DOI:
10.1039/C6PY01303A
(Paper)
Polym. Chem., 2016,
7, 6490-6499
Trityl-based alkoxyamines as NMP controllers and spin-labels†
Received
27th July 2016
, Accepted 24th September 2016
First published on 12th October 2016
Abstract
Recently, new applications of trityl-nitroxide biradicals were proposed. In the present study, attachment of a trityl radical to alkoxyamines was performed for the first time. The rate constants kd of C–ON bond homolysis in these alkoxyamines were measured and found to be similar to those for alkoxyamines without a trityl moiety. The electron paramagnetic resonance (EPR) spectra of the products of alkoxyamine homolysis (trityl-TEMPO and trityl-SG1 biradicals) were recorded, and the corresponding exchange interactions were estimated. The decomposition of trityl-alkoxyamines showed more than an 80% yield of biradicals, meaning that the C–ON bond homolysis is the main reaction. The suitability of these labelled initiators/controllers for polymerisation was exemplified by means of a successful nitroxide-mediated polymerisation (NMP) of styrene. Thus, this is the first report of a spin-labelled alkoxyamine suitable for NMP.
Introduction
Although persistent trityl radicals Ar3C˙ were discovered by Gomberg in 1900,1 only many decades later did these radicals arouse keen interest in the scientific community because of their appealing applications as probes.2–4 They are currently used as spin probes for oxymetry,2 as pH probes,5 and as spin labels for distance measurements in biomolecules.6 Very recently, the preparation of nitroxide-trityl radical dual probes was reported7 and aroused keen interest, and attracted a lot of attention as probes based on the modulation of the exchange interaction between the two spins8 and as polarizing agents for Dynamic Nuclear Polarisation-enhanced Nuclear Magnetic Resonance (DNP-NMR).9 In the same period, there was increasing interest in the practical applications of biradicals such as binitroxides because of their uses as an initiator/controller agent in Nitroxide Mediated Polymerization10–12 (NMP) and as a polarizing agent in DNP-NMR.13
In the last decade, Electron Paramagnetic Resonance (EPR) techniques experienced a tremendous development towards EPR imaging2,14 with the help of new technologies and the preparation of new spin probes such as those mentioned above leading to in vivo imaging,15 and imaging in materials.16 Moreover, in the last three decades, the chemistry of alkoxyamines R1R2NOR3 has been developed for their application to NMP as a controllers/initiators.17,18 Lately, new applications of these compounds emerged in materials sciences – as key moieties in self-healing polymers,19,20 as fluorescent switches,21,22 and as a coding system23,24 – as well as in biology as a new type of theranostic agent.25 Taking into account the valuable properties of trityl radicals as spin probes and the possible practical usefulness of alkoxyamines, and their various modes of activation – protonation, oxidation, alkylation, light irradiation – the investigation of the properties of trityl-attached alkoxyamines is a promising field. Here, we report the first preparation of several trityl-alkoxyamines (TA) based on the SG1 and TEMPO nitroxyl fragments (Fig. 1) as well as the homolysis rate constant kd of the C–ON bond, and the EPR features of the trityl-TEMPO and trityl-SG1 biradicals. Among the family of trityl paramagnets, the Finland-type radical 17 was selected because of the simplicity of its EPR signal (i.e. one peak), the suppression of the reactivity at the para position by its functionalisation,‡![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 and due to its carboxylic function suitable for many types of coupling reactions.§ The trityl radical was found to have no significant effect on kd, meaning that these alkoxyamines retained all their kinetic properties for use as a controllers/initiators in NMP, as a switch in optotronics, and as theranostic agents. Moreover, the ability of TA to control NMP was confirmed by the polymerisation of styrene with 1.
26 and due to its carboxylic function suitable for many types of coupling reactions.§ The trityl radical was found to have no significant effect on kd, meaning that these alkoxyamines retained all their kinetic properties for use as a controllers/initiators in NMP, as a switch in optotronics, and as theranostic agents. Moreover, the ability of TA to control NMP was confirmed by the polymerisation of styrene with 1.
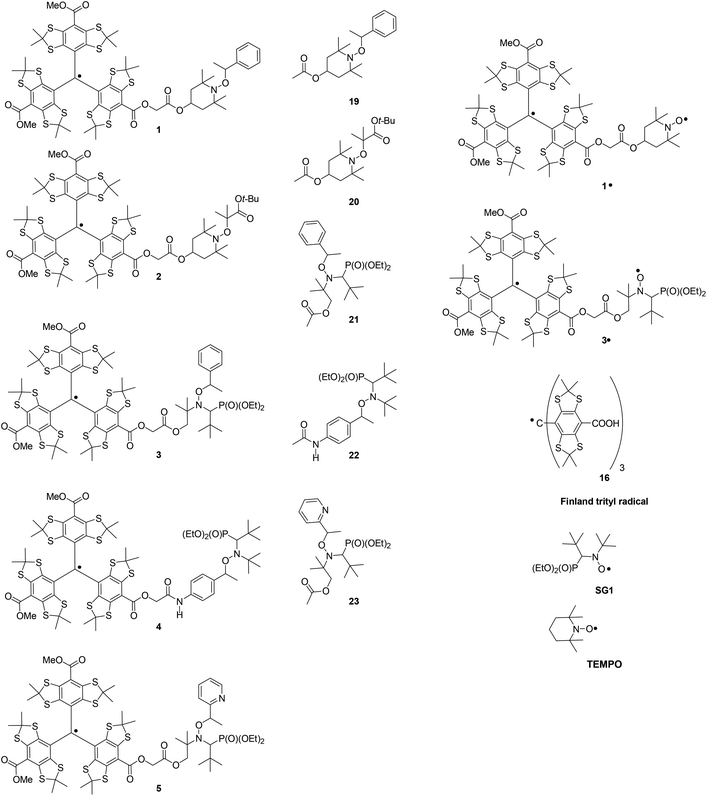 |
| | Fig. 1 Trityl-alkoxyamines and the corresponding biradicals under study. | |
Experimental section
Kinetic experiments
Homolysis rate constants kd were measured by EPR,27 as previously reported, and given by eqn (1) (Fig. S1†). Air was used as an alkyl radical scavenger for EPR experiments, respectively. Activation energies Ea were estimated using eqn (2) and the averaged frequency factor A = 2.4 × 1014 s−1.28 The values of kd and Ea are listed in Table 1.| | 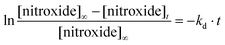 | (1) |
| | 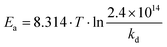 | (2) |
Table 1 Experimental homolysis rate constants kd,exp, the corresponding activation energy Ea, and the re-estimated rate constant kd at 120 °C
| TA |
Solvent |
T (°C) |
k
d,expa (10−4 s−1) |
E
ab (kJ mol−1) |
k
dc (10−4 s−1) |
|
The error on kd,exp is 5%.
Given by eqn (2), with kd values reported in the 4th column.
Re-estimated kd values at 120 °C using adapted eqn (2) and Ea values reported in the 5th column.
A diastereoisomeric mixture.
|
|
1
|
t-BuPh |
125 |
3.6 |
135.8 |
2.1 |
|
2
|
Toluene |
80 |
2.1 |
122.0 |
146.0 |
|
3
|
Toluene |
100 |
3.0d |
127.8c |
24.7 |
|
4
|
Toluene |
100 |
6.0d |
125.7c |
47.0 |
|
5
|
Toluene |
100 |
6.7d |
125.3c |
52.4 |
EPR experiments
The EPR measurements were carried out in CW mode using a Bruker EMX X-band spectrometer equipped with a BVT-2000 temperature control system.¶ The samples were degassed by 3 cycles of a freeze–pump–thaw procedure (P = 0.1 mb). The EasySpin software toolbox was used for simulation of the EPR spectra.29 To take into account the presence of various conformers of nitroxide-trityl biradicals showing slightly different spin–spin exchange interaction values, it was assumed that exchange interaction values are prone to a normal distribution with the mean value J and standard deviation ΔJ (eqn (3) and Table S1†).| | 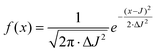 | (3) |
Polymerization experiments
A weighed amount of 1 (6.5 mg) was dissolved in 3 mL of styrene (distilled prior to the experiment). The reaction mixture was placed in a round-bottom two-neck flask equipped with a magnetic stirrer and a condenser, degassed by Ar bubbling and heated at 130 °C in an oil bath under an inert atmosphere. Sampling was performed by means of a syringe at various time intervals to monitor the evolution of the monomer conversion, molecular weight and PDI of the polymer prepared. The probes were quenched in an ice bath and stored at 4 °C prior to analysis. Monomer conversion was analysed by 1H NMR. The polymer molecular weight and distribution were analysed by gel permeation chromatography on an Agilent LC 1200 set-up equipped with an isocratic pump, a PL-mixed C GPC column, and UV and refractive index detectors. THF was used as an eluent at a flow rate of 1 ml min−1 and the column temperature was set at 35 °C. The column was calibrated by using narrow PDI polystyrene standards (Agilent).
General
Solvents and reactants were used as received. (1-Bromoethyl)benzene (97%), trifluoromethanesulfonate copper(II) (Cu(OTf)2) (98%) and 4-tert-butyl-2-(4-tert-butylpyridin-2-yl)pyridine (dTbpy) (98%) were purchased from Aldrich. tert-Butyl 2-bromo-2-methylpropanoate was purchased from Fluka. Alkoxyamines 6,308,31 and 932 were prepared according to the literature.33,34 2-Bromoacetyl bromide was obtained by a known literature method.30
Routine reaction monitoring was performed using silica gel 60 F254 TLC plates; spots were visualised by exposure to UV light and a phosphomolybdic acid solution in EtOH, followed by heating. Purification procedures were performed on chromatography columns with silica gel grade 60 (230–400 mesh). The 1H, 13C, and 31P NMR spectra were recorded with a Bruker AV-400 (1H: 400.13 MHz, 13C{1H}: 100.61 MHz) and Bruker AV-300 (1H: 300.13 MHz, 13C{1H}: 75.476 MHz) spectrometer. Deuterochloroform (CDCl3) was used as the solvent, with residual CHCl3 (δH = 7.25 ppm) or CDCl3 (δC = 77.0 ppm) being employed as an internal standard. In recording 31P-NMR spectra, 85% H3PO4 was used as an internal standard (δP = 0 ppm). The precise masses of molecular ions were determined by HRMS on a DFS Thermo scientific instrument (EI, 70 eV). The melting point was determined on an FP 900 Thermosystem microscope melting point apparatus (Mettler-Toledo International Inc., Zürich, Switzerland). Elemental analysis was performed on a Euro EA-3000 CHNS analyzer. IR spectra were recorded with a Bruker Tensor 27 FTIR spectrometer, and KBr pellets were used. Wavenumber values are given in cm−1. Electrospray ionization mass spectra ESI/MS were recorded using a hybrid quadrupole/time-of-flight Bruker micrOTOF-Q spectrometer with methanol or dichloromethane (DCM) used as a solvent and scanning the spectra in the m/z range 100–3000 in positive and negative ionization modes. Nitrogen was used as a drying gas at 220 °C and at a flow rate of 4 L min−1. The nebulizer pressure was set at 1.0 bar. The capillary voltage was set at −4.0 kV. Sample solutions were infused into the ESI source by using an LC Agilent 1200 in FIA mode (Flow Injection Analysis, 2–3 μL at a flow rate of the solvent 0.1 mL min−1).
General procedure for the preparation of 11–15
To a stirred solution of alkoxyamine (6–10, 0.1 mmol) and pyridine (0.12 mmol) in dry dichloromethane (DCM) (0.25 mL) was added a solution of 2-bromoacetyl bromide (0.13 mmol) in dichloromethane (0.30 mL). The thick caseous solid was immediately obtained, but it disappeared while the mixture was stirred under an argon atmosphere overnight at room temperature. To the resulting clear solution were added DCM (5 mL) and 0.1 M aqueous HCl (2 mL). The organic phase was separated and filtered through a short plug of silica. The filtrate was concentrated in vacuo to yield crude alkoxyamines 11 (68%), 12 (76%), 13 (96%), 14 (92%), and 15 (68%), which were used further without purification.
General procedure for the preparation of 1–5
To an alkoxyamine (11–15) were added finely powdered potassium carbonate (10 mg, 0.070 mmol), trityl 17 (50.0 mg, 0.050 mmol), dry acetone (0.300 mL) and anhydrous dimethylformamide (0.15 mL). The heterogeneous mixture was stirred at 40 °C for 24 h under an argon atmosphere, and then diluted with DCM (10 mL) and water (3 mL). The deep-green organic phase was separated, filtered through a short cotton plug and concentrated in vacuo to yield a black film. Column chromatography on a silica gel (DCM, then DCM/methanol with a gradual increase in the solvent ratio from 1:0.05 to 1:0.2 v/v) yielded TA 1–5, respectively.
Bis[8-(methoxycarbonyl)-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl]((8-(2-(4-(1-phenylethyloxyl)-3,3,5,5-tetramethyl-4-azacyclohexyloxyl)-2-oxo-ethyloxyl)carbonyl)-2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl)methyl radical 1.
0.052 g (78%), greenish-black powder, m.p. > 125 °C (decomposition). HRMS (ESI): calcd for C61H70NO9S12 [M−] 1344.1699, found 1344.167; calcd C61H71NO9S12 for [M + H+] 1345.1777, found 1345.169. IR (KBr): ν = 2953 (m), 2922 (m), 1740 (m), 1707 (vs), 1491 (m), 1452 (m), 1433 (m), 1365 (m), 1306 (m), 1274 (m), 1234 (vs), 1169 (m), 1147 (m), 1134 (s), 1111 (m), 1045 (m), 700 (m). ESR for 0.5 mM deoxygenated solution in DCM: multiplet, aH = 9.4 μT, linewidth (Gauss) 8.7 μT, g = 2.00280.
Bis[8-(methoxycarbonyl)-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl]((8-(2-(4-(1-methyl-1-tert-butoxycarbonyl-ethyloxyl)-3,3,5,5-tetramethyl-4-azacyclohexyloxyl)-2-oxo-ethyloxyl)carbonyl)-2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl)methyl radical 2.
0.063 g (91%), black powder, m.p. > 120 °C (decomposition). HRMS (ESI): calcd for C61H76NO11S12 [M−] 1382.2067, found 1382.210; calcd for C61H77NO11S12 [M + H+] 1383.2145, found 1383.217. IR (KBr): ν = 2971 (m), 2953 (m), 2924 (m), 1709 (vs), 1489 (m), 1454 (m), 1435 (m), 1365 (m), 1275 (m), 1234 (vs), 1167 (m), 1134 (s), 1113 (m). ESR for 0.5 mM deoxygenated solution in DCM: multiplet, aH = 9.4 μT, linewidth (Gauss) 8.5 μT, g = 2.00280.
Bis[8-(methoxycarbonyl)-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl]((8-(2-(3-(1-phenylethyloxyl)-4-diethoxyphosphoryl-2,2,5,5-tetramethyl-3-azahexyloxyl)-2-oxo-ethyloxyl)carbonyl)-2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl)methyl radical 3.
0.065 g (88%), greenish-black powder, m.p. > 120 °C (decomposition). HRMS (ESI): calcd for C65H81NO12PS12 [M−] 1482.2145, found 1482.251; calcd for C65H82NO12PS12 [M + H+] 1483.2223, found 1483.212; calcd for C65H81NNaO12PS12 [M + Na+] 1505.2043, found 1505.201. IR (KBr): ν = 2968 (m), 2955 (m), 2922 (m), 1709 (s), 1489 (m), 1452 (m), 1435 (m), 1365 (m), 1305 (m), 1272 (m), 1236 (vs), 1169 (m), 1149 (m), 1134 (m), 1111 (m), 1057 (m), 1026 (m), 953 (m). ESR for 0.5 mM deoxygenated solution in DCM: multiplet, aH = 9.4 μT, linewidth (Gauss) 8.7 μT, g = 2.00280.
Bis[8-(methoxycarbonyl)-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl]((8-((4-(4-diethoxyphosphoryl-4-tert-butyl-1,5,5-trimethyl-3-aza-2-oxahexyl)phenylamino)-2-oxo-ethyloxyl)carbonyl)-2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl)methyl radical 4.
0.054 g (73%), deep-green powder, m.p. > 130 °C (decomposition). HRMS (ESI): calcd for C65H83N2O11PS12 [M + H+] 1482.2383, found 1482.229; calcd for C65H82N2NaO11PS12 [M + Na+] 1504.2202, found 1504.218. IR (KBr): ν = 2956 (m), 2920 (m), 1709 (s), 1529 (m), 1487 (m), 1452 (m), 1435 (m), 1365 (m), 1271 (m), 1234 (vs), 1167 (m), 1134 (m), 1111 (m), 1053 (m), 1028 (m), 955 (m). ESR for 0.5 mM deoxygenated solution in DCM: multiplet, aH = 10.6 μT, linewidth (Gauss) 7.3 μT, g = 2.00281.
Bis[8-(methoxycarbonyl)-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl]((8-(2-(3-(1-(2-pyridyl)ethyloxyl)-4-diethoxyphosphoryl-2,2,5,5-tetramethyl-3-azahexyloxyl)-2-oxo-ethyloxyl)carbonyl)-2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl)methyl radical 5.
0.032 g (61%), black powder, m.p. > 100 °C (decomposition). HRMS (ESI): calcd for C64H80N2O12PS12 [M−] 1483.2097, found 1483.178; calcd for C64H81N2O12PS12 [M + H+] 1484.2176, found 1484.210; calcd for C64H80N2NaO12PS12 [M + Na+] 1506.1995, found 1506.189. IR (KBr): ν = 2956 (m), 2924 (m), 1745 (s), 1707 (s), 1487 (m), 1452 (m), 1434 (m), 1385 (m), 1365 (m), 1305 (m), 1273 (m), 1236 (vs), 1169 (m), 1149 (m), 1136 (m), 1111 (m), 1055 (m), 1026 (m), 957 (m), 787 (w). ESR for 0.5 mM of 5 in DCM: multiplet (splitting on 8 protons in groups of 3H, 3H, 2H), αH1 = 9.0 μT, αH2 = 9.5 μT, αH3 = 9.8 μT (respectively), linewidth (Gauss) 8.6 μT, g = 2.00281.
Preparation of tert-butyl 2-[(4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yl)oxy]-2-methylpropanoate 7.
0.70 mL (3.7 mmol) of tert-butyl 2-bromo-2-methylpropanoate was added to a Schlenk flask with 0.76 g (4.4 mmol) of 4-hydroxy-TEMPO, 0.24 g (3.9 mmol) of copper powder, 0.013 g (0.037 mmol) of Cu(OTf)2 and 0.040 g (0.15 mmol) of dTbpy. Benzene, 5 mL, was then added as a solvent, and the solution was degassed by three freeze–pump–thaw cycles. The solution was heated to 75 °C with stirring. After 5 h, all the copper powder was consumed and a beige precipitate was formed. The solution was filtered and concentrated under vacuum. The crude product was purified by column chromatography eluting with hexane/ethyl acetate (95/5 gradually increasing to 5/5). The alkoxyamine was eluted before 4-hydroxy-TEMPO and collected as a colorless fraction. After evaporation of the solvent, the dry residue was recrystallized from hexane to give 0.97 g (83%) of compound 7 as white crystals, mp 83.8 °C followed by decomposition. 1H NMR (400.13 MHz, CDCl3) δ: 4.00–3.90 (m, 1H), 1.82 (dm, 2H, Jd = 12.2 Hz), 1.46 (s, 10H), 1.41 (s, 7H), 1.20 (s, 7H), 1.07 (s, 6H). 13C{1H} NMR (100.61 MHz, CDCl3) δ: 173.9, 80.2, 79.3, 61.6, 58.8, 47.8, 32.3, 26.7, 23.3, 20.1. IR (neat): 3491 m, 3280 w, 3003 m, 2978 s, 2943 m, 2897 m, 1716 s, 1693 s, 1475 m, 1460 m, 1452 m, 1392 w, 1371 s, 1336 w, 1313 m, 1298 m, 1259 m, 1248 m, 1209 m, 1178 m, 1157 s, 1140 vs, 1080 w, 1047 s, 1034 w, 1020 w, 1009 vw, 958 m, 943 w, 930 w, 901 w, 847 m, 818 w, 773 w, 746 w, 590 w, 561 w, 542 w, 500 w, 480 vw cm−1; HRMS: calcd for C17H33NO4 [M] 315.2404; found 315.2408; Anal. calcd for C17H33NO4: C, 64.73; H, 10.54; N, 4.44. Found: C, 64.95; H, 10.56; N, 4.62.
Bis[8-(methoxycarbonyl)-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl](8-carboxyl-2,2,6,6-tetramethylbenzo[1,2-d;4,5-d′]bis[1,3]dithiol-4-yl)methyl radical 1736.
To a stirred solution of tris[(8-carboxy)-2,2,6,6-tetramethylbenzo[1,2-d:4,5-d′]bis[1,3]dithiol-4-yl]methanol 2435 (1.018 g, 1.00 mmol) and triethylamine (0.404 g, 4.00 mmol) in methanol (2.5 mL) was added a solution of freshly distilled methyl iodide (0.850 g, 6.00 mmol) in DCM (2.5 mL). The resulting mixture was stirred under an argon atmosphere at 40 °C for 36 h. 0.5 M aqueous HCl (4 mL) and DCM (5 mL) were added. The organic layer was separated, and the water phase was extracted with DCM (3 × 5 mL). The combined organic extract was washed with brine, filtered through a short silica plug, concentrated in vacuo, and dried in a desiccator to quantitatively yield triarylmethanol 25 (1.052 g). Hydrolysis of 25 into monocarboxylic acid 26 and further conversion of the diamagnetic intermediate 26 into trityl 17 were performed as reported.36 Spectroscopy data for diamagnetic substrates 25 and 26 (1H-NMR, 13C-NMR, FTIR and ESI-HRMS) and trityl 17 (FTIR, ESI-HRMS and ESR) are in agreement with the literature data.36
Results and discussion
Preparation of 1–5
Quantitative methylation of 2435 yielded 25 which was smoothly hydrolysed under controlled conditions|| into mono-acid 26. The latter, in the presence of a strong acid, afforded its cation derivative which was reduced to trityl 17 in the presence of tin chloride (Scheme 1).36 The trityl radical 17 was used for this purpose because the H-couplings of the odd electron are efficiently suppressed via the substitution of the ortho, meta and para hydrogen atoms by sulphur-centred and carbon centred substituents.
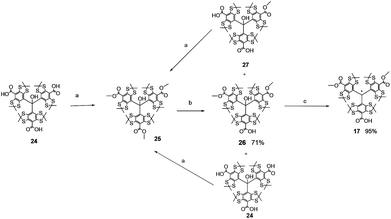 |
| | Scheme 1 Synthesis of trityl 17. (a) Methyl iodide (6 equiv.), triethylamine (4 equiv.), 40 °C, 36 h; (b) LiOH (1.55 equiv.) in THF/water, room temperature, 48 h; (c) CF3SO3H (5 equiv.) in anhydrous DCM and treated with SnCl2 (1 equiv.). | |
Thus, trityl-alkoxyamines 1–5 were prepared in two steps starting from the initial alkoxyamines 6–10, respectively, and using bromo acetyl bromide as an acetylating agent and a precursor of a spacer, and trityl radical 17 as a monofunctional nucleophile (Scheme 2). Alkoxyamine-trityl radicals 1, and 3–5 were prepared from the diastereoisomeric mixtures of 6, and 8–10, respectively. Trityl-alkoxyamines 1–5 were identified by EPR, IR, and HRMS.
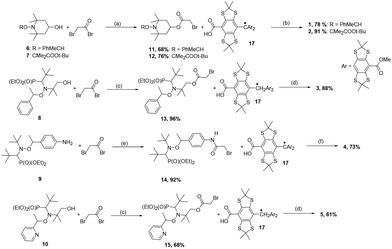 |
| | Scheme 2 Preparation of 1–5. (a) Pyridine (1.2 equiv.) in DCM, room temperature, 24 h; (b) K2CO3 in anhydrous acetone/dimethylformamide, 40 °C, 24 h, under an argon atmosphere; (c) pyridine (1.2 equiv.) in dichloromethane, room temperature, 24 h; (d) K2CO3 in anhydrous acetone/dimethylformamide, 40 °C, 24 h under argon; (e) pyridine (1.2 equiv.) in dichloromethane, room temperature, 24 h; (f) K2CO3 in anhydrous acetone/DMF, 40 °C, 24 h, under an argon atmosphere. | |
Homolysis of the C–ON bond of alkoxyamines
Rate constants kd were measured by EPR because the generated nitroxides are stable under our conditions (Table 1). As discussed in the later section, a spin–spin exchange interaction is observed between the odd electrons localised on the trityl and nitroxyl radical moieties. Nevertheless, the occurrence of this interaction does not impede the determination of kd by EPR. Gomberg's radical and some other trityl radicals are known to react slowly in a reversible manner with oxygen as displayed in Scheme 3. Because of its functionalisation at the para position, the occurrence of the reactivity displayed in Scheme 3 is expected to be very low in the Finland-type trityl radical 17.**
 |
| | Scheme 3 Reactivity of O2 with Gomberg's radical. | |
The homolysis rate constants kd for the C–ON bond in alkoxyamines are easily estimated using robust linear free energy relationships developed for nitroxyl (eqn (4))37 and alkyl (eqn (5))††32 fragments in alkoxyamines. Although long-range polar and steric effects are observed in alkoxyamines, for the sake of simplicity, it was assumed that the trityl group is far enough from the reactive centre so that it does not play any role in the bulkiness and polarity of both the nitroxyl and alkyl fragments. Consequently, it was assumed that alkoxyamines 19–23 are suitable models for estimation of the kd values for 1–5, respectively, using eqn (4) and (5). The effects of the bulkiness and polarity of the substituents attached to the nitroxyl moiety are estimated using the steric constant Es37–39 and the localised electrical Hammett constant σI.40
The polarity effect of the substituent at the para position in the aromatic ring is described by the localised electrical Hammett constants σI,p-X-C6H4.41,42 Thus, kd values were estimated at 120 °C as 2.9 × 10−4 s−1, 1.5 × 10−3 s−1, and 3.3 × 10−3 s−1 for 19 (Ea = 134.8 kJ mol−1, eqn (4)),‡‡21 (Ea = 129.4 kJ mol−1, eqn (4)),§§43 and 22 (Ea = 126.9 kJ mol−1, eqn (5)), respectively.¶¶ Although no relationships are available to estimate Ea for 20, the model of 2, the substituent scale developed a decade ago44 provides a difference in Ea, ΔEa = −12.8 kJ mol−1 from the CHMePh group to the CMe2COOR group affording Ea = 122.8 kJ mol−1 for 20. Although no relationships are available to estimate the Ea of 23, the model of 5, the replacement of styryl alkyl fragments by a pyridyl fragment is known to decrease the Ea by ca. 3 kJ mol−1 in SG1-based alkoxyamines.45,46 Thus, because the nitroxyl fragment is the same for 23 and 21, the Ea for 23 is expected to be ca. 3 kJ mol−1 lower than the Ea for 21, i.e. Ea ≈ 126.5 kJ mol−1 and kd = 3.7 × 10−3 s−1.
| | | log(kd/s−1) = −5.81(±0.09) − 2.91(±0.13)·σI − 0.85(±0.03)·Es | (4) |
| | | log(kd/s−1) = −3.1(±0.1) − 7.8(±0.4)·σI,p-X-C6H4 | (5) |
The Ea and kd values predicted by the simplified models 19–23 are in very good agreement with the experimental values reported in Table 1 for 1–5 highlighting both the validity of our assumptions and the robustness of the linear free energy relationship developed (eqn (4) and (5)), i.e. kd values differ by less than 40% and Ea less than 1.7 kJ mol−1. Moreover, as reported for other bisnitroxides,12,47,48 the spin–exchange interaction (vide supra) has no significant influence on the C–ON bond homolysis.
EPR analysis of 1˙ and 3˙
Before heating, the samples of 2 and 3 displayed only the signal of the pure trityl radical as expected (not shown). Upon heating at 100 °C for 5 hours,|||| in toluene in the presence of oxygen as an alkyl radical scavenger, for both 2 and 3, the EPR signal that was recorded at room temperature was due to three species. Considering the experimental conditions, the presence of starting materials was discarded, and the EPR signals were simulated using three new species, namely biradical 1˙ or 3˙, free trityl radical and free nitroxides TEMPO or SG1.
Although the trityl and nitroxyl radical moieties are separated by a linker of nine and eight single bonds for 1˙ and 3˙, respectively, spin–spin exchange interactions are expected to occur both in 1˙ and 3˙ due to the freedom of motion of the linker. Therefore, the value of the exchange interaction J was selected as one of the simulation parameters, i.e., J is 63 G and 170 G for 1˙ and 3˙, respectively. The accurate simulation of the EPR spectra implies the finding of all stable conformers of 1˙ and 3˙ with the corresponding J values and implements chemical exchange between all these conformers, but it is a rather complicated task. For simplicity, it is assumed that the values of spin–spin exchange interactions are subject to a normal (Gaussian) distribution with standard deviation ΔJ. The temperature dependence of the signal (Fig. S2†) showed an increase in the spin–spin exchange interaction both for 1˙ and 3˙ with increasing temperature due to an increase in the mobility of 1˙ and 3˙. A very good agreement (Fig. 2a) was achieved and the EPR hyperfine coupling constants (J value between 50 and 100 G) of 1˙ are in agreement with those reported in the literature and do not deserve more comments.49 On the other hand, as far as we know, the EPR signal of 3˙ (Fig. 2b) is the first report of a biradical combining trityl and β-phosphorylated nitroxide. The simulations afforded 14% TEMPO, 6% free trityl, and 80% 1˙ for the decomposition of 2, and 8% SG1, 4% trityl, and 88% 3˙ for the decomposition of 3. Even though more than 80% of released biradicals are observed,*** the detection of free nitroxides and free trityl points to the occurrence of side-reactions.†††
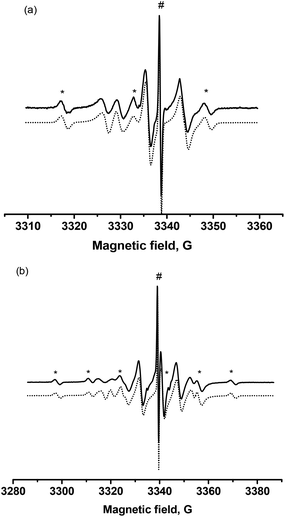 |
| | Fig. 2 EPR spectrum of the solution of (a) 1˙ and (b) 3˙ after 6 hours of heating of a 0.1 mM solution of 2 and 3 at 100 °C in toluene in oxygen saturated solution. (*) Free nitroxide, (#) free trityl radical, a solid line for the experimental spectrum, and a dotted line for the simulated spectrum. Parameters of simulation: (a) J = 63 G, ΔJ = 30 G for 1˙, and TEMPO: aN = 15.4 G, g = 2.00595; (b) J = 170 G, ΔJ = 130 G for 3˙, and SG1: aN = 13.6 G, aP = 44.9 G and g = 2.00595. For a trityl radical: g = 2.00281. | |
Under the experimental conditions (100 °C in toluene with non-reversible scavenging by O2 of the generated alkyl radicals, k1 ≈ 109 M−1 s−1),50 the homolysis of the alkoxyamines (˙T-N-R) is expected to produce only the signal of the biradicals (˙T-N˙) 1˙ and 3˙ (Scheme 5). Accordingly, the presence of free trityl and free nitroxides denotes the occurrence of side-reactions although they occur to a low extent and do not impede the determination of kd. Furthermore, to take into account these side-reactions, a 10-reaction scheme is proposed with alkoxyl and peroxyl radicals as reactive key intermediates (Scheme 5). Importantly, several species are expected to provide the same signal, that is the signal of the free nitroxide is due to N˙, X1N˙, X2N˙, and X3N˙, and the signal of the free trityl radical is due to only T˙ as the starting materials ˙TNR decomposed completely.
Although the para-positions on the aromatic rings of trityl radicals of type 17 are substituted by carboxylic acids or carboxylates, reaction with peroxyl radicals has been reported at the para position (Scheme 4), and is considered one of the main pathways of trityl radical decay.51–53 Consequently, the analogous reaction with trityl radicals carrying ester groups cannot be disregarded and might account for the decay of the trityl signal. It is noteworthy that in the case of the ˙TN˙ biradical and ˙TNR alkoxyamine, the reaction with the alkylperoxyl radical would afford a free nitroxide X1N˙ (Scheme 5). For the sake of simplicity, it is assumed that the peroxyl radicals reacted in the same way and with the same rate constants with ˙TNR and ˙TN˙, that is k4 = k5 ≈ 106 M−1 s−1.54 The dimerization of peroxyl radicals into tetroxides ROOOOR (k2 ≈ 103–106 M−1 s−1),54,55 which spontaneously and instantaneously collapse into alkoxyl radicals (k3 > 104 s−1),54 is well known and may result in unexpected chemistry.56 Therefore, reactions (6) and (7) (Scheme 5) involving alkoxyl radicals are included in the kinetic scheme to account for the generation of free trityl radicals. The reactivity is expected to take place in the linker via an H-abstraction reaction affording an alkyl radical which then decomposes into a free trityl radical and a nitroxide or alkoxyamine. For simplicity, it is assumed that the alkoxyl radicals reacted in the same way and with the same rate constants with ˙TNR and ˙TN˙, that is k6 = k7 > 106 M−1 s−1.54,57 For simplicity, as the reaction with the trityl moiety and the linker takes place far from the C–ON bond, it is assumed that the new alkoxyamines generated X1NR, and X2NR are homolysed at the same rate as the starting material ˙TNR. The same reactions with the peroxyl radicals are disregarded because the H-abstraction rate constants are expected to be low.55 Reactions (8) and (9) (Scheme 5) are expected to account for the decay of alkoxyl and peroxyl radicals in non-reactive by-products. These reactions describe a complicated scheme of decays involving several reactions. For the sake of simplicity, pseudo-first order decays and similar rate constants for the alkoxyl and peroxyl radicals, i.e., k8 = k9 = 1000 s−1 (this rate was modulated to reach the best simulation) are assumed. Under these experimental conditions, nitroxides are known to be stable in the presence of oxygen, and thus we can assume that the nitroxyl moiety of the generated biradical doesn't take part in any other reactions.
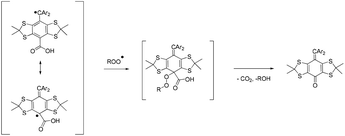 |
| | Scheme 4 Conversion of trityl carboxylic acids to diamagnetic species in oxidative reactions with alkylperoxyl radicals. | |
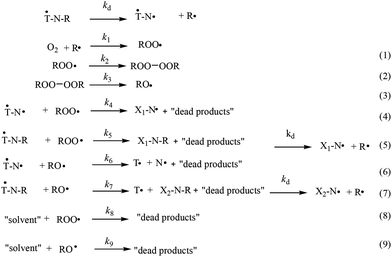 |
| | Scheme 5 The reaction scheme describing decomposition of alkoxyamines 1–5 in the presence of oxygen, and applied to account for the EPR signal reported in Fig. 2. | |
Using a reasonable set of rate constants for Scheme 5, it is possible to reach a good agreement, i.e., 14% TEMPO, 6% trityl, and 80% 1˙ in radical species, with the ratios reported in Fig. 3a. Nevertheless, these side-reactions do not impede the use of 1–5 as initiators/controllers in NMP or the use of their corresponding biradicals (or trityl radical) as probes as long as TA is not overheated for a long period. At the same parameters (Fig. 3b) 10% SG1, 1% trityl, and 89% 1˙ after 5 h are obtained, in good agreement with the EPR analysis (Fig. 2b).
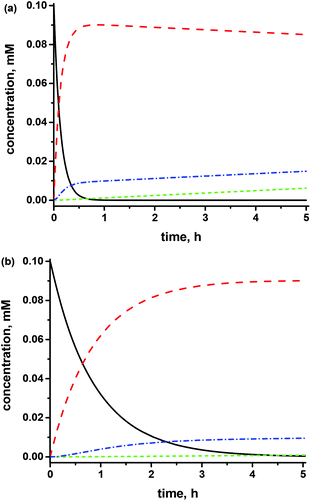 |
| | Fig. 3 Time dependence of the concentrations of 2 (a) and 3 (b), biradical 1˙ (a) and 3˙ (b), trityl and free nitroxides TEMPO (a) and SG1 (b) after the thermal decomposition of alkoxyamines as given in Scheme 5. Kinetics parameters: k1 = 109 M−1 s−1, k2 = 104 M−1 s−1, k3 = 104 s−1, k4 = k5 = 106 M−1 s−1, k6 = k7 = 3 × 106 M−1 s−1, k8 = k9 = 1000 s−1, and [O2]0 = 8 mM. (a) [2]0 = 10−4 M and kd = 2 × 10−3 s−1; (b) [3]0 = 10−4 M, k2 = 107 M−1 s−1, k8 = 1000 s−1, k9 = 106 s−1 and kd = 3 × 10−4 s−1. A dashed red line for trityl-nitroxides, a black solid line for TA, a dotted green line for the free trityl radical, and a dashed/dotted blue line for free nitroxides. | |
Polymerization of styrene initiated with 1
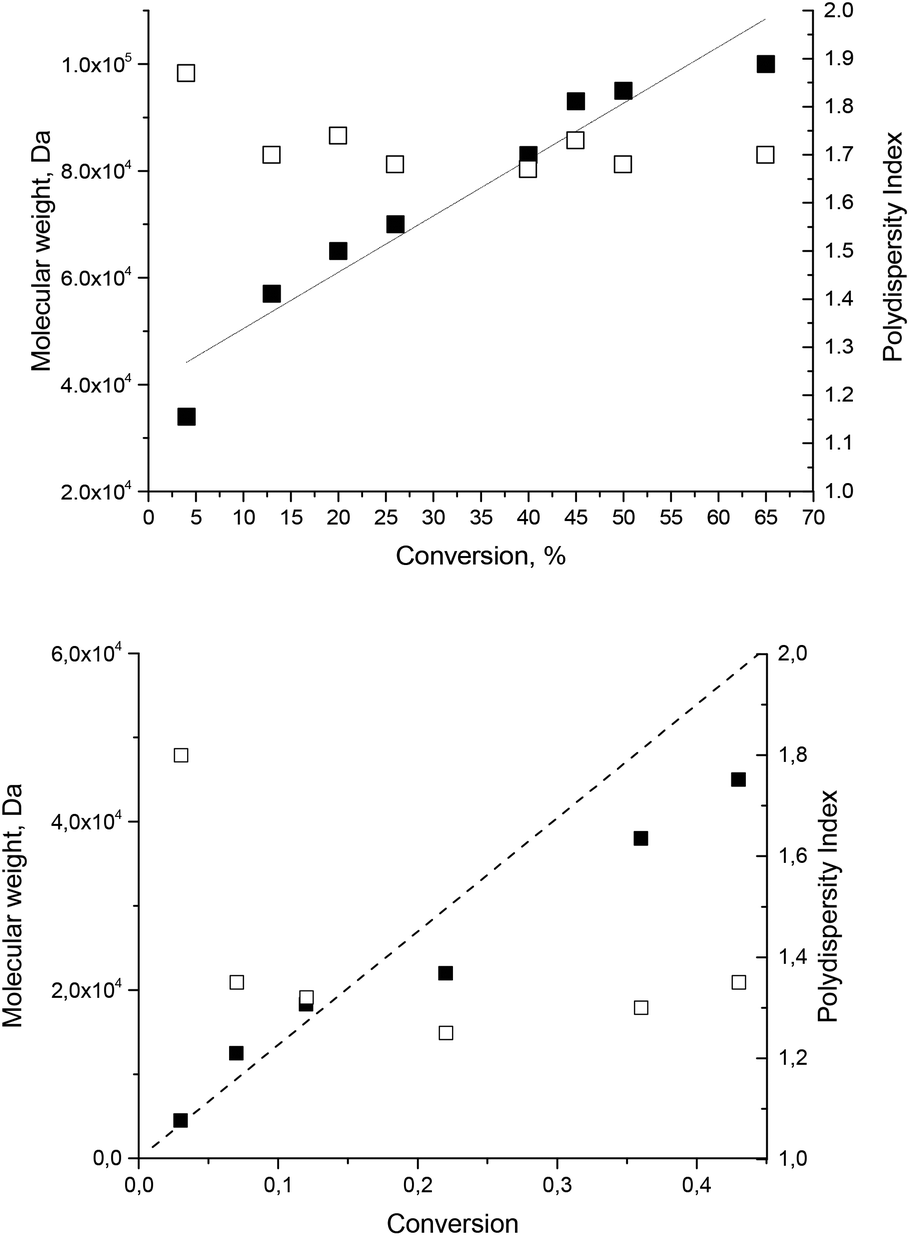
The possible usefulness of 1–5 as initiators in nitroxide mediated polymerization is highlighted by the successful controlled bulk polymerization of styrene with 1 as an initiator/controller (Fig. 4) with styrene/1 ratios of 5000:1 and 1350:1, according to the linear increase in molecular weight with conversion upon polymerisation of styrene. The polydispersity index (PDI, Fig. 4) decreases with the conversion as expected and is close to 1.5, i.e. slightly larger for the high ratio (targeted Mn of 500000 g mol−1), due to the occurrence of self-initiation,58 and lower for the small ratio (targeted Mn of 135000 g mol−1).
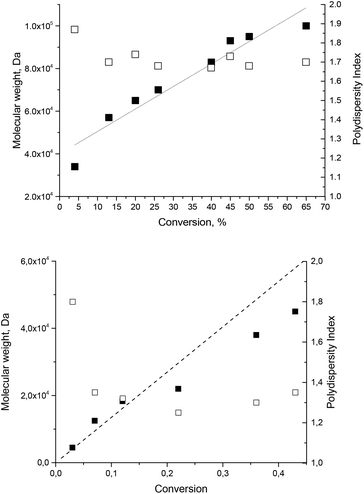 |
| | Fig. 4 Molecular weight (left axis, filled squares) and polydispersity index (right axis, open squares) evolution vs. conversion plot for bulk polymerization of styrene initiated with alkoxyamine 1 at 130 °C (top) and 125 °C (bottom) with styrene/1 ratios 5000:1 (top) and 1350:1 (bottom). A dotted line for the theoretical evolution of Mn. | |
To check the “living” character of the polymerization, end group analysis is performed. The polymer was precipitated from the reaction mixture, washed and dried. After that the polymer was dissolved in toluene (10−4 M solution based on the molecular weight). EPR is recorded to determine the concentration of the spin labels. The signal of the trityl radical (9 × 10−5 M concentration) is observed as expected. The solution saturated with oxygen is heated at 120 °C for 5 hours. Subsequent EPR showed the formation of biradical 1˙ with the double integration of the signal twice the one of the starting materials 1 as expected, meaning the decomposition of polymeric alkoxyamines occurred and proving the presence of a nitroxyl end group in the polymer. The fraction of “living” chains is estimated as 90%. The EPR spectra of the polymer sample before and after heating are displayed in Fig. S11.†
Re-initiation of the radical polymerization of bulk styrene (Fig. 5) using a polystyrene macro-initiator based on 1 confirmed nicely the reported livingness.
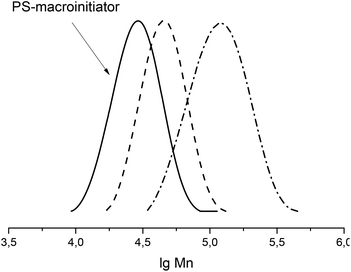 |
| | Fig. 5 Re-initiation test in bulk styrene at 125 °C using a PS-macroinitiator (Mn = 43000 g mol−1) based on 1 with a styrene/initiator ratio of 2000:1. Sampling at 90 minutes and 4 hours. | |
Taking into account the data reported in the literature, as good as livingness and controlled polymerization are expected with TA 2–5.18,28,59,60
Conclusion
A convenient two-step preparation of the first trityl-alkoxyamines is reported. The observed homolysis rate constants, kd, for trityl-alkoxyamines are similar to those for the corresponding unlabelled alkoxyamines. It is clearly shown that the trityl radical moiety has no influence on kd regardless of its position, i.e. in the alkyl fragment for 4 or in the nitroxyl fragment for 1–3 and 5 which can be externally activated on the pyridyl moiety. The preservation of the kinetic properties of the alkoxyamines opens up many opportunities for practical applications in polymer sciences, materials sciences, and in theranostics. For example, we can envision the use of trityl as an end-group to track the distribution of polymer drugs, or it may be used to dope the magnetic properties of some materials.
Acknowledgements
Alkoxyamine 10 was kindly provided by Dr J.-P. Joly. GA, PB, and SRAM thank Aix-Marseille Université, CNRS, ANR (ANR-14-CE16-0023-01) and the A*MIDEX project (ANR-11-IDEX-0001-02) funded by the “Investissements d'Avenir” French Government program, managed by the French National Research Agency (ANR), the Fondation ARC pour la recherche sur le cancer (PJA 20141201886). EGB, MVE, SRAM, DAP, EVT, VMT and SIZ are grateful to the Russian Science Foundation (15-13-20020) for financial support. The authors thank Andrey Kuzhelev for recording and simulation of the EPR spectra of compounds 1–5. NMR, high-resolution ESI/MS, and ESR experiments were carried out in the Chemical Service Center of the Siberian Branch of the RAS.
Notes and references
- M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757 CrossRef.
-
B. Epel, G. Redler, C. Pelizzari, V. M. Tormyshev and H. J. Halpern, Approaching Oxygen-Guided Intensity-Modulated Radiation Therapy, Oxygen Transport to Tissue XXXVII, V.876 of the series Advances in Experimental Medicine and Biolog, Part IV, 2015, p. 185 Search PubMed.
- E. G. Bagryanskaya, E. G. Krumkacheva, M. V. Fedin and S. R. A. Marque, Methods Enzymol., 2015, 563, 365 Search PubMed.
- I. Marin-Montesinos, J. C. Paniagua, A. Peman, M. Vilaseca, F. Luis, S. Van Doorslaer and M. Pons, Phys. Chem. Chem. Phys., 2016, 18, 3151 RSC.
- I. Dhimitruka, A. A. Bobko, C. M. Hadad, J. L. Zweier and V. V. Khramtsov, J. Am. Chem. Soc., 2008, 130, 10780 CrossRef CAS PubMed.
- G. Y. Shevelev, O. A. Krumkacheva, A. A. Lomzov, A. A. Kuzhelev, O. Y. Rogozhnikova, D. V. Trukhin, T. I. Troitskaya, V. M. Tormyshev, M. V. Fedin, D. V. Pyshnyi and E. G. Bagryanskaya, J. Am. Chem. Soc., 2014, 136, 9874 CrossRef CAS PubMed.
- Y. Liu, F. A. Villamena, Y. Song, J. Sun, A. Rockenbauer and J. L. Zweier, J. Org. Chem., 2010, 75, 7796 CrossRef CAS PubMed.
- X. Tan, Y. Song, H. Liu, Q. Zhong, A. Rockenbauer, F. A. Villamena, J. L. Zweier and Y. Liu, Org. Biomol. Chem., 2016, 14, 1694 CAS.
- G. Mathies, M. A. Caporini, V. K. Michaelis and Y. Liu, Angew. Chem., Int. Ed., 2015, 54, 11770 CrossRef CAS PubMed.
- E. Yoshida and K. Takeda, Polym. J., 2001, 33, 590 CrossRef CAS.
- N. L. Hill and R. Braslau, Macromolecules, 2005, 38, 9066 CrossRef CAS.
- A. Kaim, K. Pietrasik and T. Stokłosa, Eur. Polym. J., 2010, 46, 519 CrossRef CAS.
- A. Zagdoun, G. Casano, O. Ouari, G. Lapadula, A. J. Rossini, M. Lelli, M. Baffert, D. Gajan, L. Veyre, W. E. Maas, M. Rosay, R. T. Weber, C. Thieuleux, C. Coperet, A. Lesage, P. Tordo and L. Emsley, J. Am. Chem. Soc., 2012, 134, 2284 CrossRef CAS PubMed.
- M. Tseitlin, J. R. Biller, H. Elajaili, V. V. Khramtsov, I. Dhimitruka, G. R. Eaton and S. S. Eaton, J. Magn. Reson., 2014, 245, 150 CrossRef CAS PubMed.
-
B. Epel, G. Redler, V. M. Tormyshev and H. J. Halpern, Towards Human Oxygen Images with Electron Paramagnetic Resonance Imaging, Oxygen Transport to Tissue XXXVII, V.876 of the series Advances in Experimental Medicine and Biolog, Part VII, 2015, p. 363 Search PubMed.
- O. Lafon, M. Rosay, F. Aussenac, X. Lu, J. Trébosc, O. Cristini, C. Kinowski, N. Touati, H. Vezin and J.-P. Amoureux, Angew. Chem., Int. Ed., 2011, 50, 8367 CrossRef CAS PubMed.
- G. Audran, P. Brémond and S. R. A. Marque, Chem. Commun., 2014, 50(59), 7921 RSC.
-
Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science, ed. D. Gigmes, Series: RSC Polymer Chemistry Series, 2015 Search PubMed.
- C. Yuan, M. Z. Rong, M. Q. Zhang and Z. P. Zhang, Chem. Mater., 2011, 23, 5076 CrossRef CAS.
- Z. P. Zhang, M. Z. Rong, M. Q. Zhang and C. Yuan, Polym. Chem., 2013, 4, 4648 RSC.
- M. Becker, L. D. Cola and A. Studer, Chem. Commun., 2011, 47, 3392 RSC.
- M. Becker, L. De Cola and A. Studer, J. Mater. Chem. C, 2013, 1, 3287 RSC.
- R. K. Roy, A. Meszynska, C. E. Laure, L. Charles, C. Verchin and J.-F. C. O. Lutz, Nat. Commun., 2015, 6, 1 Search PubMed.
- L. Charles, C. Laure, J.-F. Lutz and R. K. Roy, Macromolecules, 2015, 48, 4319 CrossRef CAS.
- G. Audran, P. Brémond, J.-M. Franconi, S. R. A. Marque, P. Massot, P. Mellet, E. Parzy and E. Thiaudiere, Org. Biomol. Chem., 2014, 12, 719 CAS.
-
(a) Y. Liu, F. A. Villamena and J. L. Zweier, Chem. Commun., 2008, 4336 RSC;
(b) Y. Song, Y. Liu, C. Hemann, F. A. Villamena and J. L. Zweier, J. Org. Chem., 2013, 78, 1371 CrossRef CAS PubMed.
- S. Marque, C. Le Mercier, P. Tordo and H. Fischer, Macromolecules, 2000, 33(12), 4403 CrossRef CAS.
- D. Bertin, D. Gigmes, S. R. A. Marque and P. Tordo, Chem. Soc. Rev., 2011, 40, 2189 RSC.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42 CrossRef CAS PubMed.
- M. M. Goerger and B. S. Hudson, J. Org. Chem., 1988, 53, 3148 CrossRef CAS.
- S. Acerbis, D. Bertin, B. Boutevin and D. Gigmes, Helv. Chim. Acta, 2006, 89, 2119 CrossRef CAS.
- G. Audran, P. Brémond, J.-P. Joly, S. R. A. Marque and T. Yamasaki, Org. Biomol. Chem., 2016, 14, 3574 CAS.
- K. Matyjaszewski, B. E. Woodworth, X. Zhang, S. G. Gaynor and Z. Metzner, Macromolecules, 1998, 31, 5955 CrossRef CAS.
- R. Nicolaÿ and K. Matyjaszewski, Macromolecules, 2011, 44, 240 CrossRef.
- O. Y. Rogozhnikova, V. G. Vasiliev, T. I. Troitskaya, D. V. Trukhin, T. V. Mikhalina, H. J. Halpern and V. M. Tormyshev, Eur. J. Org. Chem., 2013, 3347 CrossRef CAS PubMed.
- This procedure is a modified version of D. Trukhin, O. Rogozhnikova, T. Troitskaya, V. Vasiliev, M. Bowman and V. Tormyshev, Synlett, 2016, 893 CAS.
- S. Marque, J. Org. Chem., 2003, 68(20), 7582 CrossRef CAS PubMed.
- T. Fujita, C. Takayama and M. Nakajima, J. Org. Chem., 1973, 38, 1623 CrossRef CAS.
- H. Fischer, A. Kramer, S. R. A. Marque and P. Nesvadba, Macromolecules, 2005, 38, 9974 CrossRef CAS.
- M. Charton, Prog. Phys. Org. Chem., 1981, 13, 119 CrossRef CAS.
- M. Charton, Adv. Mol. Struct. Res., 1999, 5, 25 CrossRef CAS.
- D. Bertin, D. Gigmes, S. R. A. Marque, S. Milardo, J. Peri and P. Tordo, Collect. Czech. Chem. Commun., 2004, 69, 2223 CrossRef CAS.
- G. Audran, P. Brémond, S. R. A. Marque and T. Yamasaki, J. Org. Chem., 2016, 81, 1981 CrossRef CAS PubMed.
- E. Beaudoin, D. Bertin, D. Gigmes, S. R. A. Marque, D. Siri and P. Tordo, Eur. J. Org. Chem., 2006, 1755 CrossRef CAS.
- P. Brémond and S. R. A. Marque, Chem. Commun., 2011, 47, 4291 RSC.
- P. Brémond, A. Koïta, S. R. A. Marque, V. Pesce, V. Roubaud and D. Siri, Org. Lett., 2012, 14, 358 CrossRef PubMed.
- S. R. A. Marque and D. Siri, Macromolecules, 2009, 42(4), 1404 CrossRef CAS.
- C. Chachaty, W. Huang, L. Marx, B. Charleux and A. Rassat, Polymer, 2003, 44, 397 CrossRef CAS.
- Y. Liu, F. A. Villamena, A. Rockenbauer, Y. Song and J. L. Zweier, J. Am. Chem. Soc., 2013, 135, 2350 CrossRef CAS PubMed.
-
Alkoxyl, carbonyloxyl, phenoxyl, and related radicals, Radical reaction rates in liquids, ed. H. Fischer, Landolt-Börnstein Springer Verlag, Berlin Heidelberg, 1997, group II, vol. 18, suppl. vol. II/13, subvolume A Search PubMed.
- C. Decroos, Y. Li, G. Bertho, Y. Frapart, D. Mansuy and J.-L. Boucher, Chem. Commun., 2009, 1416 RSC.
- C. Decroos, Y. Li, A. Soltani, Y. Frapart, D. Mansuy and J.-L. Boucher, Arch. Biochem. Biophys., 2010, 502, 74 CrossRef CAS PubMed.
- C. Decroos, T. Prangé, D. Mansuy, J.-L. Boucher and Y. Li, Chem. Commun., 2011, 47, 4805 RSC.
- K. U. Ingold, Acc. Chem. Res., 1969, 2, 1 CrossRef CAS.
-
J. Howard, Alkoxyl, carbonyloxyl, phenoxyl, and related radicals, Radical reaction rates in liquids, ed. H. Fischer, Landolt-Börnstein Springer Verlag, Berlin Heidelberg, 1997, group II, vol. 18, suppl. vol. II/13, subvolume D2 Search PubMed.
- P. Brémond, K. Kabytaev and S. R. A. Marque, Tetrahedron Lett., 2012, 53, 4543 CrossRef.
-
J. Lusztyk, Alkoxyl, carbonyloxyl, phenoxyl, and related radicals, Radical reaction rates in liquids, ed. H. Fischer, Landolt-Börnstein Springer Verlag, Berlin Heidelberg, 1997, group II, vol.18 Search PubMed, suppl. vol. II/13, subvolume A.
- D. Gigmes, D. Bertin, C. Lefay and Y. Guillaneuf, Macromol. Theory Simul., 2009, 18, 402 CrossRef CAS.
-
D. Gigmes and S. R. A. Marque, Encyclopedia of Radicals in Chemistry, Biology, and Materials, ed. C. Chatgilialoglu and A. Studer, Wiley, Chichester, U.K., 2012, p. 1813 Search PubMed.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: EPR simulation data in Table S1; Fig. S1 for decays of 1–5; EPR signal temperature-dependence in Fig. S2 and S3; characterization data in Fig. S4–10. See DOI: 10.1039/c6py01303a |
| ‡ In ref. 26, the Finland trityl radical 16 is used as a model. Its reactivity with small radicals such as methyl radical Me˙ has been reported as extremely low. Consequently, it was assumed unreactive toward the styryl radical due to its size. |
| § Other positions were unreactive due to the substitution by sulphur atoms. |
| ¶ For the characterization of 1–5 under air at 25 °C, recording parameters are: 9.3 GHz; sweep width: 0.2–0.5 mT; microwave power, 1–6 μW; modulation frequency: 100 kHz; modulation amplitude: 5 μT; time constant: 81.92–163.84 ms; sweep time: 41.94–83.89 s; number of points: 512; number of scans: 1. |
| || Hydrolysis of 25 is not selective and the dicarboxylic acid 27 and the tricarboxylic acid 24 are generated. Both are re-methylated to yield 25, and are again smoothly hydrolysed. After three cycles, triarylmethanol 26 was obtained in 71% yield. |
| ** In this case, the main coupling reaction is expected to occur at the carbonyl centre, which is severely sterically hindered, and consequently, shows negligible reactivity. |
| †† This relationship was specifically developed for the SG1 moiety carrying a para-substituted aryl fragment. |
| ‡‡ Steric constant Es is a linear combination of the steric demand r(i) of each group attached to the nitroxyl moiety (see ref. 37 and 38). Some years ago, constants for cyclic nitroxyl fragments have been developed (ref. 39), and it is assumed that r(Me2CH2CHOAcCH2CMe2) = r(R2CCH2CHOHCH2CR2) = 0.22, r(Me) = 0.00, and Es = −2.70. The electrical effect is accounted by the sum of the electrical effect of each group attached to the nitroxyl moiety (ref. 37), σI,Me = −0.01, σI,CH2CHOAcCH2 = 2 × (σI,CH2CHOAcMe/2) = σI,CH2CHOAcMe ≈ σI,CH2CHOAcMe = 0.05 (see ref. 39 and 40) and σI = 0.01. |
| §§ Recently, we showed that the leveled steric effect (see ref. 43) applies to this family of molecules. Thus, for instance, the same steric demand is assumed for the CH2OAc group as for the COOMe group, i.e. Es = −5.0 as given in ref. 37. The electrical effect is described by the sum of the electrical effect of each group attached to the nitroxyl moiety (ref. 37 and 40), σI,H = 0.00, σI,Me = σI,t-Bu = −0.01, σI,P(O)(OEt)2 = 0.32, σI,CH2OAc = 0.15, and σI = 0.43. |
| ¶¶ σI,p-XC6H4 is a linear combination of the electrical localized Hammett constant σI,X and of the delocalized Hammett constant σ0R,X (see ref. 40 and 41). σI,NHAc = 0.28, σ0R,NHAc = −0.25 and σI,p-XC6H4 = −0.08. |
| |||| After this time of heating, the concentrations in 2 and 3 are estimated, from the data reported in Table 1, as 2 × 10−20 M and 5 × 10−7 M, respectively. |
| *** The less than 20% loss is mainly due to the errors in the preparation of the samples (small amounts) and in the reference (TEMPO) used to estimate the radical concentration. Moreover, side-reactions are also possible as mentioned in the article. |
| ††† Unfortunately, we did not succeed in the experimental measurements of the generation kinetics for individual species (trityl, nitroxide, biradical) due to the complexity of the EPR spectrum, e.g. broadening of the spectral lines in the presence of oxygen dissolved in toluene and their consecutive overlapping. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Olga Yu.
Rogozhnikova
bc,
Victor M.
Tormyshev
b,
Olga Yu.
Rogozhnikova
bc,
Victor M.
Tormyshev