
The effects of the photo-induced proton generation on the assembly formation of dual-temperature and pH responsive block copolymers†
Yohei
Kotsuchibashi
a,
Tomomi
Takiguchi
bc,
Mitsuhiro
Ebara
bcd and
Takao
Aoyagi
*bc
aInternational Center for Young Scientists (ICYS) and International Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
bBiomaterials Unit, WPI-MANA, NIMS, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
cDepartment of Materials Engineering, Graduate School of Pure and Applied Sciences, University of Tsukuba, 1-1-1 Tennodai, Tsukuba, Ibaraki 305-8577, Japan
dGraduate School of Industrial Science and Technology, Tokyo University of Science, 6-3-1 Niijuku, Katsushika, Tokyo 125-8585, Japan. E-mail: aoyagi.takao@nihon-u.ac.jp; Fax: +81-3-3293-7572; Tel: +81-3-3259-0433
First published on 24th August 2016
Abstract
The effects caused by photo-induced proton generation on the assembly formation of dual-temperature/pH-responsive block copolymers are investigated. The block copolymer of P(N-isopropylacrylamide (NIPAAm)-co-N-(isobutoxymethyl)acrylamide (BMAAm))-b-P(NIPAAm-co-2-carboxyisopropylacrylamide (CIPAAm)) was synthesized by reversible addition-fragmentation chain transfer (RAFT) polymerization. The block copolymers underwent self-assembly and formed a spherical micellar structure because the P(NIPAAm-co-BMAAm) block turned hydrophobic above its lower critical solution temperature (LCST), while the electrostatic repulsion of the P(NIPAAm-co-CIPAAm) block stabilized the micelle at pH levels above the pKa of the CIPAAm units. To achieve a local and remote control of micelle aggregation, the micelle solution was exposed to UV radiation in the presence of a photoacid generator of o-nitrobenzaldehyde (NBA). When UV irradiation was limited to a specific region, we were able to restrict the proton generation to only the exposed region. Interestingly, the opaque area created by micelle aggregation was stable even when UV radiation was turned off. Finally, the photo-induced proton transfer from a micelle core was demonstrated using an NBA-encapsulated micelle. This proposed system of micelle aggregation via photo-induced proton diffusion may be beneficial not only for prompt pH changes in the nanoparticle solution, but also for the design of predictive and programmable nanocontainers for drug delivery applications.
Introduction
The fabrication of extremely small objects with precisely defined structures has recently become an attractive challenge with the introduction of intriguing synthetic techniques such as controlled living radical polymerizations (CLRPs) and click chemistries.1 Smart or stimuli-responsive nanoparticles, particularly, are a new generation of nanomaterials that exhibit reversible physicochemical changes in response to their environment. A family of typical amphiphilic block copolymers is required to use organic solvents for the preparation of nanoparticles in aqueous solution.2,3 On the other hand, water-soluble stimuli-responsive polymers can form nanoparticles without organic solvents via external environmental changes, e.g. temperature, pH, light, and target molecules.4,5 These smart nanoparticles in a completely aqueous system are finding applications in many fields, such as for drug delivery, catalysis, sensors, and surface modifications.6 Temperature-responsive micelles, for example, have been developed for the combined treatment of anticancer drugs and hyperthermia, while pH-responsive micelles have been used in drug or gene delivery to the regions of local acidosis such as solid tumors, ischemic tissues, or infected sites.7 Recently, the combination of two different stimuli-responsive blocks in a block copolymer has been reported to achieve a complex system via programmable self-assemblies.8,9 Such dual-responsive systems can provide more opportunities to tune the morphology of micelles or aggregation by two or more signals. Among them, dual-temperature-responsive block copolymers can exhibit three morphological changes in aqueous solution depending on the solution temperature: hydrophilic–hydrophilic (dissolution), amphiphilic (nanoparticle), and hydrophobic–hydrophobic (aggregation/precipitation) properties. Papers on dual-temperature-responsive block copolymers have recently been collected in comprehensive reviews.10 We have prepared several types of dual-temperature responsive block copolymers exhibiting a different lower critical solution temperature (LCST) in each block.11 The LCSTs were easily controlled via the compositions of the two copolymerized monomers, and their three morphological changes could be designed to occur at the desired solution temperature. Moreover, we have successfully developed a facile protocol to produce multi-functional assemblies with targetable and tunable shell functions by simply mixing three different block copolymers with a common temperature-responsive block.12 To add a more complex system, Savoji et al. prepared a block copolymer of poly(N-n-propylacrylamide (nPA)-co-2-(diethylamino)ethyl methacrylate (DEAEMA))-b-poly(nPA-co-N-ethylacrylamide (EA)) with dual-temperature- and pH-responsive properties.13 For the pH response, the block copolymer had cationic units, which showed a reversible vesicle surface due to a combination of the solution temperature and pH. However, the micelle precipitation occurs randomly via the hydrophobic shell interaction, which is very difficult to control the aggregated nanoparticle constructions. Moreover, local construction of the precipitated micelle is also a challenge. Many physical and chemical stimuli have been applied to induce various responses of the smart polymer systems. Physical stimuli, such as in light-responsive systems, are advantageous because they allow local and remote control.14 On the other hand, chemical stimuli, such as concentration gradients of protons, ions, and oxidizing/educing agents, are also important characteristics observed in living systems for muscle and ciliary movement, pulsatile secretion of hormones, brain waves, etc. In contrast to physical stimuli, however, changing the concentration of chemical signals rapidly and precisely at a particular location in the system is difficult because it cannot easily penetrate through materials. From this perspective, we have focused on the photoinitiated proton-releasing reaction of ‘photoacid generators (PAGs)’, the pKa of which is significantly different in an excreted state than in the ground state.15 The change in pKa results in the release of a proton in aqueous solution. Most recently, we have demonstrated a novel approach to spatio-temporal proton concentration control within vial containers, hydrogels, and devices by using a PAG.16 By utilizing a system responsive to both light and pH, local and remote signals could be simultaneously triggered for predictive, timed, explosive devices because this system permits not only local activation in the illuminated area but also gradual activation in the non-illuminated area as protons diffuse.In this study, we propose a system of remote-controlled local micelle aggregation via photoinitiated photoacid generators in dual-pH-responsive block copolymers (Fig. 1). Dual-temperature and pH responsive block copolymers of P(N-isopropylacrylamide (NIPAAm)-co-N-(isobutoxymethyl)acrylamide (BMAAm))-b-P(NIPAAm-co-2-carboxyisopropylacrylamide (CIPAAm))s were used as the model micelle. The block copolymers were composed of three kinds of acrylamide-type monomers, and the CIPAAm units that are located on the micelle shell show a pH-responsive property due to their carboxylic group. These acrylamide-type copolymers have been applied in biomaterials, which are expected to be nanoassemblies with low toxicity.6a–c It is the first report on a system of remote-controlled local micelle aggregation using dual-temperature and pH responsive block copolymers. The effects caused by photo-induced proton generation on the assembly formation of the block copolymers are investigated using 1H nuclear magnetic resonance (1H NMR), gel permeation chromatography (GPC), UV-vis spectrometry, dynamic light scattering (DLS), zeta-potential, and atomic force microscopy (AFM).
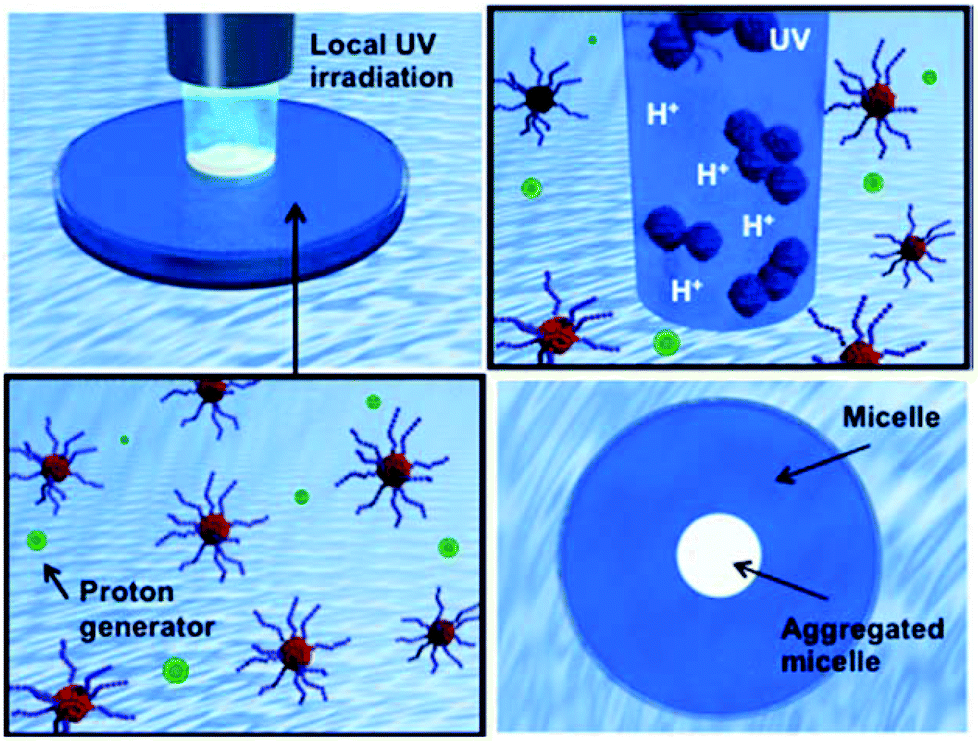 | ||
| Fig. 1 Schematic representation of a system of remote-controlled local micelle aggregation. | ||
Experimental
Materials
N-Isopropylacrylamide (NIPAAm) was kindly supplied by Kohjin Co. Ltd (Japan) and purified through recrystallization from benzene and hexane. According to the protocols published previously, 2-carboxyisopropylacrylamide (CIPAAm) was synthesized.17N-(Isobutoxymethyl)acrylamide (BMAAm) was obtained from Wako Pure Chemical Industries (Osaka, Japan) and was purified by distillation. Azobisisobutyronitrile (AIBN) was obtained from Wako Pure Chemical Industries (Japan) and was purified by recrystallization from ethanol. The chain transfer agent (CTA), cumyl dithiobenzoate (CDB), was synthesized and purified according to a previously reported protocol.18O-Nitrobenzaldehyde (NBA) was purchased from Tokyo Chemical Industry Co. Ltd (Japan) and used as received. All other chemicals and solvents were used as received. Distilled water used in this study was purified with a Millipore Milli-Q system.Preparation of P(NIPAAm-co-CIPAAm)s with different CIPAAm contents as macro-chain transfer agents (macro-CTAs)
P(NIPAAm-co-CIPAAm)s were polymerized at different CIPAAm contents as macro-chain transfer agents (macro-CTAs) by reversible addition-fragmentation chain transfer (RAFT) polymerization. A typical polymerization method of P(NIPAAm-co-CIPAAm) (CIPAAm content = 4 mol% in feed) is shown below. NIPAAm (2.63 g, 23.3 mmol), CIPAAm (152 mg, 0.97 mmol), CDB (20 mg, 7.8 × 10−2 mmol), and AIBN (5.0 mg, 3.00 × 10−2 mmol) ([NIPAAm]0/[CIPAAm]0/[CDB]0/[AIBN]0 = 298.7/12.4/1/0.38) were dissolved in 10 mL of N,N-dimethylformamide (DMF). After three freeze–pump–thaw cycles, the mixture was allowed to polymerize for 22 h at 70 °C. The polymerization container was soaked in liquid nitrogen to stop the reaction. The resulting P(NIPAAm-co-CIPAAm) was purified by reprecipitation using diethyl ether and was dried under reduced pressure.Preparation of P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s
Using the polymerized macro-CTAs of the P(NIPAAm-co-CIPAAm)s, P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s were prepared by RAFT polymerization. A typical polymerization method of the block copolymer is shown below. Macro-CTAs of P(NIPAAm-co-CIPAAm) (1.02 g, 3.61 × 10−2 mmol), NIPAAm (1.07 g, 9.43 mmol), BMAAm (304 mg, 1.93 mmol), and AIBN (2.33 mg, 1.40 × 10−2 mmol) ([NIPAAm]0/[CIPAAm]0/[Macro-CTA]0/[AIBN]0 = 261/53/1/0.39) were dissolved in 10 mL of DMF. After three freeze–pump–thaw cycles, the mixture was allowed to polymerize for 26 h at 70 °C. The polymerization container was soaked in liquid nitrogen to stop the reaction. P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s were purified by dialysis (MWCO: 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) in ethanol and distilled water, and were collected by freeze-drying as a white powder. To remove low molecular weight polymers, the P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s were further purified by ultrafiltration. The block copolymer (400 mg) was dissolved in 40 mL distilled water at 4 °C, and the temperature was increased to 25 °C to form the micelle of P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm). Ultrafiltration was used to separate the micelle and linear low molecular weight polymers at 25 °C and the assembled block copolymers were collected via freeze-drying.
000) in ethanol and distilled water, and were collected by freeze-drying as a white powder. To remove low molecular weight polymers, the P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s were further purified by ultrafiltration. The block copolymer (400 mg) was dissolved in 40 mL distilled water at 4 °C, and the temperature was increased to 25 °C to form the micelle of P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm). Ultrafiltration was used to separate the micelle and linear low molecular weight polymers at 25 °C and the assembled block copolymers were collected via freeze-drying.
NBA loading into micelle
NBA (5 or 10 mg) was completely dissolved in 5 mL of distilled water using ultrasonic cleaning equipment. Block copolymers of P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s (10 mg) were added into the NBA solution and were completely dissolved at 10 °C. The mixture solution was stirred for 10 min at 10 °C, and the solution temperature was increased up to 25 °C (1 °C/5 min) to capsulate the NBA into the micelle core. The NBA loaded micelle solution was stored for 20 h at 25 °C before purification. To purify the NBA loading micelles, the suspension was added into an ultrafiltration tube (MWCO: 100000) and was separated by centrifugation (3000 rpm, 25 °C) to remove the excess free NBA. The volume of the NBA loaded micelle suspension was concentrated to a volume of 1 mL, and 3 mL of distilled water was slowly added into the suspension at 25 °C. The diluted NBA loaded micelle suspension (4 mL) was centrifuged (3000 rpm, 25 °C). This washing process was repeated three times. Finally, the volume was adjusted to 5 mL by adding 4 mL of distilled water. To estimate the loading amount of NBA, the NBA loaded micelle suspension (2.5 mL) was added into an ultrafiltration tube (MWCO: 3000) and was centrifuged (3500 rpm, 4 °C) to separate the NBA (as the filtrate) and the dissolved block copolymers (as the residue). The residue was washed with a total of 6 mL of distilled water (3 mL, 2 times) at 4 °C, and the block copolymers were collected through freeze-drying to calculate the loading amount of NBA. The collected block copolymer was 4.8 mg. The amount of loaded NBA was calculated by HPLC. The NBA loading micelle suspension (300 μL) was mixed with methanol (700 μL) to break the micelle structures. A calibration curve of NBA was prepared using a distilled water/methanol solution (3:7 vol%) at different concentrations (0.001, 0.03, 0.06, 0.08, and 0.1 mM). The calculated NBA concentration in the micelle was 0.19 mM. According to the weight of the collected block copolymer (4.8 mg in 2.5 mL) and the NBA concentration calculated by HPLC (0.19 mM), the calculated NBA loading was 1.5 wt%.
UV light irradiation of NBA loading micelles
Upon UV irradiation, NBA can generate protons, resulting in a decrease of the solution pH. The NBA loading micelle suspension was kept at 37 °C. The absorbance (wavelength 500 nm) was set at zero before measurement. Then, UV radiation (20 mW cm−2) was applied locally to the suspension for 15 seconds. The absorbance and solution pH were measured continuously. This process was performed for 900 s.Characterization
1H NMR spectra of copolymers were recorded with a JNM-GSX300 spectrometer operating at 300 MHz (JEOL, Tokyo, Japan) to confirm successful synthesis and determine the chemical composition of the synthesized copolymers.The molecular weight and polydispersity of the synthesized copolymers were determined by gel permeation chromatography (GPC) at 40 °C (DMF including 10 mM LiBr, 1 mL min−1) with a TOSOH TSK-GEL a-2500 and a-4000 (Tosoh, Tokyo, Japan) connected to a RI-2031 refractive index detector (JASCO International Co., Ltd, Tokyo, Japan).
Transmittance/absorbance of the solution at 500 nm was continuously recorded at a heating rate of 1.0 °C min−1 by a UV-Vis spectrometer V-550 (JASCO International Co., Ltd, Tokyo, Japan). Synthesized copolymers were dissolved in an aqueous solution at the given concentration. LCSTs of copolymers were defined as the temperature at which the transmittance reached 50%.
Dynamic light scattering (DLS) was performed with a DLS-8000 series (Otsuka Electronics Co., Ltd, Osaka, Japan) using a light scattering apparatus equipped with a He–Ne laser and a temperature controller. All samples were kept at the given temperatures to reach equilibrium prior to the measurements. We obtained the diameter data using the Marquardt method (cumulative number = 80).
The zeta-potential was measured using an Electro Phonetic Light Scattering ELS-6000 (Otsuka Electronics Co., Ltd, Osaka, Japan). All samples were kept at the given temperatures to reach the equilibrium prior to measurement.
Atomic force microscopy (AFM) measurements were performed using the SPM-9500J3 (Shimadzu Co., Kyoto, Japan) in non-contact mode using a Si3N4 cantilever (spring constant; 42 N m−1). A substrate of silicon wafer was washed with ethanol and water before use. The micelles were dropped on the silicon wafers by the spin-coating method.
Separation was performed using the high performance liquid chromatography (HPLC) system (La Chrom ELITE, HITACHI, Tokyo, Japan) with a TOSOH TSK gel ODS-80TM (Tosoh, Tokyo, Japan) to measure the amount of loaded NBA in micelles. For eluent, water/methanol (3/7 vol%) was used at 15 °C with a flow rate of 0.3 mL min−1. A UV wavelength of 254 nm was selected for NBA detection.
To irradiate UV radiation into the micelle suspension, a super high-pressure Hg lamp (250 SXUI2150HQ, Ushio, Tokyo, Japan) was used (365 nm, 19.90 mW cm−2) with a heat-absorbing filter (HA-50 Hoya, Tokyo, Japan).
Results and discussion
Preparation of P(NIPAAm-co-CIPAAm)s by RAFT polymerization
First, we carefully selected a chain transfer agent (CTA) because the end group of a polymer strongly affects the solution properties. Duan et al., for example, demonstrated that the lower critical solution temperature (LCST) of P(N-isopropylacrylamide) (PNIPAAm) with a hydrophobic pyrene end group was decreased to 29.3, 26.3, 24.8, and 21.7 °C as the molecular weight Mn,MS decreased to 5000, 4200, 3400, and 3000 g mol−1. The LCST was increased again from 21.7 °C to 26 °C when the pyrene end groups were capped with hydrophilic β-cyclodextrin.19 Nakayama et al. also reported the effect of phenyl, hydroxyl, and pH responsive sulfadimethoxine (SD) end groups on the temperature-responsive properties of the amphiphilic block copolymers of P(benzyl methacrylate)-b-P(NIPAAm-co-N,N-dimethylacrylamide (DMAAm)).20,21 In RAFT polymerization, chain transfer agents (CTAs) have a structure of Z–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S)–S–R, where the Z and R groups are located at ends of the polymer chain. In this study, therefore, nonionic cumyl dithiobenzoate (CDB) was selected as the CTA to avoid the effects of the end group on the solution properties of pH response properties. First, we prepared a temperature and pH responsive P(NIPAAm-co-2-carboxyisopropylacrylamide (CIPAAm)) copolymer that forms the shell part of the micelle. The anionic CIPAAm monomer has a similar structure to NIPAAm and the P(NIPAAm-co-CIPAAm) copolymers are known to show sensitive temperature responsive properties even at high CIPAAm contents for their approximate reactive-rates compared to that of typical acrylate type monomers such as acrylic acid.22,23 P(NIPAAm-co-CIPAAm)s were prepared by RAFT polymerization with different CIPAAm contents: 4, 10, and 20 mol% in feed (Scheme S1(A)† and Table 1). The calculated CIPAAm contents of the P(NIPAAm-co-CIPAAm) from 1H NMR were 5, 8, and 20 mol% (Fig. S1†), and the molecular weights were 11000, 11100, and 9300 g mol−1 with relatively narrow Mw/Mn (1.30–1.35). The number of monomer units in the copolymers were calculated from the CIPAAm contents and the molecular weights, and were abbreviated as P(NIPAAm88-co-CIPAAm5) (CIPAAm: 5 mol%), P(NIPAAm85-co-CIPAAm7) (CIPAAm: 8 mol%), and P(NIPAAm59-co-CIPAAm15) (CIPAAm: 20 mol%). Transmittance changes of the P(NIPAAm-co-CIPAAm)s in response to a change in the solution temperature were measured at pH 2 and 12 (Fig. S2†). At pH 2, the LCSTs of P(NIPAAm-co-CIPAAm)s were at 29.4, 29.1, and 25.8 °C, with increasing CIPAAm contents of 5, 8, and 20 mol%, respectively, for the protonated CIPAAm units. On the other hand, at pH 12, all P(NIPAAm-co-CIPAAm) copolymers showed no LCST between 10 and 80 °C because of the electrostatic repulsion due to the deprotonated CIPAAm units. These results suggest that P(NIPAAm-co-CIPAAm)s show both temperature and pH responsive properties clearly. The functionalities are easily controlled via the CIPAAm content and the solution pH. Next, a P(NIPAAm-co-BMAAm) block was polymerized from the chain end of the P(NIPAAm-co-CIPAAm) macro-CTA by RAFT polymerization.
S)–S–R, where the Z and R groups are located at ends of the polymer chain. In this study, therefore, nonionic cumyl dithiobenzoate (CDB) was selected as the CTA to avoid the effects of the end group on the solution properties of pH response properties. First, we prepared a temperature and pH responsive P(NIPAAm-co-2-carboxyisopropylacrylamide (CIPAAm)) copolymer that forms the shell part of the micelle. The anionic CIPAAm monomer has a similar structure to NIPAAm and the P(NIPAAm-co-CIPAAm) copolymers are known to show sensitive temperature responsive properties even at high CIPAAm contents for their approximate reactive-rates compared to that of typical acrylate type monomers such as acrylic acid.22,23 P(NIPAAm-co-CIPAAm)s were prepared by RAFT polymerization with different CIPAAm contents: 4, 10, and 20 mol% in feed (Scheme S1(A)† and Table 1). The calculated CIPAAm contents of the P(NIPAAm-co-CIPAAm) from 1H NMR were 5, 8, and 20 mol% (Fig. S1†), and the molecular weights were 11000, 11100, and 9300 g mol−1 with relatively narrow Mw/Mn (1.30–1.35). The number of monomer units in the copolymers were calculated from the CIPAAm contents and the molecular weights, and were abbreviated as P(NIPAAm88-co-CIPAAm5) (CIPAAm: 5 mol%), P(NIPAAm85-co-CIPAAm7) (CIPAAm: 8 mol%), and P(NIPAAm59-co-CIPAAm15) (CIPAAm: 20 mol%). Transmittance changes of the P(NIPAAm-co-CIPAAm)s in response to a change in the solution temperature were measured at pH 2 and 12 (Fig. S2†). At pH 2, the LCSTs of P(NIPAAm-co-CIPAAm)s were at 29.4, 29.1, and 25.8 °C, with increasing CIPAAm contents of 5, 8, and 20 mol%, respectively, for the protonated CIPAAm units. On the other hand, at pH 12, all P(NIPAAm-co-CIPAAm) copolymers showed no LCST between 10 and 80 °C because of the electrostatic repulsion due to the deprotonated CIPAAm units. These results suggest that P(NIPAAm-co-CIPAAm)s show both temperature and pH responsive properties clearly. The functionalities are easily controlled via the CIPAAm content and the solution pH. Next, a P(NIPAAm-co-BMAAm) block was polymerized from the chain end of the P(NIPAAm-co-CIPAAm) macro-CTA by RAFT polymerization.
| Monomer unitsa | CIPAAm content in feed (mol%) | CIPAAm content in copolymerb (mol%) | BMAAm content in feed (mol%) | BMAAm content in P(NIPAAm-co-BMAAm) blockb (mol%) |
M
nc (g mol−1) |
M
w/Mnc (−) |
|---|---|---|---|---|---|---|
| a The monomer units were calculated by GPC and 1H NMR. b Determined by 1H NMR. c Determined by GPC using 10 mM LiBr DMF. | ||||||
| P(NIPAAm88-co-CIPAAm5) | 4 | 5 | — | — | 11000 |
1.33 |
| P(NIPAAm85-co-CIPAAm7) | 10 | 8 | — | — | 11100 |
1.35 |
| P(NIPAAm59-co-CIPAAm15) | 20 | 20 | — | — | 9300 | 1.30 |
| P(NIPAAm88-co-CIPAAm5)-b-P (NIPAAm58-co-BMAAm7) | 4 | 5 | 17 | 11 | 18700 |
1.56 |
| P(NIPAAm59-co-CIPAAm15)-b-P (NIPAAm43-co-BMAAm10) | 20 | 20 | 17 | 18 | 15700 |
1.65 |
Preparation of dual-temperature and pH responsive P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s
When designing a P(NIPAAm-co-CIPAAm)-b-PNIPAAm block copolymer to show dual-temperature and pH responsive properties, it may be difficult to separate two LCSTs clearly because the PNIPAAm homopolymer usually has its LCST around 32 °C, which is too close to the LCST of the polymerized P(NIPAAm-co-CIPAAm)s at pH 2 (25.8–29.4 °C in Fig. S2†). To decrease the LCST of the PNIPAAm block, therefore, N-(isobutoxymethyl)acrylamide (BMAAm), a more hydrophobic acrylamide type monomer compared to NIPAAm was selected for the lower LCST block. In our previous study, LCSTs of the P(NIPAAm-co-BMAAm)s were decreased from 33 to 18 °C with increasing the BMAAm content between 0 and 17 mol%.11c In this study, we selected 17 mol% BMAAm content in the P(NIPAAm-co-BMAAm)s to obtain the LCST around 18 °C. Characterization of the synthesized P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm) block copolymers is also shown in Table 1. The P(NIPAAm-co-BMAAm) block was polymerized from the macro-CTA of P(NIPAAm-co-CIPAAm) by RAFT polymerization (Scheme S1(B)†). The BMAAm contents in the P(NIPAAm-co-BMAAm) block were measured by 1H NMR and were 11 and 18 mol%, respectively. The molecular weights of P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm) were 18700 and 15700 g mol−1 (Mw/Mn = 1.56 and 1.65), respectively, which were higher than that of the original P(NIPAAm-co-CIPAAm)s. Using these results, the number of monomer units in the block copolymers were calculated as shown below, i.e. P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) and P(NIPAAm59-co-CIPAAm15)-b-P(NIPAAm43-co-BMAAm10).
Controlled local micelle aggregation by UV irradiation
When a bulk gel is immersed in acid solution, the first contact between the gel and hydrogen ions (H+) is the gel surface. If the bulk gels can change their properties to hydrophobic under acidic conditions, they create dehydrated skin-layers on the surface which acts as a barrier to the rapid pH response.16 To solve this problem, we have focused on a supply of protons from inside of bulk gels. O-Nitrobenzaldehyde (NBA) is a light responsive proton generator that can change the solution pH to acidic conditions by UV irradiation (Fig. 2(A)). Temperature and pH responsive bulk gels incorporating NBA have successfully achieved the rapid/efficient volume change by UV irradiation.16a–c The UV irradiation method facilitates remote-controlled drug release from the gels. In fact, many types of irradiations are applied in medical fields, such as UV, magnetic field, radioactivity treatment, and infrared irradiation.24 In this study, NBA was loaded into the micelle of P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) using a simple mixture method. The NBA and block copolymers were completely dissolved in distilled water at 10 °C and the mixture solution was warmed to 25 °C in order to capsulate the NBA into micelles. The calculated loading amount of NBA was 1.5 wt%. This amount of NBA causes a pH change from 7.0 to 3.7 if all NBA release the proton via UV irradiation. Fig. 2(B) shows the diameter of the NBA loaded micelles at 25 °C. The main diameter was 180 ± 35 nm. UV light (19.90 mW cm−2) was irradiated for 900 seconds on the NBA loaded micelle suspension. The absorbance and the solution pH were recorded every 15 seconds, as shown in Fig. 2(C). With UV irradiation, the absorbance increased. On the other hand, the solution pH decreased from 5.9 to 3.9 (Fig. S3†). These results suggest that the P(NIPAAm88-co-CIPAAm5) shell of the micelles was dehydrated by the acidic conditions triggered via UV irradiation. When the NBA was dissolved in the micelle suspension (no loading), the absorbance increase was also observed via UV irradiation (Fig. S4†). We also tried local micelle aggregation via UV irradiation (Fig. 2(D)†). A mixture solution of NBA (6 mg) and the micelle (P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7), 40 mg) was prepared in 5 mL of distilled water at 25 °C. The solution had a light blue color due to micelle formation (i.e. Tyndall phenomenon). The micelle suspension was irradiated for 3 min at 25 °C, and a white spot was locally observed at the area of irradiation. After 5 min of irradiation, the white spot gradually expanded by proton diffusion. These results suggested that remote-controlled local micelle aggregation was achieved via UV irradiation. Moreover, the construction of micelle aggregation was completely dissolved in the aqueous solution at 10 °C (10 min). This reversibility is another advantage of this system.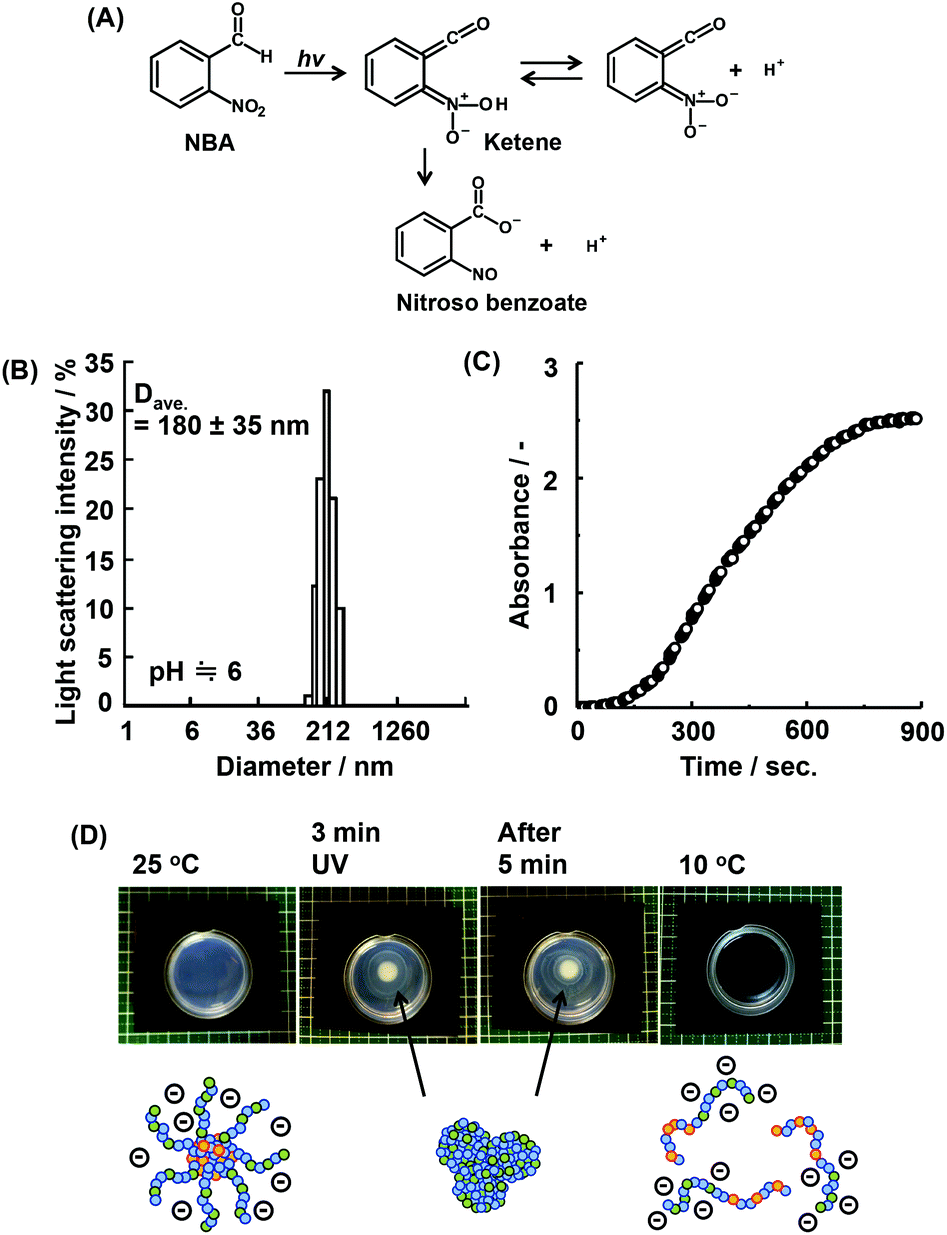 | ||
| Fig. 2 (A) Photochemistry mechanism of o-nitrobenzaldehyde (NBA). (B) Size distribution histogram for the NBA loaded micelle of P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) at 25 °C at pH 6. (C) Absorbance change of the NBA loaded micelle suspension by UV irradiation. (D) Micelle aggregation by local UV irradiation. | ||
Transmittance changes of P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s under different solution conditions
We checked the physicochemical properties of the block copolymers in detail. Fig. 3(A) shows the transmittance changes of P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) at pH 2 and pH 12. At both pH levels, the P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) showed a transmittance decreasing around 17–20 °C due to the LCST of the P(NIPAAm58-co-BMAAm7) block. At pH 2, another LCST caused by dehydration of the P(NIPAAm88-co-CIPAAm5) block was observed at 28 °C. On the other hand, at pH 2, the LCSTs of P(NIPAAm59-co-CIPAAm15)-b-P(NIPAAm43-co-BMAAm10) were 20–22 and 24 °C (Fig. S5†). The LCSTs of polymers are usually increased via decreasing the polymer concentration. The LCSTs of P(NIPAAm-co-CIPAAm) blocks in the block copolymers, however, were significantly lower than the P(NIPAAm-co-CIPAAm) copolymers with the same content of CIPAAm (Fig. S2†) in spite of the fact that the concentration of the P(NIPAAm-co-CIPAAm) block was low compared to the copolymer. It is also known that the LCST is decreased when the polymers are located on a surface, such as substrates, micelles, and particles, due to topically high concentrations. Xu et al. prepared a dual-temperature responsive block copolymer of PNIPAAm-b-P(2-(dimethylamino)ethyl methacrylate) (PDMA, LCST 40–50 °C) from a hyperbranched polyester (HBP) with topically high concentrations. The dehydrations of PNIPAAm and PDMA on the HBP were observed as the diameter decreased and they started at 20 and 35 °C, respectively. The LCSTs were lower than those of their homopolymers.25 Therefore, it is expected that P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s can form the micelle structures at temperatures between two LCSTs. On the other hand, at pH 12, the P(NIPAAm-co-CIPAAm) block showed no LCST because of electrostatic repulsion of the deprotonated CIPAAm units. The LCST of P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) was investigated in detail at different solution pH levels (pH 3.5, 4.3, 5.0, 6.0, 7.5, and 8.0) (shown in Fig. S6†). At pH 3.5, two LCSTs were clearly observed as well as pH 2 as shown in Fig. 3(A). At pH 4.3 and 5.0, the LCSTs of the P(NIPAAm88-co-CIPAAm5) block gradually increased and broadened due to the electrostatic repulsion of some deprotonated CIPAAm units. Decreasing transmittance caused by dehydration of the P(NIPAAm88-co-CIPAAm5) block was not observed over pH 6.0 due to electrostatic repulsion. These results suggest that block copolymers showed dual-temperature and pH responsive properties. Next, the dependency of assembly/aggregation of the block copolymers on the solution temperature and pH was investigated. As the solution temperature increases between two LCSTs, the block copolymers are predicted to form a micelle consisting of a dehydrated P(NIPAAm-co-BMAAm) core and a hydrated P(NIPAAm-co-CIPAAm) shell.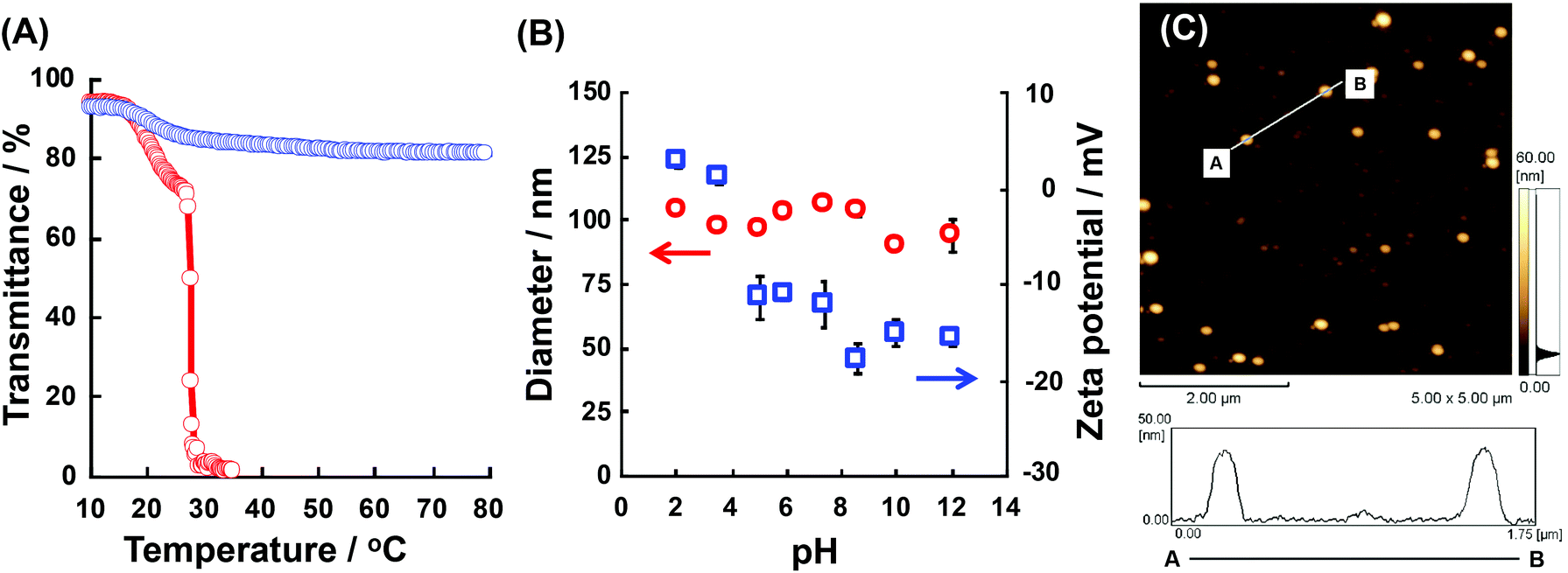 | ||
| Fig. 3 (A) Transmittance change of 0.1% w/v aqueous solution of P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) at pH 2 and 12 as a function of temperature. (B) Diameter and zeta potential of P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) (0.1% w/v) at 25 °C in different pH solutions. (C) AFM image of micelles consisting of P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7). | ||
Assembly/aggregation behavior of P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s at different solution conditions
The P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm) block copolymers are expected to form micelle structures over the LCST of the P(NIPAAm-co-BMAAm) block. Fig. 3(B) shows the diameter and zeta-potential of micelles consisting of P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) at 25 °C in various solution pH levels. The diameters were 94–106 nm, independent of the pH. On the other hand, the zeta potential was strongly affected by the solution pH. The surface charge was shifted to a negative charge with increasing solution pH. The pH responsive surface charge comes from the protonation/deprotonation of CIPAAm units in the P(NIPAAm-co-CIPAAm) block that are always located on the micelle shell. The pH-dependent shift of zeta-potential agreed with the transmittance changes in Fig. S6.† The average diameter of P(NIPAAm59-co-CIPAAm15)-b-P(NIPAAm43-co-BMAAm10) was around 116 nm at 25 °C and pH 12 (Fig. S7†). The spherical nanoparticles were also observed by AFM (Fig. 3(C)). Micelle formation was measured using 1H NMR in D2O at various solution pH levels and temperatures. The solution pH levels were adjusted via DCl or NaOD. All polymer peaks were assigned in DMSO at 25 °C (Fig. 4). In the pH 2 and 12 solutions at 25 °C (over the LCST of the P(NIPAAm-co-BMAAm) block), there were no peaks due to BMAAm units in spite of the fact that the peaks of NIPAAm and CIPAAm units were clearly observed. The disappearance of BMAAm peaks indicate that the dehydrated P(NIPAAm-co-BMAAm) block was located in the micelle core, surrounded by a hydrated P(NIPAAm-co-CIPAAm) shell. When the solution temperature was increased to 40 °C at pH 2, the peaks of P(NIPAAm-co-CIPAAm) also disappeared due to dehydration. At pH 12, on the other hand, the peaks of P(NIPAAm-co-CIPAAm) retained their signals at 40 °C because the electrostatic repulsion prevents aggregation. These results suggest that dual-temperature and pH responsive P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm)s form micelles (with a P(NIPAAm-co-BMAAm) core and a P(NIPAAm-co-CIPAAm) shell) at higher solution temperatures, and that the surface charge caused by CIPAAm units can be controlled by the solution pH (Fig. 5).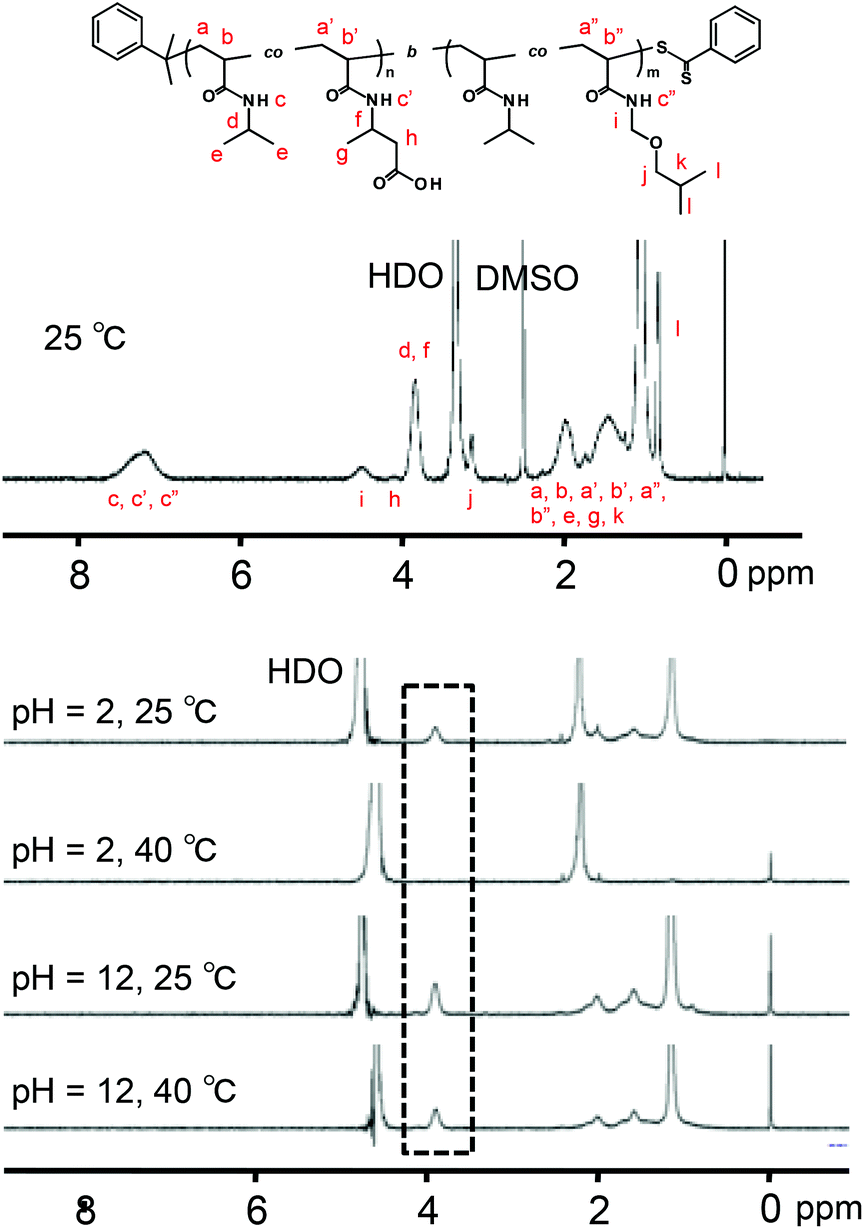 | ||
| Fig. 4 Temperature and pH dependent 1H NMR spectra of P(NIPAAm88-co-CIPAAm5)-b-P(NIPAAm58-co-BMAAm7) (1% w/v). | ||
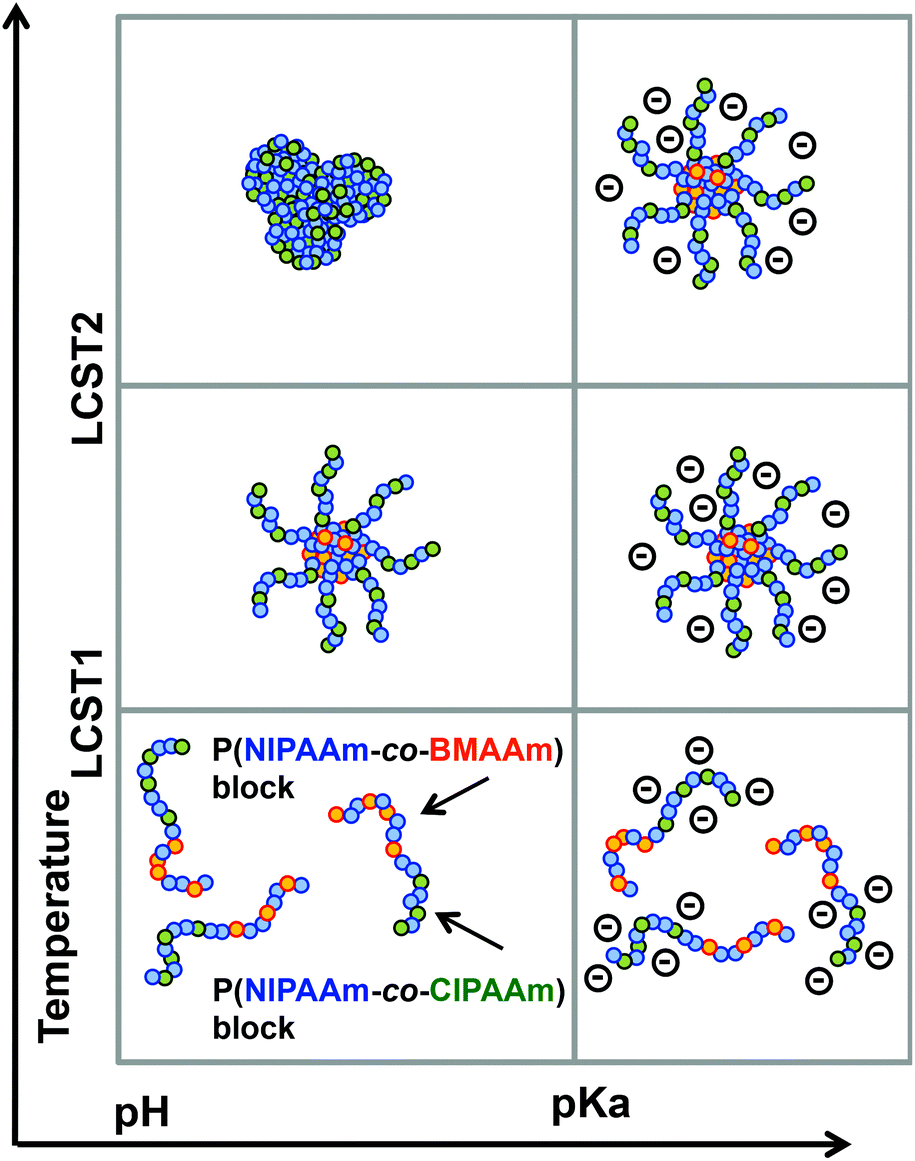 | ||
| Fig. 5 Dual-temperature and pH responsive properties of P(NIPAAm-co-CIPAAm)-b-P(NIPAAm-co-BMAAm). | ||
Conclusions
In summation, the effect of the assembly formation via photo-induced proton generation was investigated using dual-temperature/pH-responsive block copolymers of P(NIPAAm-co-BMAAm)-b-P(NIPAAm-co-CIPAAm)s. The two LCSTs of each block were controlled via the BMAAm and CIPAAm contents, respectively. When the solution temperature was increased between the LCSTs, the block copolymers formed a micelle consisting of a P(NIPAAm-co-BMAAm) core and a P(NIPAAm-co-CIPAAm) shell. The micelle suspension with a photoacid generator of NBA was used as a system of remote-controlled local micelle aggregation. By UV irradiation, a white region consisting of micelle aggregation was locally constructed in the suspension, like an island. The opaque area caused by micelle aggregation was stable, even when UV radiation was removed. Photo-induced proton transfer from the micelle core was also achieved using the NBA-encapsulated micelle. This new system of controlled local micelle aggregation in aqueous solution will be applied in biomaterials for use in live cell imaging and scaffold materials.References
- (a) J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef; (b) M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Macromolecules, 1993, 26, 2987 CrossRef CAS; (c) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS; (d) J. S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7572 CrossRef CAS; (e) J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS; (f) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (g) A. B. Lowe, Polym. Chem., 2010, 1, 17 RSC.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2001, 53, 95 CrossRef.
- K. V. Butsele, R. Jérôme and C. Jérôme, Polymer, 2007, 48, 7431 CrossRef.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- A. S. Hoffman and P. S. Stayton, Prog. Polym. Sci., 2007, 32, 922 CrossRef CAS.
- (a) K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113 CrossRef CAS; (b) K. Kataoka, G. S. Kwon, M. Yokoyama, T. Okano and Y. Sakurai, J. Controlled Release, 1993, 24, 119 CrossRef CAS; (c) G. S. Kwon and T. Okano, Adv. Drug Delivery Rev., 1996, 21, 107 CrossRef CAS; (d) F. Caruso and C. Schüler, Langmuir, 2000, 16, 9595 CrossRef CAS; (e) O. S. Wolfbeis, J. Mater. Chem., 2005, 15, 2657 RSC; (f) Q. Xie, G. Fan, N. Zhao, X. Guo, J. Xu, J. Dong, L. Zhang, Y. Zhang and C. C. Han, Adv. Mater., 2004, 16, 1830 CrossRef CAS.
- (a) D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655 CrossRef CAS PubMed; (b) Y. Li, G. H. Gao and D. S. Lee, Adv. Healthcare Mater., 2013, 3, 388 CrossRef PubMed; (c) J. Akimoto, M. Nakayama and T. Okano, J. Controlled Release, 2014, 193, 2 CrossRef CAS PubMed.
- S. Guragain, B. P. Bastakoti, V. Malgras, K. Nakashima and Y. Yamauchi, Chem. – Eur. J., 2016, 21, 13164 CrossRef PubMed.
- P. Schattling, F. D. Jochum and P. Theato, Polym. Chem., 2014, 5, 25 RSC.
- (a) I. Dimitrov, B. Trzebicka, A. H. E. Müller, A. Dworak and C. B. Tsvetanov, Prog. Polym. Sci., 2007, 32, 1275 CrossRef CAS; (b) S. Strandman and X. X. Zhu, Prog. Polym. Sci., 2015, 42, 154 CrossRef CAS.
- (a) Y. Kotsuchibashi, Y. Kuboshima, K. Yamamoto and T. Aoyagi, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6142 CrossRef CAS; (b) Y. Kotsuchibashi, K. Yamamoto and T. Aoyagi, J. Colloid Interface Sci., 2009, 336, 67 CrossRef CAS PubMed; (c) Y. Kotsuchibashi, M. Ebara, K. Yamamoto and T. Aoyagi, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4393 CrossRef CAS; (d) Y. Kotsuchibashi, M. Ebara, K. Yamamoto and T. Aoyagi, Polym. Chem., 2011, 2, 1362 RSC; (e) Y. Kotsuchibashi and R. Narain, Polym. Chem., 2014, 5, 3061 RSC; (f) Y. Kotsuchibashi, M. Ebara, A. S. Hoffman, R. Narain and T. Aoyagi, Polym. Chem., 2015, 6, 1693 RSC.
- Y. Kotsuchibashi, M. Ebara, N. Idota, R. Narain and T. Aoyagi, Polym. Chem., 2012, 3, 1150 RSC.
- M. T. Savoji, S. Strandman and X. X. Zhu, Langmuir, 2013, 29, 6823 CrossRef CAS PubMed.
- (a) F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468 RSC; (b) S. Chatani, C. J. Kloxin and C. N. Bowman, Polym. Chem., 2014, 5, 2187 RSC; (c) N. Rapoport, Prog. Polym. Sci., 2007, 32, 962 CrossRef CAS.
- (a) M. Irie, J. Am. Chem. Soc., 1983, 105, 2079 CrossRef; (b) J. Choi, N. Hirota and M. Terazima, J. Phys. Chem. A, 2001, 105, 12 CrossRef CAS; (c) S. Abbruzzetti, M. Carcelli, D. Rogolino and C. Viappiani, Photochem. Photobiol. Sci., 2003, 2, 796 RSC; (d) M. V. George and J. C. Scaiano, J. Phys. Chem., 1980, 84, 493 CrossRef.
- (a) P. Techawanitchai, M. Ebara, N. Idota, T. Asoh, A. Kikuchi and T. Aoyagi, Soft Matter, 2012, 8, 2844 RSC; (b) P. Techawanitchai, M. Ebara, N. Idota and T. Aoyagi, Colloids Surf., B, 2012, 99, 53 CrossRef CAS PubMed; (c) P. Techawanitchai, N. Idota, K. Uto, M. Ebara and T. Aoyagi, Sci. Technol. Adv. Mater., 2012, 13, 064202 CrossRef; (d) Y. Kotsuchibashi, M. Ebara, T. Sato, Y. Wang, R. Rajender, D. G. Hall, R. Narain and T. Aoyagi, J. Phys. Chem. B, 2015, 119, 2323 CrossRef CAS PubMed.
- T. Aoyagi, M. Ebara, K. Sakai, Y. Sakurai and T. Okano, J. Biomater. Sci., Polym. Ed., 2000, 11, 101 CrossRef CAS PubMed.
- S. Perrier, C. Barner-Kowollik, J. F. Quinn, P. Vana and T. P. Davis, Macromolecules, 2002, 35, 8300 CrossRef CAS.
- Q. Duan, Y. Miura, A. Narumi, X. Shen, S. Sato, T. Satoh and T. Kakuchi, J. Polym. Sci., Part B: Polym. Chem., 2006, 44, 1117 CrossRef CAS.
- M. Nakayama, Y. Kawahara, J. Akimoto, H. Kanazawa and T. Okano, Colloids Surf., B, 2012, 99, 12 CrossRef CAS PubMed.
- M. Nakayama and T. Okano, Biomacromolecules, 2005, 6, 2320 CrossRef CAS PubMed.
- T. Maeda, M. Takenouchi, K. Yamamoto and T. Aoyagi, Polym. J., 2009, 41, 181 CrossRef CAS.
- T. Maeda, T. Kanda, Y. Yonekura, K. Yamamoto and T. Aoyagi, Biomacromolecules, 2006, 7, 545 CrossRef CAS PubMed.
- D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656 RSC.
- J. Xu, S. Luo, W. Shi and S. Liu, Langmuir, 2006, 22, 989 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The structure of P(NIPAAm-co-CIPAAm). The transmittance change and diameter of dual-temperature and pH responsive block copolymers under various conditions. The absorbance change of micelle suspensions via UV irradiation under various conditions. See DOI: 10.1039/c6py01269h |
| This journal is © The Royal Society of Chemistry 2017 |