DOI:
10.1039/C6PY01254J
(Paper)
Polym. Chem., 2016,
7, 5943-5948
CO2/pH-responsive particles with built-in fluorescence read-out†
Received
20th July 2016
, Accepted 29th August 2016
First published on 6th September 2016
Abstract
A novel fluorescent monomer was synthesized to probe the state of CO2-responsive cross-linked polymeric particles. The fluorescent emission of this aminobromomaleimide-bearing monomer, being sensitive to protic environments, can provide information on the core hydrophilicity of the particles and therefore indicates the swollen state and size of the particles. The particles’ core, synthesized from DEAEMA (N,N-diethylaminoethyl methacrylate), is responsive to CO2 through protonation of the tertiary amines of DEAEMA. The response is reversible and the fluorescence emission can be recovered by simply bubbling nitrogen into the particle solution. Alternate purges of CO2 and N2 into the particles’ solution allow several ON/OFF fluorescence emission cycles and simultaneous particle swelling/shrinking cycles.
Introduction
Stimuli-responsive polymers have received great interest in recent years and have been developed for different applications such as nanotechnology, bio-materials and drug delivery.1–4 To trigger a response and/or change in properties, external stimuli such as temperature, pH, CO2 or light can be utilized.5–9 The recent interest in CO2 as a stimulus for responsive materials is due to the natural abundance of this gas and its bio-compatibility. The usage of CO2-responsive polymers is widely reported to trigger and control self-assembly morphology transitions.10–13 These responsive polymers are usually synthesized with monomers containing tertiary amine, amidine, guanidine or imidazole functional groups.14–16 These functional groups can all be protonated upon a decrease in pH which in turn can be induced by CO2. Tertiary amine-containing monomers such as DMAEMA (N,N-dimethylaminoethyl methacrylate), DEAEMA and DPAEMA (N,N-diisopropylaminoethyl methacrylate) exhibit a pH-responsive behavior, as they can react with strong and weak acids.17,18 These pH-responsive monomers are also responsive to CO2 in water, as CO2 partially dissolves in water to form an equilibrium with carbonic acid, which is a weak acid that can dissociate into HCO3−, CO32− and H+, allowing protonation of the amine monomers. The CO2 response is a reversible process where reversal is carried out by simply bubbling nitrogen or argon in the solution, which displaces carbonic acid and shifts the equilibrium toward the initial pH.19 CO2-responsive polymer assemblies can also be obtained via encapsulation of amine-bearing small molecules into non-responsive systems such as micelles.20 Further examples of CO2-responsive materials include the use of CO2/pH-responsive latexes as Pickering emulsifiers, reported by Morse and co-workers.21 The cross-linked latexes were synthesized from DEAEMA, DVB (divinylbenzene) and PEGMA (poly(ethylene glycol methacrylate)) and showed a reversible diameter increase from 230 nm to 590 nm using HCl/KOH or CO2/N2 gas purges to change the pH. The protonation of the DEAEMA cross-linked core, using HCl or CO2, increases the core hydrophilicity, which induces a swelling effect. The deprotonation of DEAEMA via N2 purging or with KOH decreases the hydrophilicity of the core and results in a reduction of the particle diameter back to its original size. Recently, Chen and co-workers also reported the preparation of CO2-responsive polymeric microgels which are composed of a DEAEMA core covalently stabilized with PEGA (poly(ethylene glycol acrylate)), cross-linked with EGDMA (ethylene glycol dimethacrylate) or BIS (methylene bis(acrylamide)).22 The different microgels obtained showed a reversible size increase upon CO2 and argon bubbling. Depending on the CO2 concentration, these microgels can reversibly swell, or swell then collapse.
Fluorescent dyes are of key interest owing to their potential use in drug delivery systems.23–25 Liang and co-workers presented the advantages of a responsive and fluorescent combination for bio-medical applications with the preparation of polymeric nanoparticles that are fluorescent, pH-responsive and biocompatible for intracellular imaging and drug delivery.26 We previously reported the synthesis of fluorescently labelled proteins and polymers with a dithiomaleimide moiety; a small functional group that does not affect the polymer or protein scaffold.27 This fluorescent functional group was also incorporated into an amphiphilic block copolymer which self-assembled into spherical micelles and can be used in nanomedicine.28 We also recently reported the one-pot synthesis of fluorescent nanogels that were covalently dyed using a dithiomaleimide methacrylate monomer.29,30 It was also demonstrated that these particles do not exhibit self-quenching at high concentration unlike commonly used fluorophores such as phloxine B. Dithiomaleimides have been previously demonstrated to be highly fluorescent when the maleimide unit is conjugated to an alkyl thiol,27,31 although dithiomaleimides in the presence of an excess of thiol can undergo substitution, which may result in loss of fluorescent properties if substituted with an aromatic thiol.32 To counter this substitution effect and thus the loss of fluorescence, we have recently developed a new class of highly emissive fluorophores, the aminobromomaleimides (ABM).33 Their fluorescence properties are environment dependent; in protic solvents a loss of fluorescence can be observed. Herein, we report the first synthesis of fluorescent CO2-responsive polymeric particles by emulsion polymerization. A novel ABM functional fluorescent monomer, present in the particle core, was utilized as a probe of the core hydrophobicity. By simple CO2 bubbling, the particles become swollen and, as a consequence of the increased hydrophilicity of the particles, their fluorescence drastically decreases. This swelling is reversible by purging the solution with nitrogen and ON/OFF cycles of fluorescence are reproducible with successive CO2/N2 purges. PDEAEMA (poly(N,N-diethylaminoethyl methacrylate)) was used as the CO2-responsive core-forming segment, with OEGMA (oligoethylene glycol methacrylate) as the hydrophilic shell-forming block.
Experimental
Materials
Dry solvents were obtained by passing over a column of activated alumina using an Innovative Technologies solvent purification system. DEAEMA was filtered through a plug of alumina prior to use and stored at 4 °C. All other chemicals were purchased from Aldrich, Fluka or Acros and used as received.
Synthetic procedures
Synthesis of the dibromomaleimide methacrylate monomer (DBMMA).
To an oven-dried round-bottom flask under an inert nitrogen atmosphere was added triphenyl phosphine (1.03 g, 1 equiv.) and dry THF (35 mL). The mixture was cooled to −78 °C before the dropwise addition of diisopropyl azodicarboxylate (DIAD) (0.769 mL, 1 equiv.). The mixture was stirred for 5 min before adding 2-hydroxyethyl methacrylate (0.475 mL, 1 equiv.), stirred for a further 5 min before adding 2,2-dimethylpropan-1-ol (0.170 g, 0.5 equiv.), and stirred a further 5 min before adding 2,3-dibromomaleimide (1.00 g, 1 equiv.). The reaction was allowed to warm to room temperature while stirring for 18 h. The solvent was removed in vacuo, and the crude mixture purified by column chromatography on silica gel using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of petroleum ether 40–60 °C and dichloromethane, to give the product as a white solid (1.037 g, 73%). The monomer was characterized by 1H NMR spectroscopy in CDCl3, 13C NMR spectroscopy and mass spectrometry, see ESI.†
:1 mixture of petroleum ether 40–60 °C and dichloromethane, to give the product as a white solid (1.037 g, 73%). The monomer was characterized by 1H NMR spectroscopy in CDCl3, 13C NMR spectroscopy and mass spectrometry, see ESI.†
Synthesis of aminobromomaleimide methacrylate (ABMMA).
DBMMA (1) (1.00 g, 1 equiv.) was dissolved in THF (50 mL). To the solution, sodium carbonate (0.720 g, 2.5 equiv.) was added and stirred. Isopropylamine (0.250 mL, 1.05 equiv.) was added dropwise to the solution, whereby an immediate color change of the solution to yellow and the formation of a white precipitate was observed. Upon complete addition of isopropylamine, the solution was left to stir for 2 h at room temperature. The solvent was removed in vacuo, the residue taken up in CH2Cl2 (150 mL), washed with water (2 × 150 mL) and dried with MgSO4. The organic layer was concentrated in vacuo and the product purified by column chromatography on silica gel using a 10:1 mixture of petroleum ether 40–60 °C and ethyl acetate to yield the product as a yellow-orange crystalline solid (0.88 g, 94%). The product was characterized by 1H NMR spectroscopy in CDCl3, 13C NMR spectroscopy, mass spectrometry and fluorescence spectroscopy, see ESI.†
General procedure for the synthesis of fluorescent particles.
OEGMA (0.11 mmol) was first dissolved in 44 mL of deionised water and then EGDMA (0.126 mmol), and ABMMA (2) (0.145 mmol) were dissolved in DEAEMA (13.5 mmol) and added dropwise to the solution. The mixture, whilst stirred, was degassed with nitrogen for 30 min and further heated at 65 °C for 30 min. The initiator, potassium persulfate (KPS) (0.093 mmol), was dissolved in water (1 mL) and degassed with nitrogen before being added to the reaction mixture. The reaction was stirred at 65 °C for 16 h under a nitrogen atmosphere. The particles were purified by exhaustive dialysis (MWCO 3.5 kDa) against deionised water.
Results and discussion
Synthesis and characterization of the fluorescent monomer
The fluorescent monomer containing the ABM functionality was synthesized in two steps, see Scheme 1. First, 2,3-dibromomaleimide methacrylate (DBMMA, 1) was synthesized by alkylation of 2-hydroxyethyl 2-methylprop-2-enoate with 2,3-dibromomaleimide using a modified Mitsunobu reaction procedure reported by Walker.34 The DBMMA monomer (1) was then functionalized via mono-substitution of the bromine with isopropylamine to obtain an aminobromomaleimide methacrylate (ABMMA, 2), following a procedure similar to that used for the synthesis of a library of aminomaleimides, previously reported by our group.33 The monomer presents excitation maxima at 247 nm and 372 nm corresponding to an emission maximum at 482 nm in 1,4-dioxane.
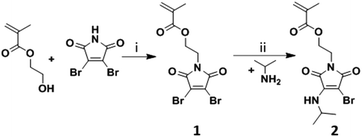 |
| | Scheme 1 Synthesis of the ABMMA monomer (2). Conditions: (i) PPh3, DIAD, 2,2-dimethylpropan-1-ol in THF; (ii) Na2CO3 in THF. | |
Synthesis and characterization of fluorescent PDEAEMA particles
Fluorescent CO2/pH-responsive particles were designed with a tertiary amine-bearing monomer (DEAEMA) and the fluorescent ABMMA monomer (1), the former allows a pH-response while the latter monomer allows a fluorescence read-out of the particles’ state. Particles were synthesized via emulsion polymerization in water and the polymerization was initiated by using potassium persulfate (KPS) as the initiator, see Scheme 2. The emulsion polymerization procedure consists of emulsifying an insoluble monomer phase in water in the presence of a stabilizing amphiphilic compound. The amphiphilic compound in this case is OEGMA, which copolymerizes with DEAEMA to form a covalently linked hydrophilic shell. The hydrophobic core is composed of PDEAEMA cross-linked with 1 wt% of EGDMA and 2 wt% of the fluorescent ABMMA monomer (2). The tertiary amines of the core-forming block allow a CO2-responsive behavior while the presence of the ABM allows a built-in fluorescence read-out. Several batches of particles were synthesized. The influence of the OEGMA molecular weight (from 360 Da to 2000 Da) on the particles' size and responsive character was studied. As a control experiment, particles were also synthesized without the fluorescent monomer in order to confirm it does not affect the particles' response to CO2. The hydrodynamic diameter of the different particles in solution in deionized water was measured by dynamic light scattering (DLS) and it was found that the particles are all in the same size range ca. 200 nm, see Table 1 and Fig. 1b, d for the DLS of particles 4 and 8, see ESI† for particles 3, 5, 6 and 7. The variation of the OEGMA molecular weight or the incorporation of ABMMA did not affect the particles’ morphology and size range. The different OEGMA utilized changed the shell's density as the degree of polymerization of OEGMA was kept the same whilst the monomer increased in size. The size of fluorescent particles with different shells (4 and 8) was also confirmed by transmission electron microscopy (TEM), see Fig. 1a and c. The particles’ spherical morphology was confirmed and the average diameter was measured to be 257 nm for particles 4 and 330 nm for particles 8. The difference between the DLS hydrodynamic diameter and the TEM average diameter occurs as a consequence of a drying effect. Fluorescence spectroscopy was performed on particles containing the fluorescent monomer (4 and 8) in solution in deionized water. The particles both present an emission maximum at 487 nm for an excitation at 375 nm, see Fig. 2a for the emission spectrum of particles 8.
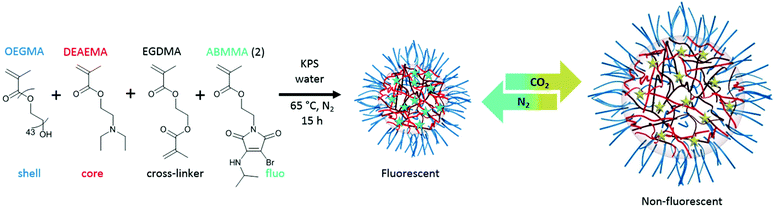 |
| | Scheme 2 Synthesis of particles (8) via emulsion polymerization in water at 65 °C and the reversible particle response upon CO2 and N2 bubbling. | |
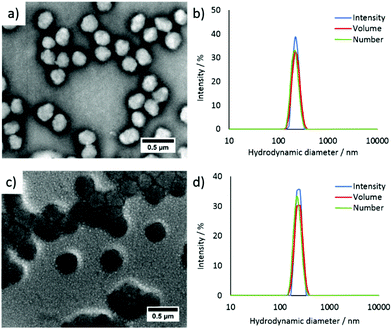 |
| | Fig. 1 (a) TEM image of particles 4, (b) DLS analysis of particles 4, (c) TEM image of particles 8, (d) DLS analysis of particles 8. TEM grids were stained with an aqueous solution of uranyl acetate, see ESI† for more details.35 | |
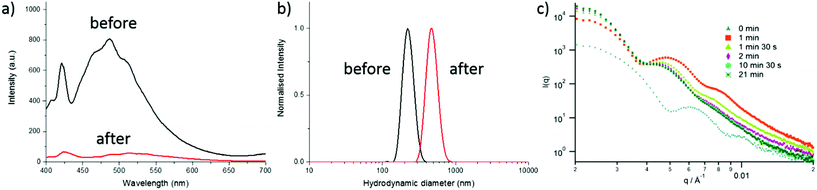 |
| | Fig. 2 (a) Fluorescence emission spectra of particles 8 before and after the first CO2 bubbling, (b) DLS of particles 8 before and after the first CO2 purge, (c) in situ size monitoring of particles 3 by SAXS over 21 min of CO2 bubbling. | |
Table 1 Characteristics of the different PDEAEMA particles synthesized
| No. |
OEGMA MW (Da) |
Fluorescent? |
Hydrodynamic diameter (nm) |
|
3
|
360 |
No |
185 |
|
4
|
360 |
Yes |
220 |
|
5
|
500 |
No |
235 |
|
6
|
950 |
No |
220 |
|
7
|
2000 |
No |
200 |
|
8
|
2000 |
Yes |
230 |
Particle size monitoring
DLS was utilized to monitor changes in the particle diameter in response to bubbling with CO2. When CO2 is bubbled in the particle solution, the amines of PDEAEMA present in the core of the particles become protonated and therefore hydrophilic. Owing to their cross-linked structure, the particles swell and increase in diameter instead of disassembling. A size increase upon CO2 bubbling was observed for all the particles, see Fig. 2b for particles 8 and ESI† for particles 3, 4, 5, 6 and 7. For example, the hydrodynamic diameter of particles 3 increased from 185 nm to 390 nm and particles 7 present a diameter increase from 200 nm to 435 nm. The size increase of the non-fluorescent particles with the small molecular weight OEGMA shell (3) was also monitored by small angle X-ray light scattering (SAXS) with an in situ CO2 purge, see Fig. 2c. The SAXS data shows an increase of the radius of gyration within minutes. Analysis of the SAXS curves over a period of 21 min indicates an increase of particle size as well as an increase of the dispersity. The initial particles could be analyzed as spherical micelles with really low core dispersity with some polymer coils accounting for the outer hydrated shell.36,37 After bubbling CO2 the dispersity of the core slightly increased (as evidenced by the loss of oscillations in the raw SAXS profiles) and a more pronounced core–shell spherical morphology was observed with an increase of core radius. An initial diameter of 180 nm is observed, and an increase of the diameter is observed over time: after 2 min of bubbling, 255 nm and after 21 min, 280 nm.
Fluorescence emission monitoring
As previously demonstrated by our group, the ABM functional group is sensitive to polar protic environments.33 Thus, the ABM-bearing monomer should be able to probe the protonation of its environment. Therefore, the increasing hydrophilicity of the core upon CO2 bubbling should quench the fluorescence emission of the particles. Fluorescence emission of the particles 8 was measured before and after CO2 bubbling, a drastic decrease of the intensity was observed at 487 nm from 810 a.u. to 50 a.u. (see Fig. 2a). The decrease in fluorescence emission upon CO2 bubbling was observed under a UV lamp (λ = 345 nm), see video in ESI.†
Reversibility of the system: CO2/N2 purge cycles
To test the reversibility of the system, the particle solution was repeatedly purged with successive cycles of CO2 and N2 bubbling. Purge cycles were monitored by fluorescence spectroscopy and DLS. The purging time was kept constant for the entire experiment, CO2 was bubbled for 15 min and N2 was bubbled for 30 min. Cycle experiments were performed on fluorescent particles with the denser OEGMA shell (8). The hydrodynamic diameter and the fluorescence emission at 487 nm of the particles (8) were measured after each CO2 or N2 purge. As shown in Fig. 3, the size of the particles alternatively increases and decreases six times. Similarly the emission intensity is reversibly quenched eight times (Fig. 4). Although particles with a less dense shell (3, 4, 5, 6) can swell with CO2, they are unable to collapse upon N2 bubbling. For example, particles 5 exhibit a diameter of 235 nm before bubbling, 430 nm after CO2 bubbling and 1000 nm after N2 purge. This irreversible swelling occurs as a consequence of the density of the shell that cannot counter the effect of core chains that irreversibly penetrate the shell during the first protonation. These hydrophobic chains get trapped in the shell when the particles shrink and their presence promotes inter-particle interactions, which leads to the formation of uncontrolled large aggregates. The formation of aggregates is significantly reduced with a denser shell made of OEGMA with a molecular weight of 2000 Da.
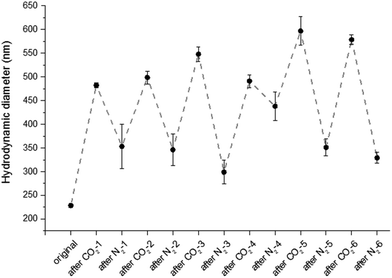 |
| | Fig. 3 Hydrodynamic diameter of the particles (8) in deionized water measured after each gas purge. | |
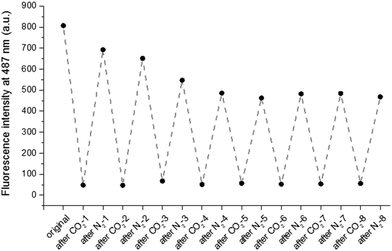 |
| | Fig. 4 Fluorescence emission intensity at 487 nm measured of the particles (8) in deionized water after each gas purge. | |
Conclusions
In conclusion, we have presented the synthesis of fluorescent CO2-responsive cross-linked polymeric particles. The fluorescent dye was incorporated in the core in the form of a novel fluorescent monomer containing an ABM functional group while the CO2-responsive character was introduced by the presence of tertiary amines. These particles swell as the core is made hydrophilic by bubbling CO2 into the solution. This change of local hydrophobic character then induces a decrease in ABM fluorescence intensity. Thus, the fluorescence intensity can be used to probe the particle core hydrophobicity and particle size.
Acknowledgements
The authors thank Dr Dafni Moatsou and Dr Mathew Robin for helpful discussions, BP and the EPSRC for funding.
Notes and references
- M. R. Hill, E. J. MacKrell, C. P. Forsthoefel, S. P. Jensen, M. Chen, G. A. Moore, Z. L. He and B. S. Sumerlin, Biomacromolecules, 2015, 16, 1276–1282 CrossRef CAS PubMed.
- I. Cobo, M. Li, B. S. Sumerlin and S. Perrier, Nat. Mater., 2015, 14, 143–159 CrossRef CAS PubMed.
- P. Theato, B. S. Sumerlin, R. K. O'Reilly and I. I. I. T. H. Epps, Chem. Soc. Rev., 2013, 42, 7055–7056 RSC.
- C.-Y. Chen and H.-L. Wang, Macromol. Rapid Commun., 2014, 35, 1534–1540 CrossRef CAS PubMed.
- M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2013, 42, 7204–7213 RSC.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- X. Xiao, S. He, M. Dan, F. Huo and W. Zhang, Chem. Commun., 2014, 50, 3969–3972 RSC.
- B. W. Liu, H. Zhou, S. T. Zhou, H. J. Zhang, A. C. Feng, C. M. Jian, J. Hu, W. P. Gao and J. Y. Yuan, Macromolecules, 2014, 47, 2938–2946 CrossRef CAS.
- B. A. Abel, M. B. Sims and C. L. McCormick, Macromolecules, 2015, 48, 5487–5495 CrossRef CAS.
- A. Darabi, P. G. Jessop and M. F. Cunningham, Chem. Soc. Rev., 2016, 45, 4391–4436 RSC.
- Q. Yan and Y. Zhao, Chem. Commun., 2014, 50, 11631–11641 RSC.
- S. Lin and P. Theato, Macromol. Rapid Commun., 2013, 34, 1118–1133 CrossRef CAS PubMed.
- E. Girard, T. Tassaing, J.-D. Marty and M. Destarac, Chem. Rev., 2016, 116, 4125–4169 CrossRef CAS PubMed.
- Q. Yan, R. Zhou, C. Fu, H. Zhang, Y. Yin and J. Yuan, Angew. Chem., Int. Ed., 2011, 50, 4923–4927 CrossRef CAS PubMed.
- P. Schattling, I. Pollmann and P. Theato, React. Funct. Polym., 2014, 75, 16–21 CrossRef CAS.
- J. Y. Quek, P. J. Roth, R. A. Evans, T. P. Davis and A. B. Lowe, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 394–404 CrossRef CAS.
- S. Shahalom, T. Tong, S. Emmett and B. R. Saunders, Langmuir, 2006, 22, 8311–8317 CrossRef CAS PubMed.
- J. Pinaud, E. Kowal, M. F. Cunningham and P. Jessop, ACS Macro Lett., 2012, 1, 1103–1107 CrossRef CAS.
- Y. Hoshino, K. Imamura, M. Yue, G. Inoue and Y. Miura, J. Am. Chem. Soc., 2012, 134, 18177–18180 CrossRef CAS PubMed.
- S. Salentinig, P. Jackson and A. Hawley, Macromolecules, 2015, 48, 2283–2289 CrossRef CAS.
- A. J. Morse, S. P. Armes, K. L. Thompson, D. Dupin, L. A. Fielding, P. Mills and R. Swart, Langmuir, 2013, 29, 5466–5475 CrossRef CAS PubMed.
- Y. Chen, T. Zhao, B. Wang, D. Qiu and N. Ma, Langmuir, 2015, 31, 8138–8145 CrossRef CAS PubMed.
- M. P. Robin and R. K. O'Reilly, Polym. Int., 2015, 64, 174–182 CrossRef CAS.
- H. Wang, M. Xu, M. Xiong and J. Cheng, Chem. Commun., 2015, 51, 4807–4810 RSC.
- J.-K. Y. Tan, J. L. Choi, H. Wei, J. G. Schellinger and S. H. Pun, Biomater. Sci., 2015, 3, 112–120 RSC.
- S. Li, K. Hu, W. Cao, Y. Sun, W. Sheng, F. Li, Y. Wu and X.-J. Liang, Nanoscale, 2014, 6, 13701–13709 RSC.
- M. P. Robin, P. Wilson, A. B. Mabire, J. K. Kiviaho, J. E. Raymond, D. M. Haddleton and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 2875–2878 CrossRef CAS PubMed.
- M. P. Robin, A. B. Mabire, J. C. Damborsky, E. S. Thom, U. H. Winzer-Serhan, J. E. Raymond and R. K. O'Reilly, J. Am. Chem. Soc., 2013, 135, 9518–9524 CrossRef CAS PubMed.
- M. P. Robin, J. E. Raymond and R. K. O'Reilly, Mater. Horiz., 2015, 2, 54–59 RSC.
- M. P. Robin and R. K. O'Reilly, Chem. Sci., 2014, 5, 2717–2723 RSC.
- Z.-L. Li, L. Sun, J. Ma, Z. Zeng and H. Jiang, Polymer, 2016, 84, 336–342 CrossRef CAS.
- A. B. Mabire, M. P. Robin, H. Willcock, A. Pitto-Barry, N. Kirby and R. K. O'Reilly, Chem. Commun., 2014, 50, 11492–11495 RSC.
- A. B. Mabire, M. P. Robin, W.-D. Quan, H. Willcock, V. G. Stavros and R. K. O'Reilly, Chem. Commun., 2015, 51, 9733–9736 RSC.
- M. A. Walker, J. Org. Chem., 1995, 60, 5352–5355 CrossRef CAS.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- D. B. Wright, J. P. Patterson, A. Pitto-Barry, A. Lu, N. Kirby, N. C. Gianneschi, C. Chassenieux, O. Colombani and R. K. O'Reilly, Macromolecules, 2015, 48, 6516–6522 CrossRef CAS.
- D. B. Wright, J. P. Patterson, A. Pitto-Barry, P. Cotanda, C. Chassenieux, O. Colombani and R. K. O'Reilly, Polym. Chem., 2015, 6, 2761–2768 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6py01254j |
|
| This journal is © The Royal Society of Chemistry 2016 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Rebecca J.
Williams
a,
Helen
Willcock
a,
Nigel
Kirby
b,
Emma
Chapman
c and
Rachel K.
O'Reilly
*a
a,
Rebecca J.
Williams
a,
Helen
Willcock
a,
Nigel
Kirby
b,
Emma
Chapman
c and
Rachel K.
O'Reilly
*a





