
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSET-LRP of acrylates catalyzed by a 1 penny copper coin†
R.
Aksakal
 ab,
M.
Resmini
*b and
C. R.
Becer
*a
ab,
M.
Resmini
*b and
C. R.
Becer
*a
aSchool of Engineering and Materials Science, Queen Mary University of London, London E1 4NS, UK. E-mail: r.becer@qmul.ac.uk
bSchool of Biological and Chemical Sciences, Queen Mary University of London, London E1 4NS, UK. E-mail: m.resmini@qmul.ac.uk
First published on 26th August 2016
Abstract
A British 1 penny coin (1 p) was used to polymerize a selection of hydrophobic and hydrophilic acrylate monomers using linear and star shaped initiators to obtain polymers even in 50 gram scale with low dispersity values (Đ = 1.05–1.11). When compared with Cu wire systems, no induction period was observed, hence demonstrating an economic and easily accessible catalyst for SET-LRP acrylates.
The ability to control chemical composition and architecture is a key priority in polymer synthesis, to obtain materials with the specific characteristics for desired applications. In the last two decades, several robust controlled radical polymerization (CRP) techniques have been developed, that enable the synthesis of well-defined polymers, with excellent control over chain length, dispersity and composition. Among the most established CRP methods reversible addition-fragmentation chain transfer (RAFT) polymerization,1,2 nitroxide mediated polymerization (NMP),3,4 atom transfer radical polymerization (ATRP)5–7 and more recently, single electron transfer living radical polymerization (SET-LRP)8,9 are the four major techniques that have been widely investigated.
SET-LRP, in particular, is a versatile method that allows excellent control over the polydispersity (PDI) with high chain end fidelity, even at high monomer conversions. Unlike the activation step in ATRP with Cu(I)X (X = Br or Cl), the mechanism proposed for SET-LRP, requires the disproportionation of Cu(I)X to Cu(0) and Cu(II)X2, in a polar solvent (i.e. DMSO, H2O or alcohols), and an N-donor ligand (i.e. Me6TREN or PMDETA).10,11 Despite various hypotheses the detailed mechanism of SET-LRP is yet to be fully understood.12–15 Detailed optimization of reaction conditions and the choice of catalyst system are essential requirements in order to obtain the desired polymer. Many reports have already identified the importance of the ligand16 choice and the deactivator concentration,17,18 effect of solvent,19 initiator type,20–22 and other additives23 as essential parameters to be considered. The choice of metal used as a catalyst in the polymerisation is also crucial. In comparison to other zero valent metals, Cu is the most widely used catalyst,24 obtained from various sources and in different formats. Elegant examples describe the successful use of Cu powder,25 Cu pellets,26 Cu tubing,27,28 Cu plate,29 Cu wire30 and in situ generated Cu(0) particles under aqueous SET-LRP conditions.
In aqueous SET-LRP, the pre-disproportionation of Cu(I)Br in water in the presence of Me6TREN22,23 results in highly active, in situ generated Cu(0) particles, which provided the shortest polymerization periods. By employing this system, multi-block copolymers of various acrylamides were synthesized within 3.5 hours.31 Our group has also recently demonstrated the first example of synthesis of star shaped penta-block copolymers via aqueous SET-LRP completed in just under 90 minutes.32 On the other hand, especially in the case of Cu wire, pellets and plate, the pre-activation of the Cu surface plays a crucial role for the reactivity and outcome of the polymerization.33 For pre-activation, typically HCl or hydrazine is employed to remove Cu oxides from the surface, which increases the polymerization rate and minimizes the induction period. Interestingly, there are conflicting reports in the literature regarding the mechanistic explanation that justifies the presence of an induction period.34,35
In this work, we present our findings on the polymerization of various hydrophilic and hydrophobic acrylates using a British 1 penny coin as an alternative and readily available copper source. A linear (EBiB, ethyl-2-bromo isobutyrate) and a 4-arm star initiator (PE-Br4) have been utilized to carry out kinetic investigation on the SET-LRP of methyl acrylate (MA), ethyl acrylate (EA), di(ethyleneglycol) ethyl ether acrylate (eDEGA) and oligo(ethylene glycol) methyl ether acrylate (OEGA) (Scheme 1). The polymers were obtained with very good control over dispersity (Đ < 1.11) and in close agreement between theoretical and experimental molecular weight (Table 1). Moreover, the feasibility of using a coin as a copper source for the polymerization of EA has been demonstrated for different chain lengths (DPn = 20–80) as well as presented the SET-LRP of EA in relatively large scale (50 g).
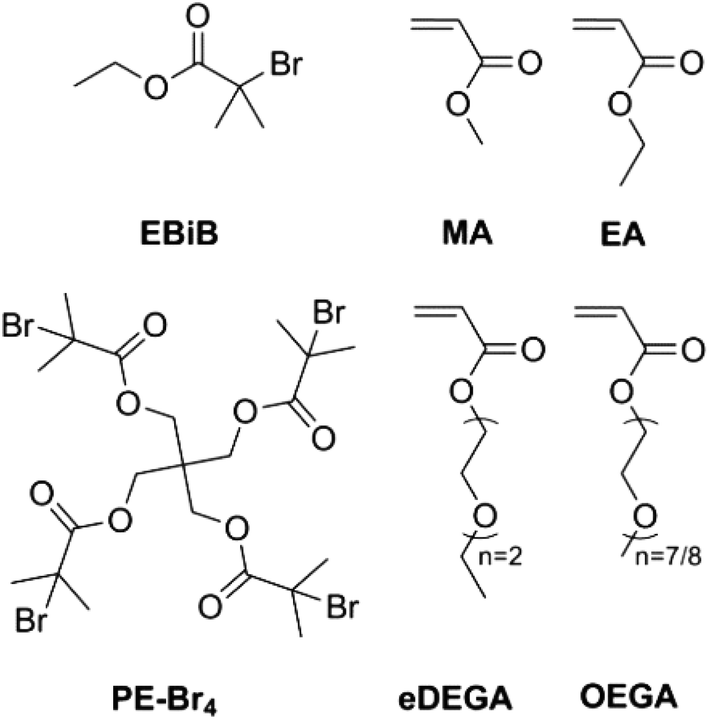 | ||
| Scheme 1 Initiators and monomers used in this work. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]:[CuBr2]:[Me6TREN] = 20:1:0.1:0.19 at 25 °C in DMSO for 3 h
:[I]:[CuBr2]:[Me6TREN] = 20:1:0.1:0.19 at 25 °C in DMSO for 3 h
| Polymer | Initiator | Monomer (DP = 20) | M n,theo (g mol−1) |
M
n,GPCa (g mol−1) |
PDIa | ρ (%) | ΔmCu(0) (mg) |
|---|---|---|---|---|---|---|---|
| a THF with NEt3 eluent, linear PMMA standards. b Conversion was measured by GC. c Conversion was measured by 1H NMR spectroscopy. | |||||||
| P1 | EBiB | MA | 1900 | 2000 | 1.10 | 97c | 6.5 |
| P2 | EBiB | EA | 2200 | 2600 | 1.10 | 99b | 7.1 |
| P3 | EBiB | eDEGA | 3900 | 4500 | 1.11 | 98b | 7.6 |
| P4 | EBiB | OEGA480 | 9200 | 9200 | 1.07 | 93c | 8.1 |
| P5 | PE-Br4 | MA | 2400 | 1600 | 1.07 | 98c | 7.9 |
| P6 | PE-Br4 | EA | 2700 | 2600 | 1.08 | 99b | 7.5 |
| P7 | PE-Br4 | eDEGA | 4400 | 4400 | 1.09 | 97b | 7.6 |
| P8 | PE-Br4 | OEGA480 | 8400 | 8300 | 1.06 | 80c | 7.5 |
In order to avoid star–star polymer coupling, initial polymerizations of MA initiated by PE-Br4 were carried out at different monomer concentrations (i.e. 1:1, 1:4 and 1:10 v/v monomer:solvent). When a MA:DMSO ratio of 1:1 (v/v) was used, significant products derived from coupling reactions were observed in the GPC traces, despite a relatively low MWD (Fig. S2–S4,†Mn,GPC = 4200 g mol−1, PDI = 1.18). The amount of coupling was at minimum when either ratios of 1:4 or 1:10 (v/v) were used. However, the conversion of the latter was determined to be 87% by 1H NMR spectroscopy, whereas the ratio of 1:4 resulted in a conversion of 97% already after 3 h (Fig. S3†). Based on these results, the monomer:solvent ratio was kept at 1:4 (v/v) for the following reactions and the polymerization kinetics were investigated for up to 3 hours.
The SET-LRP kinetics for P1–P4, which are catalysed by 1 p coin at 25 °C in DMSO are shown in Fig. 1. The reactions were initiated with the linear initiator (EBiB), whereas all molar ratios were kept constant as [Monomer]:[EBiB]:[Me6TREN]:[CuBr2] = 20:1:0.19:0.1. The data suggest that when the linear portion of the curves (the first 60 minutes) for P1, P2 and P3 are analyzed, very similar kappp values are obtained with no evidence of any induction period (Fig. 1).
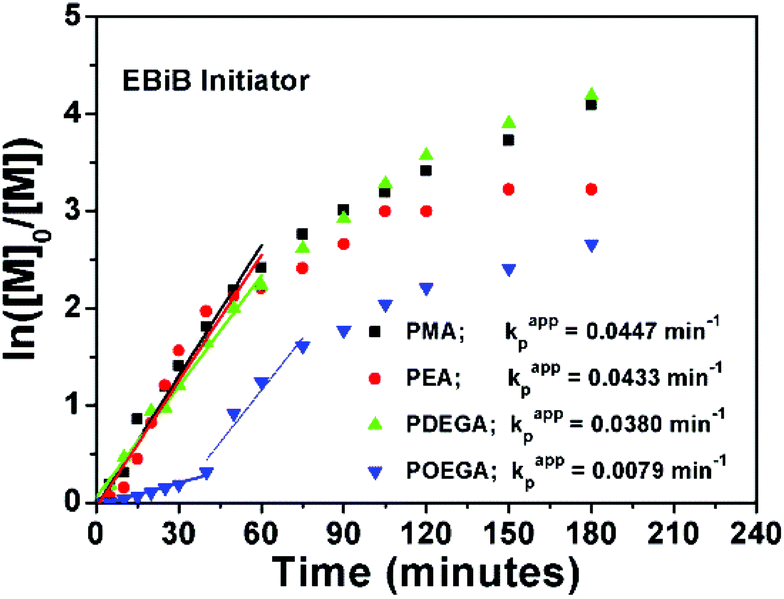 | ||
| Fig. 1 Ln([M]0/[M]) vs. time plot for P1–P4 with the corresponding kappp values. | ||
On the other hand, a significant deviation from the general trend was observed for P4 in the first 30 minutes (ρ = 17%), which was attributed to the known induction period of (OEGA)n monomers, bearing long side chains.36 The second linear regime for P4 between 30–60 minutes, relates to the overall values obtained for P1–P3 in the first hour. After an hour of reaction period, all polymerization reactions tend to display a second linear regime, in which the rate constant decreases until quantitative conversion is reached. This is due to the low concentration of the monomer left at later stages of the polymerization. P4 follows a similar trend as P1–P3, after the induction period. Although very good control over the MWDs was maintained, high molecular weight shoulders have become evident in the obtained GPC traces for P3 and P4 (Fig. 2, GPC traces C–D). Moreover, the data indicates that as bimolecular termination takes place the active chain end concentration decreases, thus effectively leading to a higher ligand and CuBr2 to dormant species ratio, which in turn slows down the reaction (i.e. persistent radical effect).37 Nevertheless, all polymerizations reached almost quantitative conversions within 3 hours, regardless of the monomers chosen. Furthermore, analysis of P1 by 1H NMR spectroscopy and MALDI-ToF MS showed a high degree of end group-fidelity (Fig. 3 and see Fig. S5† for full spectrum).
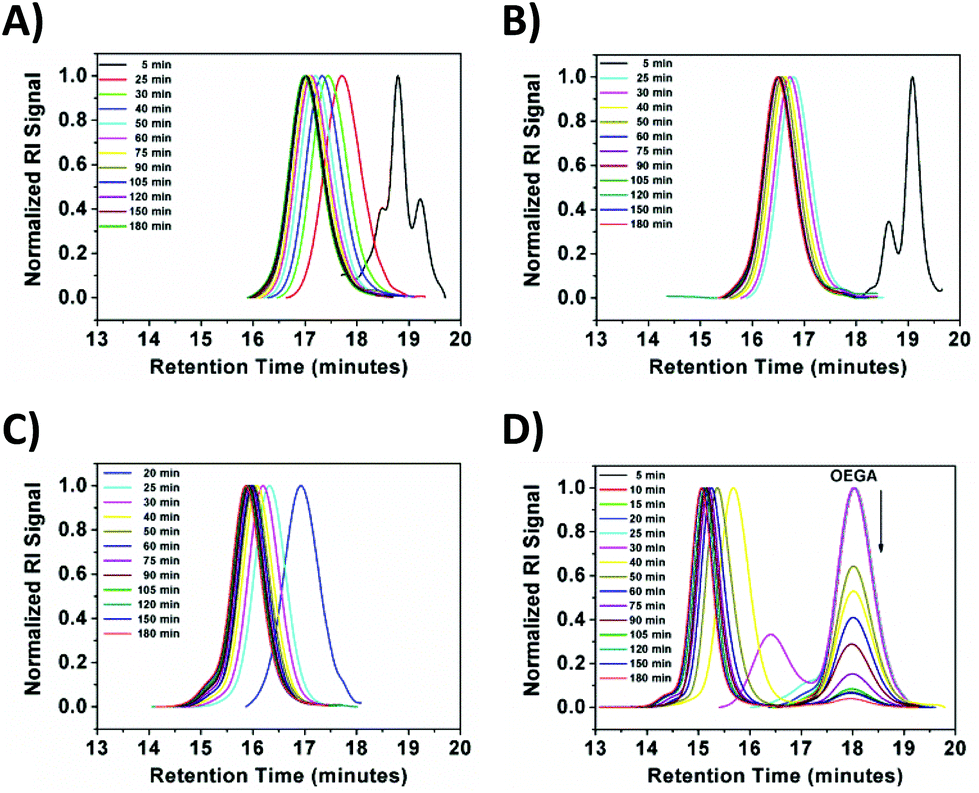 | ||
| Fig. 2 GPC traces of the obtained polymers (A) P1, (B) P2, (C) P3 and (D) P4 using the EBiB initiator. | ||
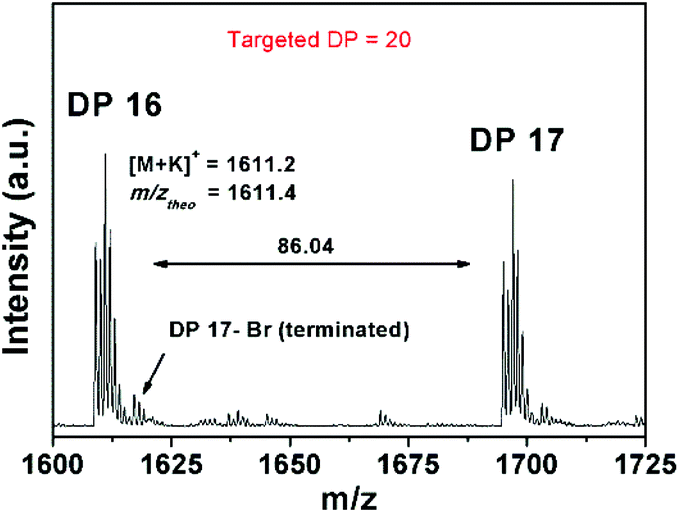 | ||
| Fig. 3 Expanded MALDI-ToF spectrum of PMA20 (P1) obtained via Cu coin mediated SET-LRP in DMSO. | ||
By comparing the broad signal of CH–Br (ω-terminus) between 4.09–4.00 ppm and CH3–CH2– (initiator) between 3.92–3.80 ppm, the end group fidelity was calculated to be 81% by 1H NMR spectroscopy (Fig. S6†). It is known that the end group fidelity can still be improved even further by varying the ligand and/or CuBr2 concentration in such SET-LRP systems.
The increase in molecular weight (Mn) with higher conversion shows a similar trend to the one shown in Fig. 1. Up to a conversion of about 30%, the experimental Mn values measured for P4 are not in agreement with the theoretical values (Fig. 4, blue line). This might be due to difference in the hydrodynamic volume of POEGA in comparison to PMMA standards. For all other values a linear evolution of Mn with respect to monomer conversion was observed within very close approximation to the theoretical values.
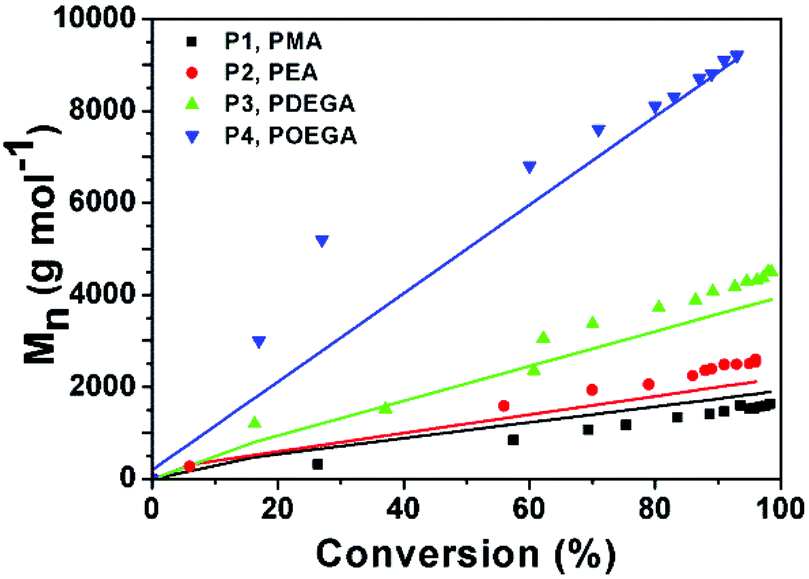 | ||
| Fig. 4 M n vs. conversion plot for P1–P4. Coloured symbols represent Mn obtained from GPC; lines represent theoretical Mn calculated from corresponding conversions. | ||
Similarly to the case with the linear initiator, when a branched initiator was utilized the same molar ratios of [Monomer]:[PE-Br4]:[Me6TREN]:[CuBr2] = 20:1:0.19:0.1 were used. Here only the amount of the initiator was varied, in order to keep the concentration of the monomer and catalyst system to solvent constant. Fig. S11–14,† shows the obtained GPC traces for the polymers P5–P8. All polymerizations reached to quantitative conversions, where as a conversion of 80% for P8 was obtained (Mn,GPC = 8300 g mol−1, PDI = 1.06) in 3 hours.
By using a four-arm star initiator for P5–P8, the effective ligand concentration per initiating site has been decreased to a fourth. Although comparable kappp values could not be obtained from the Ln([M]0/[M]) vs. time plot (P5–P8, Fig. 5), a significantly slower conversion was observed for the first 30 minutes, when compared to the linear P1–P4 polymerizations.
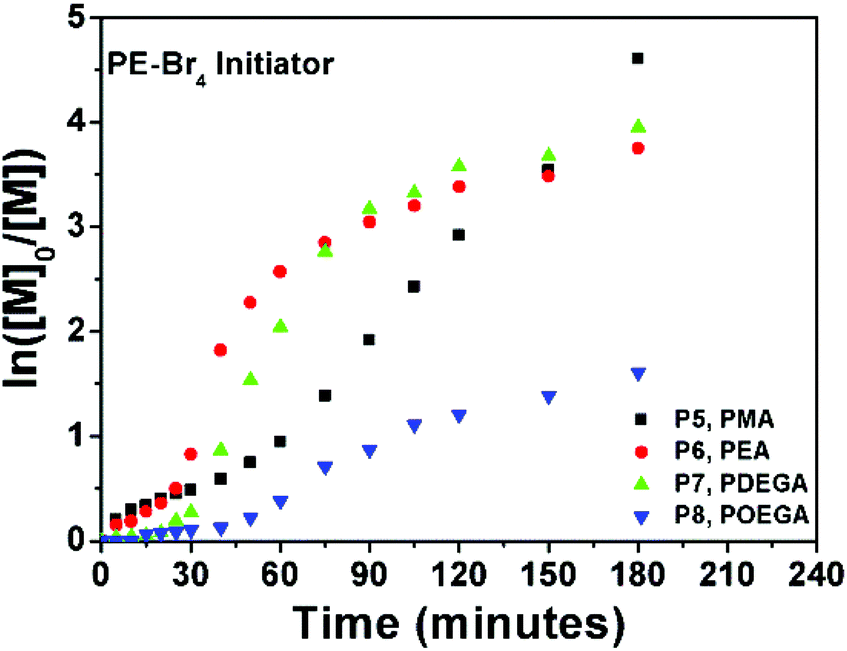 | ||
| Fig. 5 Ln([M]0/[M]) vs. time plot for P5–P8 obtained via 1 penny mediated SET-LRP using PE-Br4 as an initiator. | ||
Contrary to the trend observed with the linear initiator where a decrease in the polymerization rate occurs (Fig. 1), monomer consumption tends to speed up after 30 minutes for P5–P8. The increase in rate for P5–P7 is significantly more evident than the increase for P8, which can be explained with the induction time for OEGA. Furthermore, the evolutions of the molecular weights obtained from GPC analysis are found to be in excellent agreement with the theoretical molecular weights (Fig. S7†). For instance, with the exception of polymer P8, all molecular weights during the polymerization follow a linear trend whilst growing. For P8, a rapid increase in Mn is observed up to 12% monomer consumption (Mn,GPC = 3300 g mol−1, PDI = 1.05), which is followed by a slower linear increase up to 80% conversion. Nevertheless the MWD remains low throughout the polymerization, which can be taken as a reliable indication that good control is maintained, with no apparent coupling reactions taking place.
Furthermore, EA was polymerized using EBiB at DP = 40 and 80 (P9 and P10, respectively) under the same conditions. Good control was retained over polymerization (PDI 1.07–1.10) even at high DP and negligible amount of coupling reactions were observed (Fig. 6). Kinetic investigation of the polymerization shows identical behaviour to P2 (DPn = 20). Finally, an attempt was done to obtain a polymer on large scale by polymerizing EA (DPn = 80) on a 50 g scale (P11) using a single 1p coin. Within 3 hours, monomer conversion has already reached to 90% (Mn,GPC = 7400 g mol−1, PDI = 1.06). At this point, the polymerization was allowed to proceed for 16 hours to reach to full conversion (Mn,GPC = 8300 g mol−1, PDI = 1.06, Fig. S8†).
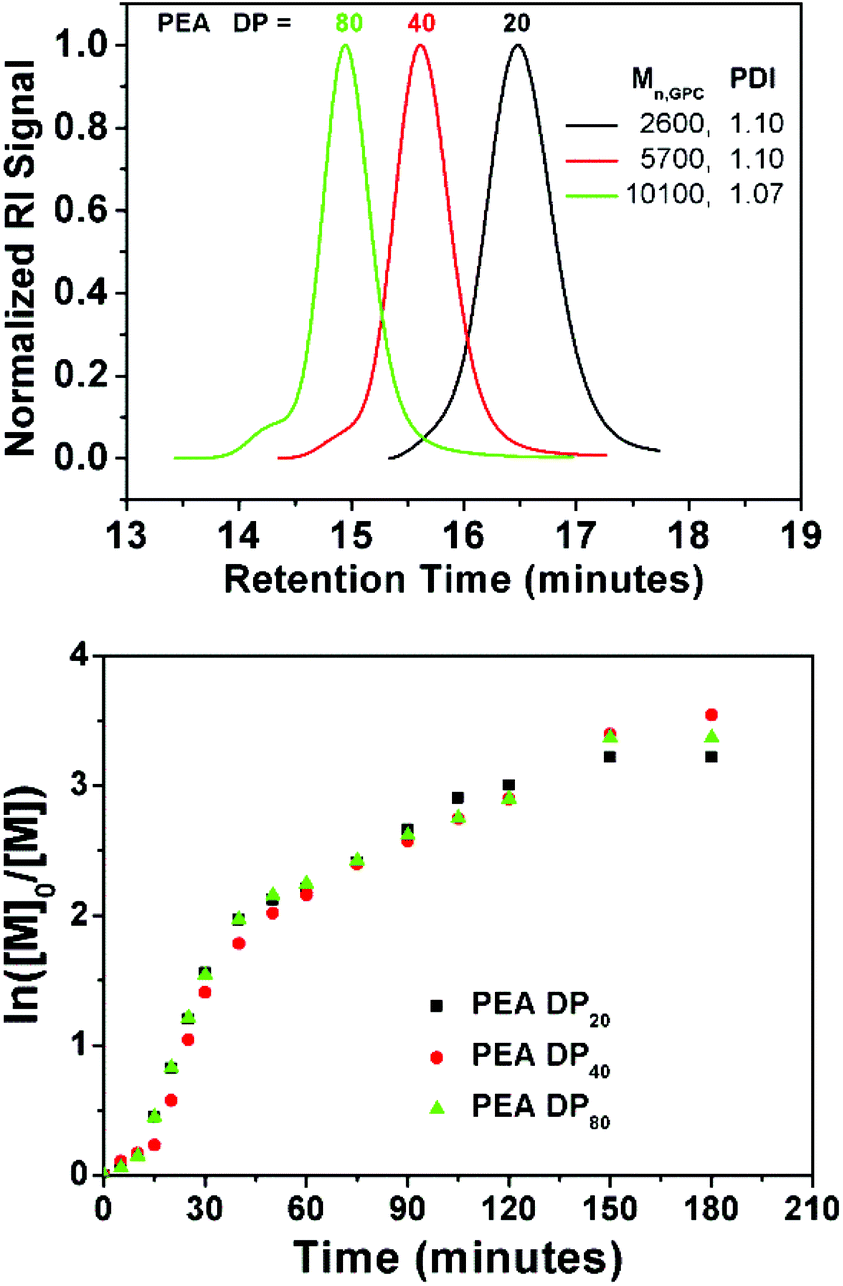 | ||
| Fig. 6 GPC traces of the obtained PEA at different DPn, P2 = DP20, P9 = DP40 and P10 = DP80 (Top). Ln([M]0/[M]) vs. time plot for P2, P9 and P10 obtained via 1 penny mediated SET-LRP using EBiB as an initiator (Bottom). | ||
For each polymerization reaction, the weight change of the copper coin was monitored, by noting the difference in mass of the copper coin at t = 0 and right after the polymerization was stopped at t = 3 h. Before pre-activation, the copper coin was immersed into a beaker containing HCl (2–3 times), then thoroughly washed with deionised H2O and acetone before drying under nitrogen. The initial weights of the Cu coins were found to be around 3.5 and 3.6 g. After this, the coins were pre-activated as usual (see ESI 3.2,† Experimental procedure). When the polymerizations occurred for 3 hours, the Cu coins were immediately removed from the reaction medium and rinsed with acetone and dried under nitrogen, prior to weighing. Negligible mass losses in comparison to the initial weight of the Cu coins were obtained. The mass changes were found to be between 6.5 to 8.1 mg (Δmcoin < 0.2%) and are listed in Table 1. Interestingly, when one of our initial attempts to polymerize eDEGA with EBiB failed due to an issue with the Schlenk line (Fig. S9†), we have also observed that the mass change of 1.7 mg took place at a monomer conversion of 7% (after 3 h), which suggests that Cu is only consumed when SET-LRP occurs.
It should be noted that there are two different types of 1 p coins in circulation. From their first issue in 1971 until 1992, the composition of a 1 p coin consisted of 97% Cu, 2.5% Zn and 0.5% Sn. Hereafter, Cu plated steel coins were introduced (94% mild steel from Fe, C and Mn, and 6% Cu).38 We have found that both coins can be used equally, providing almost identical polymerization results (Fig. S10†).
Moreover, for comparison purposes a Cu wire is used with an equal surface area to a Cu coin. The total surface area of a standard British 1 p coin (diameter = 20.3 mm, thickness = 1.65 mm)38 equals to that of a cylindrical copper wire (9.58 cm, 0.25 mm diameter, ESI eqn (1)†). Therefore, EA was polymerized with EBiB using a 9.6 cm (diameter = 0.25 mm, P12) Cu wire under the same polymerization conditions as for P2. We have then investigated the effect of a shorter (5 cm in length) Cu wire (P13) for comparison, as this is a widely employed standard Cu wire length in the literature. As expected, induction periods were observed for P12 and P13, of 25 and 40 minutes, respectively.34 Nicolas et al. attributed this initial slow rate of polymerization to autocatalysis by studying the initiator conversion, which is not applicable for this study as only the monomer concentration was monitored.35 On the contrary, when a coin with the same surface area was employed the kinetics for P2 provided a conversion of 6% in 5 minutes, with a linear increase in the kinetic plot (Fig. 7). In order to investigate if the acceleration in the polymerization is due to the uneven surface of the Cu coin, we have utilized an extra long (20 cm in length) Cu wire (P14). However, an induction period of 20 minutes was also observed, which indicates that a higher surface area of Cu is not the main reason for eliminating the induction period.
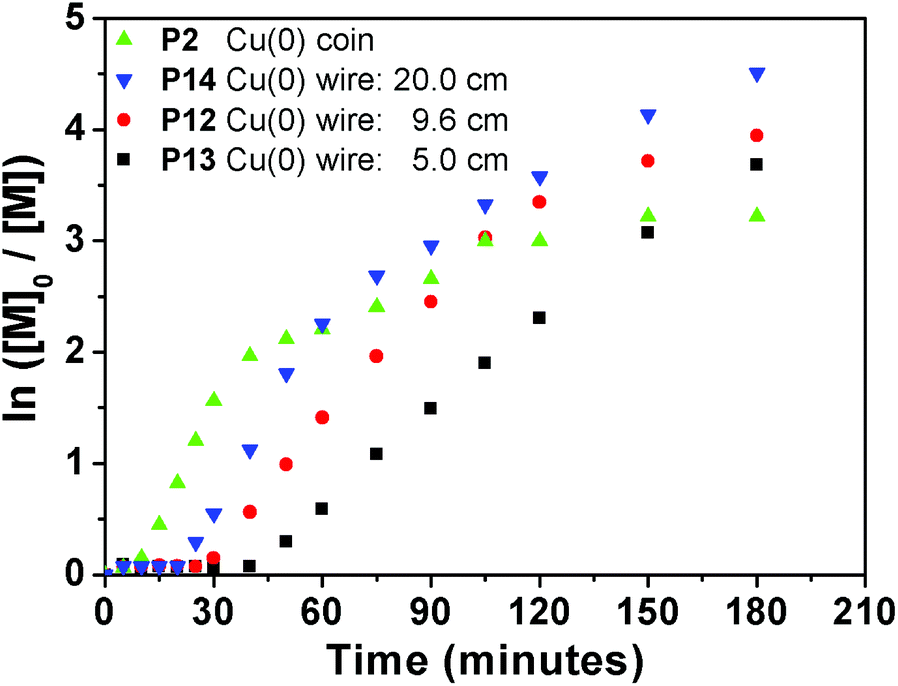 | ||
| Fig. 7 Ln([M]0/[M]) vs. time plot for P2, P12 and P13 obtained via copper mediated SET-LRP using EBiB as initiator. | ||
In conclusion, a series of well defined polymers were obtained via 1 penny coin catalyzed SET-LRP of MA, EA, DEGA and OEGA using both a linear (EBiB) and a 4-arm star initiator (PE-Br4). All obtained polymers were characterized in details via GPC, 1H NMR and MALDI-ToF MS techniques. Although some of the polymers displayed minor bimolecular termination to some extent, linear PMA and PEA displayed no coupling reactions at all (PDI = 1.10). Furthermore, in order to demonstrate the scope of this protocol, we have shown the polymerization of PEA at various degrees of polymerizations (DP = 20, 40 and 80) as well as synthesized PEA80 at a relatively large scale (50 g) with excellent control over MWD and in good agreement between theoretical and experimental molecular weight. Furthermore, a Cu wire with an equal surface area was used for polymerization to demonstrate a comparison to a more widely used system in the literature. The obtained results indicate that, a Cu coin can be utilized as a cheap, convenient and readily available alternative source to Cu wire for Cu(0) mediated polymerizations of acrylates.
References
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS
.
- D. J. Keddie, Chem. Soc. Rev., 2014, 43, 496–505 RSC
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661–3688 CrossRef CAS PubMed
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS
- V. Percec, A. V. Popov, E. Ramirez-Castillo, M. Monteiro, B. Barboiu, O. Weichold, A. D. Asandei and C. M. Mitchell, J. Am. Chem. Soc., 2002, 124, 4940–4941 CrossRef CAS PubMed
- V. Percec, T. Guliashvili, J. S. Ladislaw, A. Wistrand, A. Stjerndahl, M. J. Sienkowska, M. J. Monteiro and S. Sahoo, J. Am. Chem. Soc., 2006, 128, 14156–14165 CrossRef CAS PubMed
- B. M. Rosen and V. Percec, Chem. Rev., 2009, 109, 5069–5119 CrossRef CAS PubMed
- N. Zhang, S. R. Samanta, B. M. Rosen and V. Percec, Chem. Rev., 2014, 114, 5848–5958 CrossRef CAS PubMed
- D. Konkolewicz, Y. Wang, M. Zhong, P. Krys, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 8749–8772 CrossRef CAS
- D. Konkolewicz, Y. Wang, P. Krys, M. Zhong, A. A. Isse, A. Gennaro and K. Matyjaszewski, Polym. Chem., 2014, 5, 4396–4417 RSC
- F. Alsubaie, A. Anastasaki, V. Nikolaou, A. Simula, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 5517–5525 CrossRef CAS
- F. Alsubaie, A. Anastasaki, V. Nikolaou, A. Simula, G. Nurumbetov, P. Wilson, K. Kempe and D. M. Haddleton, Macromolecules, 2015, 48, 6421–6432 CrossRef CAS
- A. Anastasaki, C. Waldron, P. Wilson, R. McHale and D. M. Haddleton, Polym. Chem., 2013, 4, 2672–2675 RSC
- L. C. Ding W, Y. Sun, H. Luan, T. Yu and G. Qu, Polym. Bull., 2011, 67, 7 CrossRef
- A. Anastasaki, C. Waldron, P. Wilson, C. Boyer, P. B. Zetterlund, M. R. Whittaker and D. Haddleton, ACS Macro Lett., 2013, 2, 896–900 CrossRef CAS
- A. Anastasaki, A. J. Haddleton, Q. Zhang, A. Simula, M. Droesbeke, P. Wilson and D. M. Haddleton, Macromol. Rapid Commun., 2014, 35, 965–970 CrossRef CAS PubMed
- N. H. Nguyen, J. Kulis, H.-J. Sun, Z. Jia, B. van Beusekom, M. E. Levere, D. A. Wilson, M. J. Monteiro and V. Percec, Polym. Chem., 2013, 4, 144–155 RSC
- L. Voorhaar, S. Wallyn, F. E. Du Prez and R. Hoogenboom, Polym. Chem., 2014, 5, 4268–4276 RSC
- S. Fleischmann and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2236–2242 CrossRef CAS
- M. Gavrilov, T. J. Zerk, P. V. Bernhardt, V. Percec and M. J. Monteiro, Polym. Chem., 2016, 7, 933–939 RSC
- A. Anastasaki, V. Nikolaou and D. M. Haddleton, Polym. Chem., 2016, 7, 1002–1026 RSC
- G. Lligadas, B. M. Rosen, C. A. Bell, M. J. Monteiro and V. Percec, Macromolecules, 2008, 41, 8365–8371 CrossRef CAS
- M. E. Levere, I. Willoughby, S. O'Donohue, A. de Cuendias, A. J. Grice, C. Fidge, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 1086–1094 RSC
- N. Chan, M. F. Cunningham and R. A. Hutchinson, Macromol. Rapid Commun., 2011, 32, 604–609 CrossRef CAS PubMed
- J. A. Burns, C. Houben, A. Anastasaki, C. Waldron, A. A. Lapkin and D. M. Haddleton, Polym. Chem., 2013, 4, 4809–4813 RSC
- T. Zhang, Y. Du, F. Muller, I. Amin and R. Jordan, Polym. Chem., 2015, 6, 2726–2733 RSC
- B. C. Soeriyadi, A. H. Nyström, P. B. Zetterlund and M. R. Whittaker, J. Am. Chem. Soc., 2011, 133, 4 CrossRef PubMed
- F. Alsubaie, A. Anastasaki, P. Wilson and D. M. Haddleton, Polym. Chem., 2015, 6, 406–417 RSC
- R. Aksakal, M. Resmini and C. R. Becer, Polym. Chem., 2016, 7, 171–175 RSC
- N. H. Nguyen and V. Percec, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5109–5119 CrossRef CAS
- Y. Gao, T. Zhao, D. Zhou, U. Greiser and W. Wang, Chem. Commun., 2015, 51, 14435–14438 RSC
- S. Harrisson, P. Couvreur and J. Nicolas, Macromolecules, 2012, 45, 7388–7396 CrossRef CAS
- A. Simula, G. Nurumbetov, A. Anastasaki, P. Wilson and D. M. Haddleton, Eur. Polym. J., 2015, 62, 294–303 CrossRef CAS
- H. Fischer, Chem. Rev., 2001, 101, 3581–3610 CrossRef CAS PubMed
- The Royal Mint, Coin Designs and Specifications, http://www.royalmint.com (accessed on: 01.08.2016).
Footnote |
| † Electronic supplementary information (ESI) available: Polymerization kinetics and additional NMR, MALDI and GPC traces. See DOI: 10.1039/c6py01295g |
| This journal is © The Royal Society of Chemistry 2016 |