DOI:
10.1039/C6PY01296E
(Paper)
Polym. Chem., 2016,
7, 6450-6456
Synthesis of α,ω-heterotelechelic PVP for bioconjugation, via a one-pot orthogonal end-group modification procedure†
Received
25th July 2016
, Accepted 27th August 2016
First published on 29th August 2016
Abstract
A simple one pot orthogonal procedure for synthesizing α-aldehyde, ω-thiol heterotelechelic poly(N-vinylpyrrolidone) (PVP) is introduced. Firstly we designed a xanthate chain transfer agent with an acetal protected aldehyde functionality in the leaving group, then we synthesized α-acetal ω-xanthate end-functional PVP, via a RAFT-mediated polymerization. The end-groups were modified via a facile, modular protocol, by first aminolysing the ω-xanthate end-groups to form thiols, using an excess of a primary amine, and subsequently acidifying the reaction medium to simultaneously convert excess primary amine to its (non-interfering) quaternary ammonium salt form, as well as effect the acid-catalysed deprotection of the acetal into an aldehyde functionality, to access the α-aldehyde, ω-thiol-PVP. Finally, we demonstrated the utility of these end-groups by performing conjugations with model small molecules. This study establishes a facile procedure for accessing different and bio-relevant end-functional groups with a biocompatible vinyl polymer, suitable for making drug delivery vehicles.
Introduction
Bioconjugates of synthetic vinyl polymers have many biomedical applications, including as drug delivery vehicles.1–4 Often the conjugates are prepared by coupling the reactive end-groups of telechelic polymers with complementary functional groups in bioactive compounds or bio(macro)molecules.1,2 In this regard, heterotelechelic polymers are particularly interesting because the different end-groups can often be addressed orthogonally.5–7 Synthetic strategies for preparing telechelic polymers have been enhanced through advances made in reversible deactivation radical polymerization (RDRP), which enable routine access to precise polymers, with defined end-groups usable in bioconjugate formation.3,8–13 One of the main RDRP processes is the reversible addition fragmentation chain transfer (RAFT) technique, mediated by thiocarbonyl thio compounds, commonly referred to as RAFT agents.14 RAFT agents generally consist of a leaving (R) group, and a thiocarbonyl thio functional (Z) group, both retained in the final polymer as α and ω end-groups, respectively. Functional groups incorporated into the R or Z groups, are retained as α and ω end-functionalities, respectively. Z groups are however unstable to nucleophiles, are often coloured and degrade into odorous compounds. Therefore, the in situ synthesis of end-functional polymers is traditionally conducted with R-group functional RAFT agents, for example, bearing activated esters,15,16 activated disulfides,17 biotin,6 or azide functionalities,18 yielding α-end telechelic functional polymers, for use in bioconjugate formation. The ω-end thiocarbonyl thio moieties are readily converted into thiols by treating the polymer with nucleophiles, mainly primary amines.17,19 Thiol end-groups have numerous functionalization possibilities, including click-type Michael addition reactions with acrylate and maleimide functionalities and disulfide formation, e.g. with cysteine residues of peptides or proteins.20,21
Whilst the synthesis of mono-telechelic functional polymers is a mature paradigm, there are still only a few reports on the synthesis of α,ω-heterotelechelic polymers. One of these approaches entails using RAFT agents with functionalised R and Z groups to directly synthesise α,ω-heterotelechelic functional polymers.22 The nucleophilic and thermal instability of thiocarbonyl thio moieties, however, makes the ω-end-functionality inherently unstable, encumbering this approach. Other methods utilise a combination of functionalised R-groups, for α-end-functionality; and either aminolysis of the Z group to form a thiol,5,23 or the use of radical–radical coupling, with concomitant Z group removal,6,7 to install ω-functionality. Heterotelechelic α-amine, ω-thiol poly(N-vinylpyrrolidone) (PVP), has been obtained by simply treating PVP, synthesized using O-ethyl S-(phthalimidomethyl) xanthate RAFT agent, with hydrazine, to effect both the hydrazinolysis of the phthalamidomethyl R group, to an amine, and the aminolysis of the ω-xanthate chain end to a thiol at the same time.24 There is a lot of scope, however, to introduce other combinations of bio-relevant end-functionalities using facile, preferably one pot, and orthogonal procedures, in order to further enhance this technology. Orthogonally addressable α,ω-heterotelechelic functionalities such as aldehydes and thiols interest us, because they have many applications in bioconjugate formation.4,21,25 Aldehydes and thiols, react readily with amine and thiol functionalities, respectively, which are found in lysine and cysteine residues, respectively, of peptides and proteins, under relatively undemanding conditions. PVP, which can be synthesized via the RAFT-mediated polymerization of N-vinylpyrrolidone (NVP),26 with xanthate RAFT agents, is an established biocompatible polymer,27,28 and an attractive choice in the design of α,ω-heterotelechelic polymers for bioconjugate formation.25,29
Herein we introduce a facile, one-pot and orthogonal method converting RAFT-made α-acetal, ω-xanthate PVP, into α-aldehyde, ω-thiol heterotelechelic PVP. Firstly, we polymerized NVP, via RAFT-mediated polymerization using a triazole based xanthate RAFT agent incorporating an acetal-protected aldehyde in the R group. Along the way we optimised the structure of the triazole based R group, to obtain good control over the RAFT polymerization of NVP; as well as the structure of the triazole linked acetal, in order to enhance its acid catalysed deprotection into an aldehyde. Then we developed a facile, one-pot two-step protocol for aminolysing the Z group, and then deprotecting the acetal into aldehyde functionality via acid catalysis. Finally we illustrated the use of these functional end-groups in bioconjugate formation, and stimuli (pH) responsive release, through the use of model reactions with small organic molecules.
Results and discussion
In previous work, in our group, we used RAFT agents with a (1-phenyl-1H-1,2,3-triazol-4-yl)methyl (triazole based) leaving group to, successfully, control the polymerizations of vinyl acetate, NVP, n-butyl acrylate and styrene.30 We used this approach to introduce a functional group, i.e. an acetal protected aldehyde, into the R group of RAFT agent 1 (Fig. 1), synthesised via the copper-catalyzed azide alkyne coupling reaction (Scheme S1†).18,30 NVP was subsequently polymerized using RAFT agent 1, targeting two degrees of polymerisation (DP), 100 and 200, respectively (entries 1 and 2, Table 1). The obtained polymers, however, exhibited high Đ values, of 1.91 and 1.73, respectively, much higher than expected for a well-behaved RDRP process. We attributed this to a poor leaving group ability of RAFT agent 1's R group, compared to the PVP macroradical, causing hybrid behaviour.30 In our earlier successful RAFT polymerizations, with triazole-based RAFT agents, the triazole was directly linked (conjugated) to a phenyl ring, on the N-1 of the triazole,30 which probably complemented the triazole's pseudo-aromatic resonance, thereby improving its leaving group ability. RAFT agent 1, however, contains a methylene group as a substituent on the N-1 of the triazole, which cannot offer the same resonance stabilisation as the phenyl moiety in our earlier work. We therefore introduced a methyl substituent into the R group (i.e. RAFT agent 2, Scheme S2†), so as to form a more stable secondary radical during the polymerization reaction, with a better leaving group ability. Pleasingly, the polymerisation of NVP using RAFT agent 2 provided much better control as evidenced by the low Đ values (entries 3–5, Table 1), affirming our earlier hypothesis that the poor control exhibited by RAFT agent 1 was due to the poor quality of its leaving group. Subsequent 1H NMR spectroscopic analysis confirmed the retention of α-acetal (methylene ‘g’ at 4.47 ppm and methine ‘h’ at 5.18 ppm) and ω-xanthate (methylene ‘f’ at 4.60 ppm) end-groups in the polymers (Fig. 2). The ratio of α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ω-end groups, calculated using the signals for protons ‘f’:‘g’, or ‘f’:‘h’ was above 0.9, indicating a high degree of telechelic functionalization. Additionally, Mn values calculated from 1H NMR spectroscopy (Mn, NMR) were in good agreement with theoretical Mn values, (entries 3–5, Table 1) indicating very good end group retention.
:ω-end groups, calculated using the signals for protons ‘f’:‘g’, or ‘f’:‘h’ was above 0.9, indicating a high degree of telechelic functionalization. Additionally, Mn values calculated from 1H NMR spectroscopy (Mn, NMR) were in good agreement with theoretical Mn values, (entries 3–5, Table 1) indicating very good end group retention.
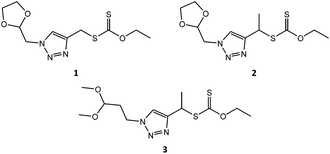 |
| | Fig. 1 RAFT agent structures. | |
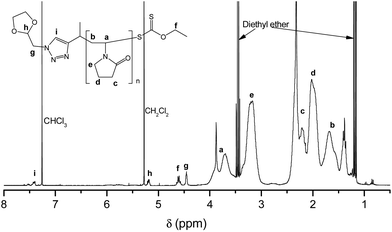 |
| | Fig. 2 Representative 1H NMR spectrum of PVP, in CDCl3, synthesised using RAFT agent 2. | |
Table 1 RAFT-mediated polymerization of NVP
| Entry |
RAFT agent |
[M]:[RAFT]:[AIBN] |
α
(%) |
Time (h) |
M
n, theo (g mol−1) |
M
n, SECb (g mol−1) |
M
n, NMRc (g mol−1) |
Đ
|
|
Conversions, determined by 1H NMR spectroscopy.34
M
n, SEC determined by SEC in DMAc relative to PMMA standards.
M
n, NMR determined by 1H NMR spectroscopy.35
It was not possible to determine end groups because chain ends were not retained quantitatively.
|
| 1 |
1
|
100:1:0.20 |
60 |
5 |
7000 |
6400 |
n/ad |
1.91 |
| 2 |
1
|
200:1:0.20 |
62 |
5 |
14000 |
12700 |
n/ad |
1.73 |
| 3 |
2
|
200:1:0.20 |
26 |
15 |
6100 |
4100 |
6000 |
1.20 |
| 4 |
2
|
200:1:0.25 |
24 |
15 |
5700 |
3500 |
5500 |
1.17 |
| 5 |
2
|
200:1:0.33 |
25 |
15 |
6000 |
3900 |
5900 |
1.19 |
| 6 |
3
|
50:1:0.25 |
60 |
20 |
3700 |
2700 |
3900 |
1.24 |
| 7 |
3
|
100:1:0.25 |
60 |
20 |
7000 |
5300 |
7300 |
1.16 |
As part of the RAFT agent structure optimisation, we also tested the acid catalysed deprotection of the cyclic acetal R group functionality, into aldehyde, since acetals are known to readily convert into aldehydes with acid catalysis.31 Surprisingly, the cyclic acetal of model compound 4 remained intact upon treatment with 1 M HCl in acetone at room temperature for 24 h (Scheme 1), as evidenced by TLC analysis and 1H NMR spectroscopic analysis of the crude reaction mixture. Repetition of the reaction under refluxing conditions resulted in structural degradation. We tried a number of deprotection conditions, e.g. using p-tosyl sulfonic acid or 10% iodine in acetone, without any success.32,33 It is known that acetals close to amino groups are more resistant to acid catalysed deprotection,36 therefore we reasoned that the failed deprotection of 4 was probably caused by the close proximity of the acetal to the triazole-ring. As shown in Scheme 2, it is likely that the triazole ring's N-3 atom, being the most basic,37 will be rapidly protonated in acidic media (4B). For hydrolysis to occur, one of the acetal oxygen atoms needs to be protonated to allow oxocarbenium ion formation (4D), but this would result in a doubly charged species (4C) incurring a high thermodynamic penalty. Thus the reaction probably goes no further than (4B), and forcing the conditions simply promotes acid catalysed breakdown of the starting material. The added fact that cyclic acetals are more stable than their acyclic counterparts probably enhances the difficulty in achieving hydrolysis on this compound.38 Consequently, we made two changes, in order to increase the acid lability of the triazole linked acetal, firstly we increased the distance between the triazole and the acetal, by including an extra methylene group and secondly we switched to using a linear acetal.
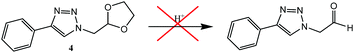 |
| | Scheme 1 Attempted acid catalysed deprotection of model acetal compound 4; 1 M HCl in acetone. | |
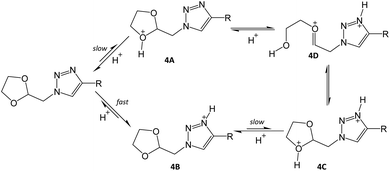 |
| | Scheme 2 Proposed mechanism of unsuccessful acetal deprotection. | |
Model compound 5 was then synthesized (Scheme S4†), and when treated with 1 M HCl in acetone, the acetal was successfully deprotected into an aldehyde in ∼4 h (Fig. S1†). Subsequently we synthesized RAFT agent 3 (Scheme S5†), bearing a linear acetal R group functionality, with a longer spacer between the triazole and the acetal. The RAFT-mediated polymerization of NVP, with RAFT agent 3, yielded well-defined polymers, with low Đ (entries 6 and 7, Table 1). We obtained monomodal SEC traces (Fig. 3a and b), and Mn increased linearly as a function of conversion (Fig. 3c) pointing to the living character of the polymerization. Also Đ values were consistently low, Đ < 1.4 (Fig. 3c). The discrepancy between the experimental and theoretical Mn values, in Fig. 3c, was attributed to the use of PMMA standards in the SEC calibration. 1H NMR spectroscopic analysis also confirmed the retention of α-acetal (methylene ‘g’ at 4.37 ppm, and methine ‘h’ at 4.47 ppm) and ω-xanthate (methylene ‘f’ at 4.60 ppm) end-groups in the polymers (Fig. 4). The ratio of α:ω-end groups, calculated using the signals for protons ‘f’:‘g’, (or ‘f’:‘h’) was also above 0.9, and, Mn, NMR values agreed reasonably well with theoretical Mn values, (entries 6 and 7, Table 1) indicating a very good end group retention.
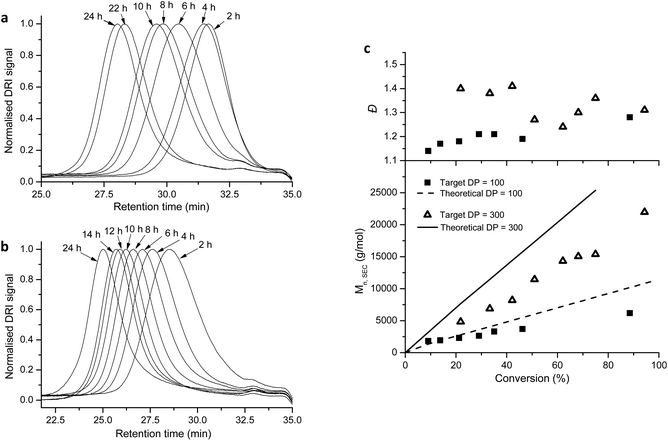 |
| | Fig. 3 SEC traces for the RAFT-mediated polymerization of NVP, with RAFT agent 3, for target DP = 100 (a), and 300 (b), and the corresponding plots of Mn and Đ, as a function of conversion (c). | |
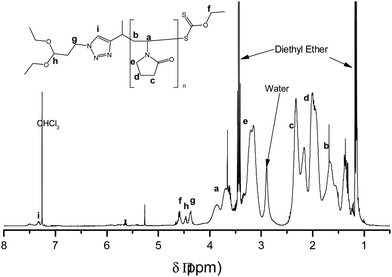 |
| | Fig. 4 Representative 1H NMR spectrum of PVP, in CDCl3, synthesised using RAFT agent 3. | |
One-pot sequential deprotection
Our envisioned one-pot, deprotection strategy entails aminolysis of the ω-xanthate end-group into a thiol, first, and then (after lowering the pH of the reaction mixture) effecting the acid catalysed deprotection of the α-acetal moiety into an aldehyde functionality. The acidic conditions utilised in acetal deprotection, also serve to quaternize excess primary amine, used for aminolysis, forming a salt that precipitates out of the reaction mixture in organic solvent, helping to prevent a deleterious side reaction, i.e. Schiff base formation between excess amine and deprotected aldehyde. We tested the proposed deprotection procedure on RAFT made PVP (entry 6, Table 1), by treating the acetone solution of the polymer with n-hexylamine, and then lowering the pH with HCl in dioxane, and stirring for 4 h (Scheme 3). Afterwards the polymer was purified by dialysis. 1H NMR spectroscopy analysis of the product showed the disappearances of the ω-xanthate's signal f, at 4.60 ppm, due to the aminolysis, and the acetal's signal h, at 4.48 ppm, due to the acid catalysed acetal deprotection, which was complemented by the appearance of the α-aldehyde end-group's proton signal (i) at 9.79 ppm (Fig. 5). The signal of the methylene protons ‘g’ also shifted slightly downfield from 4.4 ppm to 4.6 ppm, presumably due to electron withdrawing effects of the aldehyde. Further analysis was conducted using SEC. Fig. 6 shows the SEC traces before and after end-group modification, recorded with RI and UV detectors. The UV detector was set at 280 nm to allow for selective detection of the ω-xanthate chain end moiety. The elution curve, with RI detection, showed a bimodal peak for the end-group modified sample, which we attribute to disulfide formation between thiol end-groups of two polymer chains. Disulfide formation occurred because we did not take any precautions to prevent oxidising conditions, which also affirmed the successful formation of ω-thiol end-groups. The UV recorded SEC trace, of the modified polymer shows no peak in the molecular weight region of the polymer, unlike with the sample before modification, confirming the removal of the xanthate end-groups. Put together, the 1H NMR spectroscopy and SEC analysis evidence a successful one-pot, orthogonal end-group modification and synthesis of α-aldehyde, ω-thiol-PVP.
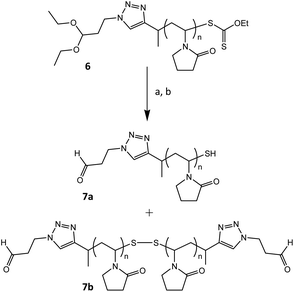 |
| | Scheme 3 One pot deprotection of RAFT made PVP (entry 6, Table 1); (a) n-hexylamine, acetone, r.t.; (b) 4 M HCl in dioxane, acetone, r.t. | |
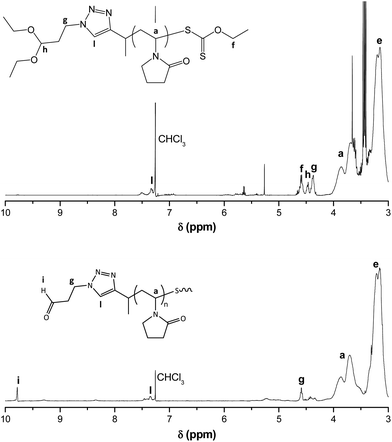 |
| | Fig. 5 Comparison of 1H NMR spectra of RAFT made PVP, (entry 6, Table 1) (top) and end-modified, aldehyde functional PVP (bottom), both in CDCl3. | |
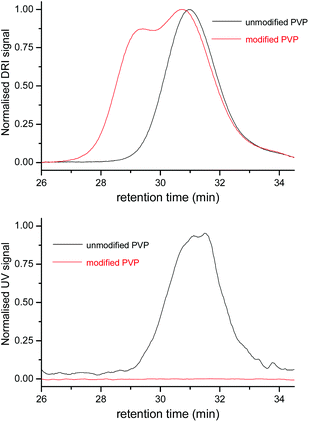 |
| | Fig. 6 SEC traces comparing the DRI (top) and UV (bottom) signals of unmodified PVP (6, Scheme 3) and end-modified PVP (7a/b, Scheme 3). | |
Model conjugation reactions and stimuli responsive release
The overall goal of this research program is to synthesize polymer drug conjugates (using hydrophobic drugs) with the ability to undergo self-assembly into micellar-type structures, decorated with targeting ligands on the outside, for cell specific recognition and uptake, where the drug will then be released in response to a specific stimulus, i.e. pH. With this in mind we tested the coupling efficiency of the end-groups with model small molecule compounds to mimic conjugate formation. We used glycine-DL-serine (Gly-DL-Ser) as a model compound to mimic the targeting ligand and conjugated it to the polymer's α-chain end, by reductive amination (Scheme 4). Analysis by 1H NMR spectroscopy showed the disappearance of the aldehyde signal (Fig. 7), presumably due to the formation of the PVP–peptide conjugate (9) via reductive amination.
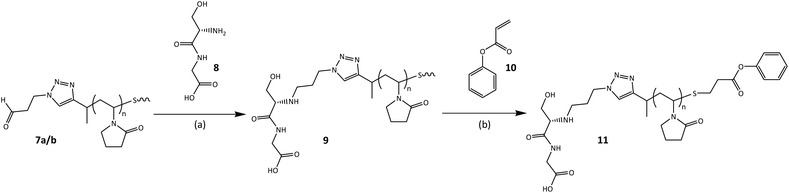 |
| | Scheme 4 Model conjugation reactions of α-aldehyde, ω-thiol functional PVP; with a model peptide (Gly-DL-Ser) and phenyl acrylate, utilising reductive amination and Michael addition chemistry, respectively; (a) sodium borate buffer (pH = 9.1), NaBH3CN, r.t.; and (b) NaBH4, triethylamine, r.t. | |
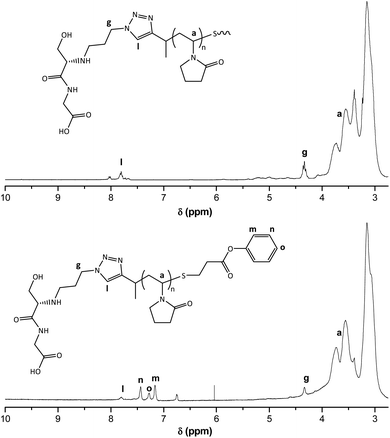 |
| | Fig. 7
1H NMR spectra of α-dipeptide-PVP conjugate (top), and the α-peptide, ω-phenolate-PVP conjugate (bottom), both in DMSO-d6. | |
The reactivity of the ω-thiol was tested by performing a thiol-Michael addition click reaction between the PVP–peptide conjugate (9) with a phenyl acrylate, in order to link PVP with the phenyl moiety via a β-thio propionate. This linkage is particularly interesting because it is cleavable under mildly acidic conditions such as those found in endosomes (pH 5.5), but it is stable at blood pH (7.4), allowing for selective release of the drug after endocytotic uptake.39 The reaction was carried out in a 50% aqueous DMF solution, with respective ratios of [PVP-SH]:[phenyl acrylate]:[triethylamine (catalyst)] of 1:1.5:9.40 The PVP-SH was formed in situ by reduction with sodium borohydride.241H NMR spectroscopy analysis, of the purified product (11), revealed the appearance of aromatic protons assignable to phenyl ring, indicating a successful thiol-Michael addition click reaction (Fig. 7).
Finally, we evaluated the acid lability of the β-thio propionate by placing the polymer conjugate (11) in an aqueous solution buffered at pH 5.5. We followed the progress of the reaction by withdrawing samples, at intervals for TLC analysis. The release kinetics of phenyl moiety were then profiled by performing colour intensity measurements of the TLC plates, using the digitizing software UN-SCAN-IT (Silk Scientific Inc), in order to obtain a (reasonably) quantitative interpretation of the TLC analysis over time. A similar experiment was performed on a conjugate kept in phosphate buffer (pH 7.4) as a control. As shown in Fig. 8, the acid catalysed cleavage of the β-thio propionate ester linker, releasing the phenyl moiety, was ∼90% complete in just over 5 h. Although the accuracy of the TLC study is not considered to be great, the release of the phenolate ion had a similar profile to that reported by Oishi et al. as determined via SEC.39 Consequently, this experiment confirmed the acid lability of the β-thio propionate ester linker.
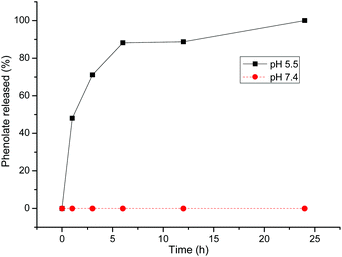 |
| | Fig. 8 Phenol release profile. | |
Conclusions
In conclusion, we have introduced a facile one-pot, orthogonal strategy for converting an α-acetal, ω-xanthate PVP into α-aldehyde, ω-thiol heterotelechelic PVP, by first aminolysing the ω-xanthate chain end, using a primary amine, and then immediately lowering the pH, to simultaneously capture excess primary amine and deprotect the acetal into an aldehyde, to afford the desired α-aldehyde, ω-thiol heterotelechelic PVP. The PVP was synthesized using a triazole-based xanthate RAFT agent. It was found that the triazole-ring alone as a substituent on the α-carbon of the R-group, was not enough to stabilise the radical during polymerisation of NVP, yielding a poorly defined PVP. Good control over molar mass and Đ was obtained after the leaving group was converted into a secondary radical. We also found that a cyclic acetal in close proximity to the nitrogen-containing triazole functionality was impossible to deprotect by acid catalysis, presumably due to unwanted protonation of the N3 atom of the triazole ring. This could be overcome by increasing the chain length between the acetal moiety and the triazole-ring (nitrogen source) and by making use of a linear acetal functionality. Finally we demonstrated the utility of the functional groups by conjugation with model small molecules. The α-aldehyde end-group was used to couple glycine-DL-serine, via reductive amination, whilst the ω-thiol end-group was utilised in a thiol-Michael addition click reaction to conjugate phenyl acrylate via an acid labile β-thio propionate ester linker. We also confirmed the acid lability of that linker. This development, of a modular, one pot protocol for functionalising polymer chain ends is very important for enhancing the versatility of biocompatible vinyl polymers for biomedical applications. Further efforts are currently directed at using the α-aldehyde, ω-thiol heterotelechelic PVP to prepare viable drug delivery systems.
Acknowledgements
This work is based on the research supported by the South African Research Chairs Initiative of the Department of Science and Technology (DST) and National Research Foundation (NRF) of South Africa (Grant No. 46855).
Notes and references
- G. N. Grover and H. D. Maynard, Curr. Opin. Chem. Biol., 2010, 14, 818 CrossRef CAS PubMed.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143 CrossRef CAS PubMed.
- J.-F. Lutz and H. G. Börner, Prog. Polym. Sci., 2008, 33, 1 CrossRef CAS.
- M. A. Gauthier and H.-A. Klok, Chem. Commun., 2008, 2591 RSC.
- P. J. Roth, F. D. Jochum, R. Zentel and P. Theato, Biomacromolecules, 2010, 11, 238 CrossRef CAS PubMed.
- K. L. Heredia, G. N. Grover, L. Tao and H. D. Maynard, Macromolecules, 2009, 42, 2360 CrossRef CAS PubMed.
- K. L. Heredia, L. Tao, G. N. Grover and H. D. Maynard, Polym. Chem., 2010, 1, 168 RSC.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5093 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008, 29, 1172 CrossRef CAS.
- K. L. Heredia, Z. P. Tolstyka and H. D. Maynard, Macromolecules, 2007, 40, 4772 CrossRef CAS.
- C. Boyer, V. Bulmus, T. P. Davis, V. Ladmiral, J. Liu and S. Perrier, Chem. Rev., 2009, 109, 5402 CrossRef CAS PubMed.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18 CrossRef CAS PubMed.
- B. D. Fairbanks, P. A. Gunatillake and L. Meagher, Adv. Drug Delivery Rev., 2015, 91, 141 CrossRef CAS PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2009, 62, 1402 CrossRef CAS.
- M. Bathfield, F. D'Agosto, R. Spitz, M.-T. Charreyre and T. Delair, J. Am. Chem. Soc., 2006, 128, 2546 CrossRef CAS PubMed.
- K. T. Wiss, O. D. Krishna, P. J. Roth, K. L. Kiick and P. Theato, Macromolecules, 2009, 42, 3860 CrossRef CAS.
- K. L. Heredia, T. H. Nguyen, C.-W. Chang, V. Bulmus, T. P. Davis and H. D. Maynard, Chem. Commun., 2008, 3245 RSC.
- S. R. Gondi, A. P. Vogt and B. S. Sumerlin, Macromolecules, 2007, 40, 474 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149 RSC.
- P. J. Roth, C. Boyer, A. B. Lowe and T. P. Davis, Macromol. Rapid Commun., 2011, 32, 1123 CrossRef CAS PubMed.
- M. H. Stenzel, ACS Macro Lett., 2013, 2, 14 CrossRef CAS.
- C. Boyer, J. Liu, V. Bulmus, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Macromolecules, 2008, 41, 5641 CrossRef CAS.
- P. J. Roth, M. Haase, T. Basché, P. Theato and R. Zentel, Macromolecules, 2010, 43, 895 CrossRef CAS.
- O. Shimoni, A. Postma, Y. Yan, A. M. Scott, J. K. Heath, E. C. Nice, A. N. Zelikin and F. Caruso, ACS Nano, 2012, 6, 1463 CrossRef CAS PubMed.
- G. Pound, J. M. McKenzie, R. F. M. Lange and B. Klumperman, Chem.
Commun., 2008, 3193 RSC.
- D. C. Wan, K. Satoh, M. Kamigaito and Y. Okamoto, Macromolecules, 2005, 38, 10397 CrossRef CAS.
- B. Nair, Int. J. Toxicol., 1998, 17, 95 CrossRef CAS.
- F. Haaf, A. Sanner and F. Straub, Polym. J., 1985, 17, 143 CrossRef CAS.
- A. N. Zelikin, G. K. Such, A. Postma and F. Caruso, Biomacromolecules, 2007, 8, 2950 CrossRef CAS PubMed.
- N. Akeroyd, R. Pfukwa and B. Klumperman, Macromolecules, 2009, 42, 3014 CrossRef CAS.
-
T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 3rd edn, 1999 Search PubMed.
- D. S. Dodd, A. C. Oehlschlager, N. H. Georgopapadakou, A. M. Polak and P. G. Hartman, J. Org. Chem., 1992, 57, 7226 CrossRef CAS.
- J. Sun, Y. Dong, L. Cao, X. Wang, S. Wang and Y. Hu, J. Org. Chem., 2004, 69, 8932 CrossRef CAS PubMed.
- A. Ilchev, R. Pfukwa, L. Hlalele, M. Smit and B. Klumperman, Polym. Chem., 2015, 6, 7945 RSC.
- G. Pound, F. Aguesse, J. B. McLeary, R. F. M. Lange and B. Klumperman, Macromolecules, 2007, 40, 8861 CrossRef CAS.
-
P. J. Kocienski, Protecting groups, Thieme Stuttgart, 3rd edn, 2005 Search PubMed.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522 RSC.
- T. H. Fife and L. K. Jao, J. Org. Chem., 1965, 30, 1492 CrossRef CAS.
- M. Oishi, S. Sasaki, Y. Nagasaki and K. Kataoka, Biomacromolecules, 2003, 4, 1426 CrossRef CAS PubMed.
- G.-Z. Li, R. K. Randev, A. H. Soeriyadi, G. Rees, C. Boyer, Z. Tong, T. P. Davis, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1196 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01296e |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence










