assembly of adamantane-functionalised poly(acrylic acid) (PAA-AD), adamantane-functionalized PEG (PEG-AD) and β-cyclodextrin-conjugated polyacrylic acid (PAA-CD). The fluorescence of the PySSCou is weak due to photoinduced electron transfer (PET) between the pyridine group and the coumarin moiety. However, upon exposure to thiol-containing compounds such as cysteine, homocysteine, GSH and DTT, the disulphide in the PySSCou is cleaved, abolishing the PET process and leading to a fluorescent turn-on. The authors demonstrate that these particles can be used to sense thiol-containing molecules down to micromolar quantities.Liu and coworkers have also developed GSH responsive materials with possible application in fluorescence imaging.171 In this case, poly(amidoamines) with disulphide linkages in the backbone and pendant PEG chains and cholesterol groups were prepared. The polymers could be self-assembled into nanoparticles in the presence of TPE-MI, an essentially non-fluorescent compound which becomes fluorescent upon reaction with thiols. The authors demonstrate that exposure to millimolar levels of GSH leads to destabilisation of the micelles and precipitation, and to fluorescent turn-on. Importantly, the utility of the particles in sensing intracellular thiols is also demonstrated in MCF-7 and HepG2 cells.
Johnson and coworkers have recently reported the use of branched-bottlebrush polymers for in vivo MRI and fluorescence imaging (Fig. 12).172 In this system, the authors employed nitroxides as organic radical contrast agents (ORCAs) for MRI, and Cy5.5 as a fluorescent probe. These moieties were incorporated into a polymer with PEG branches and a polynorbornene backbone. The materials were shown to provide significant contrast enhancement. Moreover, in their native state the nitroxides efficiently quenched the Cy5.5 fluorescence, but upon reduction with ascorbate a significant increase in fluorescence intensity was observed. Although exposure to GSH alone did not yield a significant increase in fluorescence intensity, exposure to GSH with ascorbate substantially increased the fluorescent signal. These results provide an excellent platform for the further development of redox responsive imaging agents.
 | ||
| Fig. 12 Preparation of branched-bottlebrush polymers for in vivo MRI and fluorescence imaging as described by Sowers et al. (a) Structures of branched macromonomers used. (b) General procedure for branched-bottlebrush polymer synthesis using ROMP. Reproduced with permission from Kim et al., Nature Commun., 2014, 5, 5460. © 2014 Nature Publishing Group. | ||
GSH-responsive materials have also been employed in the preparation of 19F NMR probes.173 In this case, a fluorinated polyhedral oligosilsesquioxane (F-POSS) was first prepared by reacting octaammonium polyhedral oligomeric silsesquioxane with ethyl trifluoroacetate, and then conjugating to a silica nanoparticle using a disulphide containing linker. The resulting materials were exposed to 1 mM glutathione (in either oxidised (GSSG) or reduced (GSH) form) and to a solution containing glutathione reductase and oxidised glutathione. An 19F signal was only detected when there was reduced GSH present to facilitate cleavage of the F-POSS from the silica nanoparticle. Moreover, the system was also effective for sensing glutathione reductase activity in the solution containing oxidised GSH (GSSG) and the enzyme. These results indicate the usefulness of GSH-responsive systems in potential sensing applications.
Aside from the extensive literature on release of chemotherapeutic agents and RNA, there is emerging interest in materials which are capable of releasing signalling molecules through interaction with thiol containing molecules. For instance, Foster and Matson have reported on the preparation of polymers incorporating pendant thiooxime functionality, and demonstrated that these materials are able to release H2S upon exposure to a thiol-containing molecule.174 The materials are synthesized by via RAFT polymerization of 2-(4-formylbenzoyloxy)ethyl methacrylate, which is subsequently used to conjugate S-aroylthiohydroxylamine pendants in the presence of trifluoroacetic acid. The resulting materials were shown to release H2S in the presence of a thiol-containing molecule (cysteine or GSH). We have also investigated H2S releasing materials, although in our example the H2S releasing pendant was an aryl thioamide incorporated via thionation of a benzonitrile containing polymeric precursor (Fig. 13).175 These materials were observed to release H2S hydrolytically and in the presence of a thiol stimulus. Although cysteine was used in these studies, it is highly likely that the materials would also be reactive to GSH. These materials were then used to stimulate certain kinase dependent signalling pathways (ERK and PKC signalling), thereby demonstrating that H2S release from the synthesized polymers was at a rate and quantum suitable for stimulating observable biochemical events.
 | ||
| Fig. 13 Synthesis of macromolecular H2S donors with aryl thioamide functionality as described by Ercole et al. Reagents and conditions: (i) 3-bromo-1-propanol, K2CO3, and DMF; (ii) methacrylic anhydride, TEA, and DCM; (iii) MMA, 70 °C, 1,4-dioxane; (iv) MMA, CPPMA, 70 °C, 1,4-dioxane; (v) polymer end-group removal, 1-ethylpiperidine hypophosphite, AIBN, toluene, 80 °C; (vi) thionation, NaHS, DMF, MgCl2·6H2O. CPPMA = 3-(4-cyanophenoxy) propyl methacrylate; OEGMA = oligo(ethylene glycol) methyl ether methacrylate, with average Mn = 300 g mol−1; MMA = methyl methacrylate. Reproduced with permission from Ercole et al., Biomacromolecules, 2016, 17, 371–383. © 2016 American Chemical Society. | ||
Talleli and Vicent have also reported an interesting system in which a glutathione stimulus is employed in a system suitable for polymer masked–unmasked protein therapy (PUMPT) (Fig. 14).176 Specifically, the authors first prepare pyridyl disulfide functionalised poly(glutamic acid) (PD-PGA) by first reacting native PGA with 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl morpholinium (DMTMM) chloride, and then stirring with pyridyl dithiolethylamine for 18 hours. Separately, the authors modified a proportion of lysine residues in lysozyme with N-succinimidyl-S-acetylthiopropionate (SATP) to provide a model protein with pendant thioester functionality. By combining the lysozyme and PD-PGA in different ratios (5:1, 2.5:1 and 0.5:1), the authors were able to mask the lysozyme activity to differing extents. Importantly, the resulting conjugates were stable in micromolar concentrations of GSH (20 μM, mimicking the bloodstream) but could be dissociated by exposure to millimolar concentrations (4 or 10 mM, mimicking the intracellular environment). Moreover, exposure to GSH at 10 mM also enabled recovery of lysozyme activity to varying extents. This interesting approach holds some promise for the delivery of anticancer enzymes, or for therapies in other disorders where enzyme replacement therapy may be beneficial.
 | ||
| Fig. 14 Masking protein activity with disulphide conjugated polymer and subsequent unmasking under reducing conditions. Schematic representation of the synthesis of PGA–lysozyme conjugates. (A) Modification of poly(glutamic acid) (PGA) with pyridyl dithiol via DMTMM chloride activation to form pyridyl disulphide functionalised PGA (PD PGA), (B) modification of lysozyme with N-succinimidyl-S-acetylthiopropionate (SATP), and (C) conjugation of SATP-functionalised lysozyme to PD PGA in the presence of a deacetylation solution that yields thiolated protein. Adapted from Talleli and Vicent, Biomacromolecules, 2014, 15, 4168–4177. ©2014 American Chemical Society. | ||
Della Vecchia et al. have recently reported an interesting phenomenon in which polycysteinyldopamine (PCDA) promotes the autooxidation of GSH.177 The authors prepared PCDA by allowing 5-S-cysteinyldopamine (CDA) to autoxidize at a concentration of 10 mM in sodium bicarbonate (0.05 M). Dip-coated quartz, glass, plastic and metal were effectively coated with PCDA under these conditions. The resulting polymer, either in suspension or thin film form, efficiently promoted the oxidation of GSH in phosphate buffer at pH 8.5, leading to a depletion of between 65 and 80% in less than 3 hours. This was compared to polydopamine, which had little impact on the GSH concentration. Interestingly, the effect was not observed when the solution was deoxygenated, indicating an essential role for O2 in the autooxidative effect. While these materials are not responsive to GSH per se, they nevertheless indicate an interesting interaction with the molecule which may warrant further investigation for potential sensing applications.
6. Conclusions and future outlook
Clearly, the potential for using elevated GSH concentration as a release trigger for smart drug delivery systems has motivated the synthesis of a great many new materials. Of these, the vast majority employ disulphide (or diselenide) linkages as GSH responsive moieties. Disulfides can be readily incorporated into polymers through fairly straightforward chemistries, and this has undoubtedly contributed to the substantial array of materials that have been reported. For instance, a polymer with carboxylic acid functionality can be easily crosslinked with GSH responsive linkers through reaction with cystamine, using simple amide coupling mediated by carbodiimides or N-hydroxysuccinimidyl esters. Alternatively, polymers synthesized by free radical polymerization can be readily modified with disulfides through the incorporation of N,N′-bis(acryloyl)cystamine in the polymerization. Disulfides can also be readily incorporated by using an activated disulphide (like pyridyl disulphide) as a precursor to the desired disulphide linkage. Given the ease with which these chemistries can be applied, there is little wonder that so many different disulphide containing materials have been developed. Materials with a disulphide at the junction of two blocks, or in the core of a micelle, or linking a therapeutic agent directly have provided great scope for investigating these materials in vitro. These studies provide a good evidence base for suggesting that disulphide-containing materials may be useful in developing next generation drug delivery vehicles, whether it be for the delivery of chemotherapeutic agents or genetic material such as pDNA or siRNA.While there is no shortage of in vitro evidence for the utility of disulphide containing materials in drug delivery, the in vivo evidence base is less well-formed. There have been far fewer investigations of the use of disulphide containing materials in animal models, and in many cases in vivo testing is a minor element of paper more generally concerned with material synthesis. Moreover, there is a lack of studies in which the controls provide clear, unambiguous evidence for the advantages of incorporating a disulphide. As such, while the usefulness of GSH responsive polymers has been clearly demonstrated in vitro, the jury remains out on whether this will translate to clear clinical benefits when the same materials are applied to animals or humans. Moreover, how GSH-responsive materials might impact on the extracellular concentrations of GSH is also worthy of some investigation. It is one thing to say that particles are stable in the bloodstream, but are they impacting the blood chemistry in possibly deleterious ways? This issue is generally neglected by researchers in studies where the focus in firmly on delivery as opposed to the potential biochemical impact of the materials.
Of course, as we have demonstrated, there are numerous other strategies for imparting GSH responsive behaviour aside from simply incorporating a disulphide linkage into the polymer structure. For example, exploiting the competitive coordination of GSH with certain elements may hold some promise, particularly in the delivery of platinum based drugs. Along the same lines, the use of gold nanoparticles in the preparation of GSH responsive materials may offer numerous possibilities due to the strong associations between gold and thiols: there is undoubtedly scope for further investigations in this area. Moreover, materials for which GSH can be used to trigger phase transitions provide scope for further work, as do systems where GSH-responsive polymers are combined with mesoporous particles or supramolecular materials. The recent reports in which H2S releasing materials are stimulated by thiols also suggest that there may be other, hitherto unexplored applications of GSH responsive materials that are quite distinct from traditional drug delivery applications. Altogether, we conclude that there is a promising future for the development of GSH responsive materials, with work on disulphide and diselenide based drug delivery systems likely to continue alongside the emergence of exciting new materials that respond to GSH in new ways.
Acknowledgements
This research was funded by the Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (project number CE140100036). TPD wishes to acknowledge the award of an Australian Laureate Fellowship (FL140100052). The authors acknowledge significant financial and infrastructure support from the Monash Institute of Pharmaceutical Sciences.References
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- R. K. Jain and T. Stylianopolous, Nat. Rev. Clin. Oncol., 2010, 7, 653–664 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- W. Cheng, L. Gu, W. Ren and Y. Liu, Mater. Sci. Eng., C, 2014, 45, 600–608 CrossRef CAS PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 CrossRef CAS PubMed.
- L. Miao, C. M. Lin and L. Huang, J. Controlled Release, 2015, 219, 192–204 CrossRef CAS PubMed.
- G. K. Such, Y. Yan, A. P. R. Johnston, S. T. Gunawan and F. Caruso, Adv. Mater., 2015, 27, 2278–2297 CrossRef CAS PubMed.
- F. Meng, W. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS PubMed.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- A. Latorre and Á. Somoza, Curr. Top. Med. Chem., 2014, 14, 2662–2671 CrossRef CAS PubMed.
- C. C. Song, F. S. Du and Z. C. Li, J. Mater. Chem. B, 2014, 2, 3413–3426 RSC.
- C. V. Smith, D. P. Jones, T. M. Guenthner, L. H. Lash and B. H. Lauterburg, Toxicol. Appl. Pharmacol., 1996, 140, 1–12 CrossRef CAS PubMed.
- D. Montero, C. Tachibana, J. R. Winther and C. Appenzeller-Herzog, Redox Biol., 2013, 1, 508–513 CrossRef CAS PubMed.
- R. Cheng, F. Feng, F. Meng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2011, 152, 2–12 CrossRef CAS PubMed.
- P. J. Roth, C. Boyer, A. B. Lowe and T. P. Davis, Macromol. Rapid Commun., 2011, 32, 1123–1143 CrossRef CAS PubMed.
- J. A. Syrett, D. M. Haddleton, M. R. Whittaker, T. P. Davis and C. Boyer, Chem. Commun., 2011, 47, 1449–1451 RSC.
- J. Xu, L. Tao, C. Boyer, A. B. Lowe and T. P. Davis, Macromolecules, 2010, 43, 20–24 CrossRef CAS.
- Z. Jia, J. Liu, C. Boyer, V. Bulmus and T. P. Davis, Biomacromolecules, 2009, 10, 3253–3258 CrossRef CAS PubMed.
- S. H. Yu, J. M. Hu, F. Ercole, N. P. Truong, T. P. Davis, M. R. Whittaker and J. F. Quinn, ACS Macro Lett., 2015, 4, 1278 CrossRef CAS.
- V. Bulmus, M. Woodward, L. Lin, N. Murthy, P. Stayton and A. Hoffman, J. Controlled Release, 2003, 93, 105–120 CrossRef CAS PubMed.
- M. Bencini, E. Ranucci, P. Ferruti and A. Manfredi, Macromol. Rapid Commun., 2006, 27, 1060–1066 CrossRef CAS.
- C. Li, J. Madsen, S. P. Armes and A. L. Lewis, Angew. Chem., Int. Ed., 2006, 45, 3510–3513 CrossRef CAS PubMed.
- Y. Z. You, C. Y. Hong and C. Y. Pan, Adv. Funct. Mater., 2007, 17, 2470–2477 CrossRef CAS.
- X. Wang, L. Liu, Y. Luo, H. Shi, J. Li and H. Zhao, Macromol. Biosci., 2012, 12, 1575–1582 CrossRef CAS PubMed.
- H. Yang, X. Li, Y. Lan, T. Tian, J. Cui, T. Zhu, D. Shen and G. Li, J. Mater. Chem. C, 2013, 1, 6120–6128 RSC.
- L. Liu, L. Wu, J. Tan, L. Wang, Q. Liu, P. Liu and L. Liu, Polym. Chem., 2015, 6, 3934–3941 RSC.
- A. N. Zelikin, J. F. Quinn and F. Caruso, Biomacromolecules, 2006, 7, 27–30 CrossRef CAS PubMed.
- A. N. Zelikin, Q. Li and F. Caruso, Chem. Mater., 2008, 20, 2655–2661 CrossRef CAS.
- S. F. Chong, A. Sexton, R. De Rose, S. J. Kent, A. N. Zelikin and F. Caruso, Biomaterials, 2009, 30, 5178–5186 CrossRef CAS PubMed.
- S. F. Chong, R. Chandrawati, B. Städler, J. Park, J. Cho, Y. Wang, Z. Jia, V. Bulmus, T. P. Davis, A. N. Zelikin and F. Caruso, Small, 2009, 5, 2601–2610 CrossRef CAS PubMed.
- B. Y. Li and D. T. Haynie, Biomacromolecules, 2004, 5, 1667–1670 CrossRef CAS PubMed.
- M. Ejaz, H. Yu, Y. Yan, D. A. Blake, R. S. Ayyala and S. M. Grayson, Polymer, 2011, 52, 5262–5270 CrossRef CAS.
- T. Dispinar, W. Van Camp, L. J. De Cock, B. G. De Geest and F. E. Du Prez, Macromol. Biosci., 2012, 12, 383–394 CrossRef CAS PubMed.
- D. J. Phillips and M. I. Gibson, Biomacromolecules, 2012, 13, 3200–3208 CrossRef CAS PubMed.
- M. J. Summers, D. J. Phillips and M. I. Gibson, Chem. Commun., 2013, 49, 4223–4225 RSC.
- D. J. Phillips, J. P. Patterson, R. K. O'Reilly and M. I. Gibson, Polym. Chem., 2014, 5, 126–131 RSC.
- F. Yang, J. Wang, J. Hou, H. Guo and C. Liu, Biomaterials, 2013, 34, 1514–1528 CrossRef CAS PubMed.
- A. R. Durney, S. Kawaguchi, G. Pennamon and H. Mukaibo, Mater. Lett., 2014, 133, 171–174 CrossRef CAS.
- Z. A. Nurpeissov, S. G. Alimkhanova, R. A. Mangazbayeva, Y. M. Shaikhutdinov, G. A. Mun and V. V. Khutoryanskiy, Eur. Polym. J., 2015, 69, 132–139 CrossRef.
- Y. Liang and K. L. Kiick, Biomacromolecules, 2016, 17, 601–614 CrossRef CAS PubMed.
- B. Begines, F. Zamora, M. Violante de Paz, K. Hakkou and J. A. Galbis, J. Appl. Polym. Sci., 2015, 132, 41304 CrossRef.
- S. Son, E. Shin and B. Kim, Macromolecules, 2015, 48, 600–609 CrossRef CAS.
- H. Chen, J. Jia, X. Duan, Z. Yang and J. Kong, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2374–2380 CrossRef CAS.
- J. Gan, X. Guan, J. Zheng, H. Guo, K. Wu, L. Liang and M. Lu, RSC Adv., 2016, 6, 32967–32978 RSC.
- X. Liu, J. He, D. Hu, Y. Niu, X. Xia and Y. Lu, RSC Adv., 2014, 4, 48897–48900 RSC.
- N. Ma, Y. Li, H. Xu, Z. Wang and Xi Zhang, J. Am. Chem. Soc., 2010, 132, 442–442 CrossRef CAS PubMed.
- L. Wang, W. Cao, Y. Yi and H. Xu, Langmuir, 2014, 30, 5628–5636 CrossRef CAS PubMed.
- Y. Zhou, K. Jie, B. Shi and Y. Yao, Chem. Commun., 2015, 51, 11112–11114 RSC.
- H. Xu, W. Cao and X. Zhang, Acc. Chem. Res., 2013, 46, 1647–1658 CrossRef CAS PubMed.
- S. Cajot, N. Lautram, C. Passirani and C. Jérôme, J. Controlled Release, 2011, 152, 30–36 CrossRef CAS PubMed.
- K. Rahimian, Y. Wen and J. K. Oh, Polymer, 2015, 72, 387–394 CrossRef CAS.
- M. Zhao, A. Biswas, B. Hu, K. Joo, P. Wang, Z. Gu and Y. Tang, Biomaterials, 2011, 32, 5223–5230 CrossRef CAS PubMed.
- R. Bird, T. Freemont and B. R. Saunders, Soft Matter, 2012, 8, 1047–1057 RSC.
- K. Liang, G. K. Such, Z. Zhu, S. J. Dodds, A. P. R. Johnston, J. Cui, H. Ejima and F. Caruso, ACS Nano, 2012, 6, 10186–10194 CrossRef CAS PubMed.
- A. Harada, R. Matsuki, S. Ichimura, E. Yuba and K. Kono, Molecules, 2013, 18, 12168–12179 CrossRef CAS PubMed.
- H. Urakami, J. Hentschel, K. Seetho, H. Zeng, K. Chawla and Z. Guan, Biomacromolecules, 2013, 14, 3682–3688 CrossRef CAS PubMed.
- B. L. Tardy, H. H. Dam, M. M. J. Kamphuis, J. J. Richardson and F. Caruso, Biomacromolecules, 2014, 15, 53–59 CrossRef CAS PubMed.
- L. F. Han, Q. B. Chen, Z. T. Hu, J. G. Piao, C. Y. Hong, J. J. Yan and Y. Z. You, Macromol. Rapid Commun., 2014, 35, 649–654 CrossRef CAS PubMed.
- K. Wang, H. Peng, K. J. Thurecht, S. Puttick and A. K. Whittaker, Polym. Chem., 2014, 5, 1760–1771 RSC.
- A. Cunningham, N. R. Ko and J. K. Oh, Colloids Surf., B, 2014, 122, 693–700 CrossRef CAS PubMed.
- D. Basak, R. Bej and S. Ghosh, Polym. Chem., 2015, 6, 6465–6474 RSC.
- S. Bian, J. Zheng, X. Tang, D. Yi, Y. Wang and W. Yang, Chem. Mater., 2015, 27, 1262–1268 CrossRef CAS.
- L. Jia, D. Cui, J. Bignon, A. Di Cicco, J. Wdzieczak-Bakala, J. Liu and M. Li, Biomacromolecules, 2014, 15, 2206–2217 CrossRef CAS PubMed.
- K. Kempe, S. L. Ng, S. T. Gunawan, K. F. Noi and F. Caruso, Adv. Funct. Mater., 2014, 24, 6187–6194 CrossRef CAS.
- R. Liu, X. Zhao, T. Wu and P. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef CAS PubMed.
- R. Liu, Y. Zhang and P. Feng, J. Am. Chem. Soc., 2009, 131, 15128–15129 CrossRef CAS PubMed.
- C. Giménez, C. de la Torre, M. Gorbe, E. Aznar, F. Sancenón, J. R. Murguía, R. Martínez-Máñez, M. D. Marcos and P. Amorós, Langmuir, 2015, 31, 3753–3762 CrossRef PubMed.
- Z. Xie, H. Gong, M. Liu, H. Zhu and H. Sun, J. Biomater. Sci., Polym. Ed., 2016, 27, 55–68 CrossRef CAS PubMed.
- Y. Wang, N. Han, Q. Zhao, L. Bai, J. Li, T. Jiang and S. Wang, Eur. J. Pharm. Sci., 2015, 72, 12–20 CrossRef CAS PubMed.
- H. Kim, S. Kim, C. Park, H. Lee, H. J. Park and C. Kim, Adv. Mater., 2010, 22, 4280–4283 CrossRef CAS PubMed.
- D. Xiao, H. Z. Jia, N. Ma, R. X. Zhuo and X. Z. Zhang, Nanoscale, 2015, 7, 10071–10077 RSC.
- D. Xiao, H. Z. Jia, J. Zhang, C. W. Liu, R. X. Zhuo and X. Z. Zhang, Small, 2014, 10, 591–598 CrossRef CAS PubMed.
- Q. L. Li, S. H. Xu, H. Zhou, X. Wang, B. Dong, H. Gao, J. Tang and Y. W. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 28656–28664 Search PubMed.
- M. Zhang, J. Liu, Y. Kuang, Q. Li, H. Chen, H. Ye, L. Guo, Y. Xu, X. Chen, C. Li and B. Jiang, J. Mater. Chem. B, 2016, 4, 3387–3397 RSC.
- B. Chang, D. Chen, Y. Wang, Y. Chen, Y. Jiao, X. Sha and W. Yang, Chem. Mater., 2013, 25, 574–585 CrossRef CAS.
- H. Li, J. Z. Zhang, Q. Tang, M. Du, J. Hu and D. Yang, Mater. Sci. Eng., C, 2013, 33, 3426–3431 CrossRef CAS PubMed.
- G. F. Luo, W. H. Chen, Y. Liu, Q. Lei, R. X. Zhuo and X. Z. Zhang, Sci. Rep., 2014, 4, 6064 CrossRef CAS PubMed.
- M. Quesada, C. Muniesa and P. Botella, Chem. Mater., 2013, 25, 2597–2602 CrossRef CAS.
- Y. Wang, C. Y. Hong and C. Y. Pan, Biomacromolecules, 2013, 14, 1444–1451 CrossRef CAS PubMed.
- Z. Huang, H. Cang, R. Huang, Z. Cai and H. Zhang, Polym. Sci., Ser. B, 2014, 56, 883–894 CrossRef CAS.
- Y. C. Wang, F. Wang, T. M. Sun and J. Wang, Bioconjugate Chem., 2011, 22, 1939–1945 CrossRef CAS PubMed.
- Y. Wu, H. Kuang, Z. Xie, X. Chen, X. Jing and Y. Huang, Polym. Chem., 2014, 5, 4488–4498 RSC.
- H. C. Kim, E. J. Kim, T. L. Ha, S. W. Jeong, S. G. Lee, S. J. Lee and B. Lee, Colloids Surf., B, 2015, 127, 206–212 CrossRef CAS PubMed.
- W. Cao, Y. Li, Y. Yi, S. Ji, L. Zeng, Z. Sun and H. Xu, Chem. Sci., 2012, 3, 3403–3408 RSC.
- Y. Wang, G. Wu, X. Li, J. Chen, Y. Wang and J. Ma, J. Mater. Chem., 2012, 22, 25217–25226 RSC.
- Y. Guo, Y. Zhang, J. Li, Y. Zhang, Y. Lu, X. Jiang, X. He, H. Ma, S. An and C. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 5444–5453 CAS.
- J. Ding, F. Shi, C. Xiao, L. Lin, L. Chen, C. He, X. Zhuang and X. Chen, Polym. Chem., 2011, 2, 2857–2864 RSC.
- S. T. Liu, H. Y. Tuan-Mu, J. J. Hu and J. S. Jan, RSC Adv., 2015, 5, 87098–87107 RSC.
- F. Shi, J. Ding, C. Xiao, X. Zhuang, C. He, L. Chen and X. Chen, J. Mater. Chem., 2012, 22, 14168–14179 RSC.
- A. N. Koo, K. H. Min, H. J. Lee, S. U. Lee, K. Kim, I. C. Kwon, S. H. Cho, S. Y. Jeong and S. C. Lee, Biomaterials, 2012, 33, 1489–1499 CrossRef CAS PubMed.
- X. Chang, L. Liu, Y. Guan and C. M. Dong, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2000–2010 CrossRef CAS.
- N. Ma, H. Xu, L. An, J. Li, Z. Sun and X. Zhang, Langmuir, 2011, 27, 5874–5878 CrossRef CAS PubMed.
- B. Liu, X. Zhang, Y. Chen, Z. Yao, Z. Yang, D. Gao, Q. Jiang, J. Liu and Z. Jiang, Polym. Chem., 2015, 6, 1997–2010 RSC.
- Q. Yang, L. Tan, C. He, B. Liu, Y. Xu, Z. Zhu, Z. Shao, B. Gong and Y. M. Shen, Acta Biomater., 2015, 17, 193–200 CrossRef CAS PubMed.
- N. Song, W. Liu, Q. Tu, R. Liu, Y. Zhang and J. Wang, Colloids Surf., B, 2011, 87, 454–463 CrossRef CAS PubMed.
- Y. Yao, H. Xu, C. Liu, Y. Guan, D. Xu, J. Zhang, Y. Su, L. Zhao and J. Luo, RSC Adv., 2016, 6, 9082–9089 RSC.
- Z. Xu, S. Liu, Y. Kang and M. Wang, ACS Biomater. Sci. Eng., 2015, 1, 585–592 CrossRef CAS.
- B. Khorsand, G. Lapointe, C. Brett and J. K. Oh, Biomacromolecules, 2013, 14, 2103–2111 CrossRef CAS PubMed.
- X. Zhao and P. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 166–174 CAS.
- S. Samarajeewa, R. Shrestha, M. Elsabahy, A. Karwa, A. Li, R. P. Zentay, J. G. Kostelc, R. B. Dorshow and K. L. Wooley, Mol. Pharm., 2013, 10, 1092–1099 CrossRef CAS PubMed.
- Y. Zhang, Q. Qu, M. Li and Y. Zhao, Asian J. Org. Chem., 2015, 4, 226–232 CrossRef CAS.
- N. R. Ko and J. K. Oh, Biomacromolecules, 2014, 15, 3180–3189 CrossRef CAS PubMed.
- D. H. Nguyen, J. W. Bae, J. H. Choi, J. S. Lee and K. D. Park, J. Bioact. Compat. Polym., 2013, 28, 341–354 CrossRef CAS.
- T. Cai, Y. Chen, Y. Wang, H. Wang, X. Liu, Q. Jin, S. Agarwal and J. Ji, Macromol. Chem. Phys., 2014, 215, 1848–1854 CrossRef CAS.
- Y. Zhuang, H. Deng, Y. Su, L. He, R. Wang, G. Tong, D. He and X. Zhu, Biomacromolecules, 2016, 17, 2050–2062 CrossRef CAS PubMed.
- A. Guerry, S. Cottaz, E. Fleury, J. Bernard and S. Halila, Carbohydr. Polym., 2014, 112, 746–752 CrossRef CAS PubMed.
- L. Wu, L. Zhang, G. Shi and C. Ni, Mater. Sci. Eng., C, 2016, 61, 278–285 CrossRef CAS PubMed.
- C. Chen, P. Zheng, Z. Cao, Y. Ma, J. Li, H. Qian, W. Tao and X. Yang, Biomater. Sci., 2016, 4, 412–417 RSC.
- Y. J. Pan, D. Li, S. Jin, C. Wei, K. Y. Wu, J. Guo and C. C. Wang, Polym. Chem., 2013, 4, 3545–3553 RSC.
- Y. Wang, J. Nie, B. Chang, Y. Sun and W. Yang, Biomacromolecules, 2013, 14, 3034–3046 CrossRef CAS PubMed.
- Y. Tian, S. Bian and W. Yang, Polym. Chem., 2016, 7, 1913–1921 RSC.
- S. Jin, D. Li, P. Yang, J. Guo, J. Q. Lu and C. Wang, Colloids Surf., A, 2015, 484, 47–55 CrossRef CAS.
- R. Li, F. Feng, Y. Wang, X. Yang, X. Yang and V. C. Yang, J. Colloid Interface Sci., 2014, 429, 34–44 CrossRef CAS PubMed.
- J. Zeng, P. Du, L. Liu, J. Li, K. Tian, X. Jia, X. Zhao and P. Liu, Mol. Pharmaceutics, 2015, 12, 4188–4199 CrossRef CAS PubMed.
- A. Li, H. Ma, S. Feng and J. Liu, RSC Adv., 2016, 6, 33138–33147 RSC.
- K. Tian, J. Zeng, X. Zhao, L. Liu, X. Jia and P. Liu, Colloids Surf., B, 2015, 134, 188–195 CrossRef CAS PubMed.
- B. Xue, V. Kozlovskaya, F. Liu, J. Chen, J. F. Williams, J. Campos-Gomez, M. Saeed and E. Kharlampieva, ACS Appl. Mater. Interfaces, 2015, 7, 13633–13644 CAS.
- W. Cao, Y. Li, Y. Yi, S. Ji, L. Zeng, Z. Sun and H. Xu, Chem. Sci., 2012, 3, 3403–3408 RSC.
- J. Luan, W. Shen, C. Chen, K. Lei, L. Yu and J. Ding, RSC Adv., 2015, 5, 97975–97981 RSC.
- Y. Tao, J. Han, C. Ye, T. Thomas and H. Dou, J. Mater. Chem., 2012, 22, 18864–18871 RSC.
- C. W. Lin, K. Y. Lu, S. Y. Wang, H. W. Sung and F. L. Mi, Acta Biomater., 2016, 35, 280–292 CrossRef CAS PubMed.
- H. C. Kim, E. J. Kim, S. G. Lee, S. J. Lee, H. Kim and S. W. Jeong, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 582–589 CrossRef CAS.
- R. H. Utama, Y. Jiang, P. B. Zetterlund and M. H. Stenzel, Biomacromolecules, 2015, 16, 2144–2156 CrossRef CAS PubMed.
- X. Zhao, L. Liu, X. Li, J. Zeng, X. Jia and P. Liu, Langmuir, 2014, 30, 10419–10429 CrossRef CAS PubMed.
- Y. Wu, H. Kuang, Z. Xie, X. Chen, X. Jing and Y. Huang, Polym. Chem., 2014, 5, 4488–4498 RSC.
- R. Nahire, M. K. Haldar, S. Paul, A. Mergoum, A. H. Ambre, K. S. Katti, K. N. Gange, D. K. Srivastava, K. Sarkar and S. Mallik, Biomacromolecules, 2013, 14, 841–853 CrossRef CAS PubMed.
- R. Nahire, M. K. Haldar, S. Paul, A. H. Ambre, V. Meghnani, B. Layek, K. S. Katti, K. N. Gange, J. Singh, K. Sarkar and S. Mallik, Biomaterials, 2014, 35, 6482–6497 CrossRef CAS PubMed.
- W. Chen, F. Meng, R. Cheng, C. Deng, J. Feijen and Z. Zhong, J. Controlled Release, 2015, 210, 125–133 CrossRef CAS PubMed.
- S. Wang, Y. Zhou, W. Guan and B. Ding, J. Nanopart. Res., 2009, 11, 909–916 CrossRef CAS.
- Y. Zhuang, Y. Su, Y. Peng, D. Wang, H. Deng, X. Xi, X. Zhu and Y. Lu, Biomacromolecules, 2014, 15, 1408–1418 CrossRef CAS PubMed.
- Y. Su, Y. Hu, Y. Du, X. Huang, J. He, J. You, H. Yuan and F. Hu, Mol. Pharmaceutics, 2015, 12, 1193–1202 CrossRef CAS PubMed.
- S. Yin, J. Huai, X. Chen, Y. Yang, X. Zhang, Y. Gan, G. Wang, X. Gu and J. Li, Acta Biomater., 2015, 26, 274–285 CrossRef CAS PubMed.
- Y. Cao, M. Gao, C. Chen, A. Fan, J. Zhang, D. Kong, Z. Wang, D. Peer and Y. Zhao, Nanotechnology, 2015, 26, 115101 CrossRef PubMed.
- S. Zhao, D. Wang, K. Zhang and H. Zhang, Int. J. Polym. Mater. Polym. Biomater., 2015, 64, 792–799 CrossRef CAS.
- X. Chuan, Q. Song, J. Lin, X. Chen, H. Zhang, W. Dai, B. He, X. Wang and Q. Zhang, Mol. Pharmaceutics, 2014, 11, 3656–3670 CrossRef CAS PubMed.
- Q. Yan, Y. Yang, W. Chen, J. Hu and D. Yang, Mater. Sci. Eng., C, 2016, 58, 580–585 CrossRef CAS PubMed.
- Q. Zhang, J. He, M. Zhang and P. Ni, J. Mater. Chem. B, 2015, 3, 4922–4932 RSC.
- Q. Zhang, M. R. Vakili, X. F. Li, A. Lavasanifar and X. C. Le, Biomaterials, 2014, 35, 7088–7100 CrossRef CAS PubMed.
- C. F. Riber, A. A. A. Smith and A. N. Zelikin, Adv. Healthcare Mater., 2015, 4, 1887–1890 CrossRef CAS PubMed.
- A. Kock, K. Zuwala, A. A. A. Smith, P. Ruis-Sanchis, B. M. Wohl, M. Tolstrup and A. N. Zelikin, Chem. Commun., 2014, 50, 14498–14500 RSC.
- P. Ruis-Sanchis, B. M. Wohl, A. A. A. Smith, K. Zuwala, J. Melchjorsen, M. Tolstrup and A. N. Zelikin, Adv. Healthcare Mater., 2015, 4, 65–68 CrossRef PubMed.
- K. Zuwala, A. A. A. Smith, M. Tolstrup and A. N. Zelikin, Chem. Sci., 2016, 7, 2353–2358 RSC.
- A. N. Koo, K. H. Min, H. J. Lee, S. U. Lee, K. Kim, I. C. Kwon, S. H. Cho, S. Y. Jeong and S. C. Lee, Biomaterials, 2012, 33, 1489–1499 CrossRef CAS PubMed.
- C. Shi, X. Guo, Q. Qu, Z. Tang, Y. Wang and S. Zhou, Biomaterials, 2014, 35, 8711–8722 CrossRef CAS PubMed.
- L. He, D. Li, Z. Wang, W. Xu, J. Wang, H. Guo, C. Wang and J. Ding, Polymers, 2016, 8, 36, DOI:10.3390.
- S. V. Lale, A. Kumar, S. Prasad, A. C. Bharti and V. Koul, Biomacromolecules, 2015, 16, 1736–1752 CrossRef CAS PubMed.
- S. Jin, J. Wan, L. Meng, X. Huang, J. Guo, L. Liu and C. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 19843–19852 CAS.
- K. Hu, H. Zhou, Y. Liu, Z. Liu, J. Liu, J. Tang, J. Li, J. Zhang, W. Sheng, Y. Zhao, Y. Wu and C. Chen, Nanoscale, 2015, 7, 8607–8618 RSC.
- Y. W. Hu, Y. Z. Du, N. Liu, X. Liu, T. T. Meng, B. L. Cheng, J. B. He, J. You, H. Yuan and F. Q. Hu, J. Controlled Release, 2015, 206, 91–100 CrossRef CAS PubMed.
- P. S. Kulkarni, M. K. Haldar, R. R. Nahire, P. Katti, A. H. Ambre, W. W. Muhonen, J. B. Shabb, S. K. R. Padi, R. K. Singh, P. P. Borowicz, D. K. Shrivastava, K. S. Katti, K. Reindl, B. Guo and S. Mallik, Mol. Pharmaceutics, 2014, 11, 2390–2399 CrossRef CAS PubMed.
- S. Guo, L. Lv, Y. Shen, Z. Hu, Q. He and X. Chen, Sci. Rep., 2016, 6, 21459 CrossRef CAS PubMed.
- E. Pérez, R. Olmo, C. Teijón, E. Muníz, N. Montero, J. M. Teijón and M. D. Blanco, Colloids Surf., B, 2015, 136, 222–231 CrossRef PubMed.
- M. Meyer, C. Dohmen, A. Philipp, D. Kiener, G. Maiwald, C. Scheu, M. Ogris and E. Wagner, Mol. Pharm., 2009, 6, 752–762 CrossRef CAS PubMed.
- X. J. Cai, H. Q. Dong, W. J. Xia, H. Y. Wen, X. Q. Li, J. H. Yu, Y. Y. Li and D. L. Shi, J. Mater. Chem., 2011, 21, 14639–14645 RSC.
- X. Cai, C. Dong, H. Dong, G. Wang, G. M. Pauletti, X. Pan, H. Wen, I. Mehl, Y. Li and D. Shi, Biomacromolecules, 2012, 13, 1024–1034 CrossRef CAS PubMed.
- H. J. Kim, M. Oba, F. Pittella, T. Nomoto, H. Cabral, Y. Matsumoto, K. Miyata, N. Nishiyama and K. Kataoka, J. Drug Targeting, 2012, 20, 33–42 CrossRef PubMed.
- Y. Ping, D. Wu, J. N. Kumar, W. Cheng, C. L. Lay and Y. Liu, Biomacromolecules, 2013, 14, 2083–2094 CrossRef CAS PubMed.
- K. An, P. Zhao, C. Lin and H. Liu, Int. J. Mol. Sci., 2014, 15, 9067–9081 CrossRef CAS PubMed.
- S. An, D. He, E. Wagner and C. Jiang, Small, 2015, 11, 5142–5150 CrossRef CAS PubMed.
- L. Jia, Z. Li, D. Zhang, Q. Zhang, J. Shen, H. Guo, X. Tian, G. Liu, D. Zheng and L. Qi, Polym. Chem., 2013, 4, 156–165 RSC.
- L. Nuhn, L. Braun, I. Overhoff, A. Kelsch, D. Schaeffel, K. Koynov and R. Zentel, Macromol. Rapid Commun., 2014, 35, 2057–2064 CrossRef CAS PubMed.
- Q. Lei, Y. X. Sun, S. Chen, S. Y. Qin, H. Z. Jia, R. X. Zhuo and X. Z. Zhang, Macromol. Biosci., 2014, 14, 546–556 CrossRef CAS PubMed.
- J. Kim, H. Kim and W. J. Kim, Small, 2016, 12, 1184–1192 CrossRef CAS PubMed.
- J. Zhang, M. Niemelä, J. Westermarck and J. M. Rosenholm, Dalton Trans., 2014, 43, 4115–4126 RSC.
- G. Cheng, Y. He, L. Xie, Y. Nie, B. He, Z. Zhang and Z. Gu, Int. J. Nanomed., 2012, 7, 3991–4006 CAS.
- D. Yue, G. Cheng, Y. He, Y. Nie, Q. Jiang, X. Cai and Z. Gu, J. Mater. Chem. B, 2014, 2, 7210–7221 RSC.
- K. Wang, Q. Hu, W. Zhu, M. Zhao, Y. Ping and G. Tang, Adv. Funct. Mater., 2015, 25, 3380–3392 CrossRef CAS.
- X. Ma, C. Teh, Q. Zhang, P. Borah, C. Choong, V. Korzh and Y. Zhao, Antioxid. Redox Signaling, 2014, 21, 707–722 CrossRef CAS PubMed.
- J. Xu, A. Singh and M. M. Amiji, BMC Cancer, 2014, 14, 75 CrossRef PubMed.
- C. Y. Ang, S. Y. Tan, Y. Lu, L. Bai, M. Li, P. Li, Q. Zhang, S. T. Selvan and Y. Zhao, Sci. Rep., 2014, 4, 7057 CrossRef CAS PubMed.
- W. Cheng, G. Wang, X. Pan, Y. Zhang, B. Z. Tang and Y. Liu, Macromol. Biosci., 2014, 14, 1059–1066 CrossRef CAS PubMed.
- M. A. Sowers, J. R. McCombs, Y. Wang, J. T. Paletta, S. W. Morton, E. C. Dreaden, M. D. Boska, M. F. Ottaviani, P. T. Hammond, A. Rajca and J. A. Johnson, Nat. Commun., 2014, 5, 5460 CrossRef PubMed.
- K. Tanaka, N. Kitamura and Y. Chujo, Bioorg. Med. Chem., 2012, 20, 96–100 CrossRef CAS PubMed.
- J. C. Foster and J. B. Matson, Macromolecules, 2014, 47, 5089–5095 CrossRef CAS.
- F. Ercole, F. M. Mansfeld, M. Kavallaris, M. R. Whittaker, J. F. Quinn, M. L. Halls and T. P. Davis, Biomacromolecules, 2016, 17, 371–383 CrossRef CAS PubMed.
- M. Talleli and M. J. Vicent, Biomacromolecules, 2014, 15, 4168–4177 CrossRef PubMed.
- N. Della Vecchia, R. Marega, M. Ambrico, M. Iacomino, R. Micillo, A. Napolitano, D. Bonifazi and M. d'Ischia, J. Mater. Chem. C, 2015, 3, 6525–6531 RSC.
| This journal is © The Royal Society of Chemistry 2017 |